Recent Advances in Mitochondria-Targeted Gene Delivery


Abstract
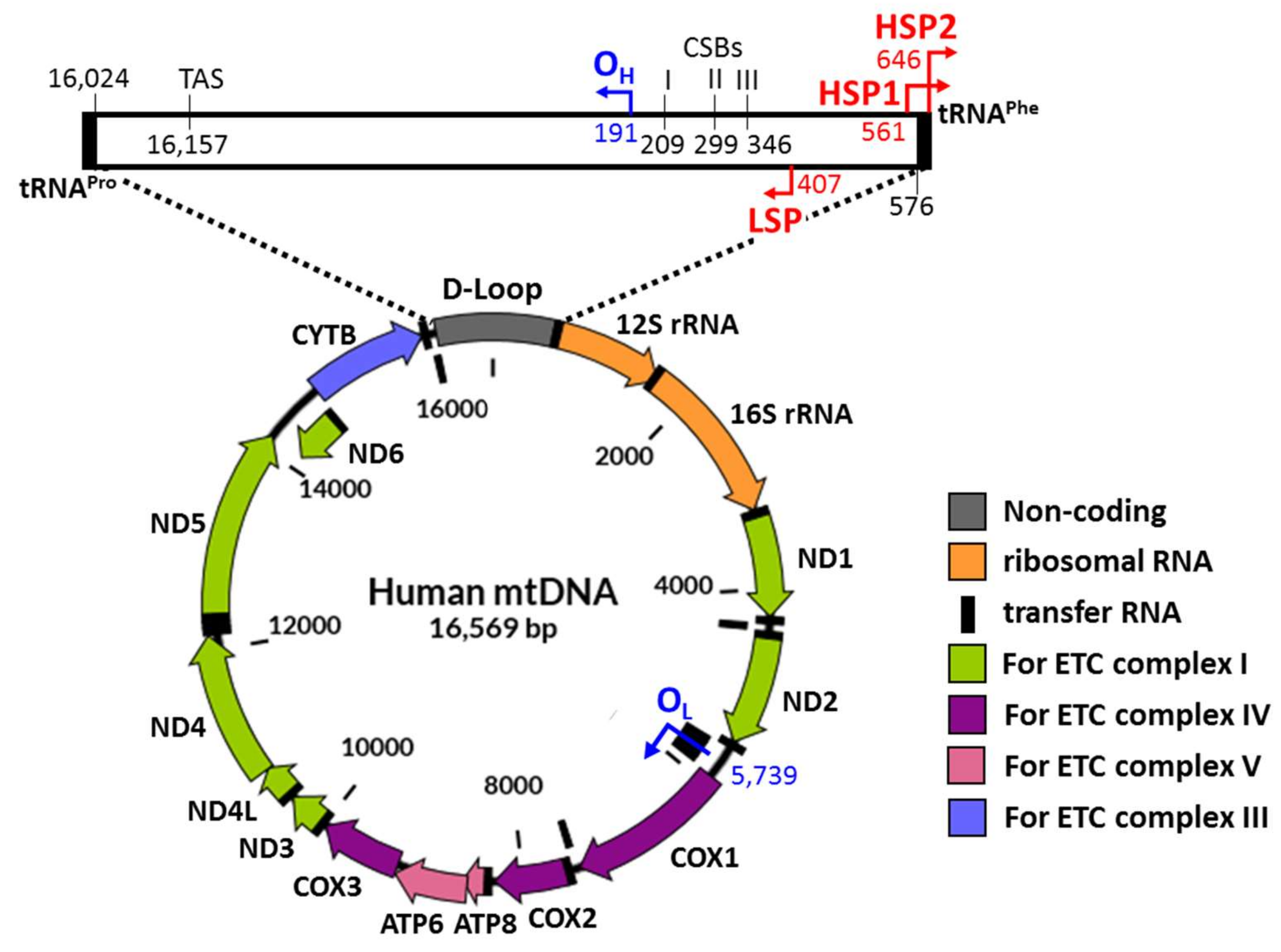
1. Introduction
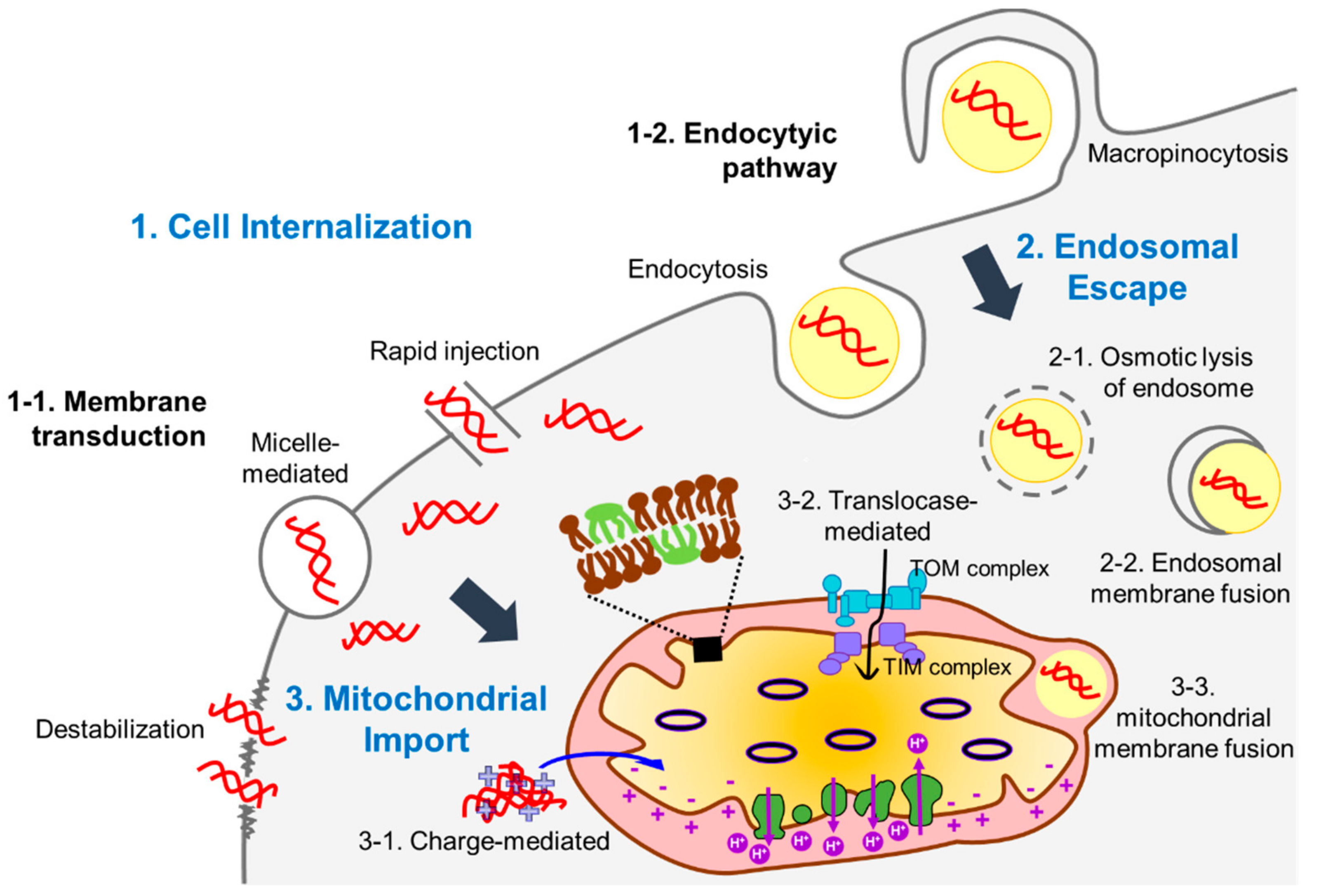
2. Physical Approaches
3. Chemical Approaches
4. Biological Approaches
5. Combinatorial Approaches
6. Cargo DNAs
7. Applications as Disease Therapies
8. Concluding Remarks
Funding
Conflicts of Interest
References
- Robin, E.D.; Wong, R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J. Cell. Physiol. 1988, 136, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Legros, F.; Malka, F.; Frachon, P.; Lombes, A.; Rojo, M. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 2004, 117 Pt 13, 2653–2662. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Yasuzaki, Y.; Yamada, Y.; Kanefuji, T.; Harashima, H. Localization of exogenous DNA to mitochondria in skeletal muscle following hydrodynamic limb vein injection. J. Control. Release 2013, 172, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Yasuzaki, Y.; Yamada, Y.; Fukuda, Y.; Harashima, H. Condensation of plasmid DNA enhances mitochondrial association in skeletal muscle following hydrodynamic limb vein injection. Pharmaceuticals 2014, 7, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Yasuzaki, Y.; Yamada, Y.; Ishikawa, T.; Harashima, H. Validation of Mitochondrial Gene Delivery in Liver and Skeletal Muscle via Hydrodynamic Injection Using an Artificial Mitochondrial Reporter DNA Vector. Mol. Pharm. 2015, 12, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, N.; Fox, T.D. Directed alteration of Saccharomyces cerevisiae mitochondrial DNA by biolistic transformation and homologous recombination. Methods Mol. Biol. 2007, 372, 153–166. [Google Scholar] [PubMed]
- Cardoso, A.M.; Morais, C.M.; Cruz, A.R.; Cardoso, A.L.; Silva, S.G.; do Vale, M.L.; Marques, E.F.; de Lima, M.C.P.; Jurado, A.S. Gemini Surfactants Mediate Efficient Mitochondrial Gene Delivery and Expression. Mol. Pharm. 2015, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Salvado, R.; Sousa, F.; Queiroz, J.; Costa, D. Development of mitochondrial targeting plasmid DNA nanoparticles: Characterization and in vitro studies. Colloid Surf. A 2015, 480, 287–295. [Google Scholar] [CrossRef]
- Santos, J.; Sousa, F.; Queiroz, J.; Costa, D. Rhodamine based plasmid DNA nanoparticles for mitochondrial gene therapy. Colloids Surf. B Biointerfaces 2014, 121, 129–140. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.G.M.; Rammohan, R.; Cheng, S.M.; Torchilin, V.P.; Weissig, V. DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J. Control. Release 2003, 92, 189–197. [Google Scholar] [CrossRef]
- Lyrawati, D.; Trounson, A.; Cram, D. Expression of GFP in the mitochondrial compartment using DQAsome-mediated delivery of an artificial mini-mitochondrial genome. Pharm. Res. 2011, 28, 2848–2862. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Lizano, C.; Torchilin, V.P. Selective DNA release from DQAsome/DNA complexes at mitochondria-like membranes. Drug Deliv. 2000, 7, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Jung, M.K.; Song, S.J.; Green, E.S.; Lee, S.; Park, H.S.; Jeong, S.H.; Han, J.; Mun, J.Y.; Ko, K.S.; et al. Functional nanosome for enhanced mitochondria-targeted gene delivery and expression. Mitochondrion 2017, 37, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Kogure, K.; Akita, H.; Yamada, Y.; Harashima, H. Multifunctional envelope-type nano device (MEND) as a non-viral gene delivery system. Adv. Drug Deliv. Rev. 2008, 60, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Fukuda, Y.; Harashima, H. An analysis of membrane fusion between mitochondrial double membranes and MITO-Porter, mitochondrial fusogenic vesicles. Mitochondrion 2015, 24, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Furukawa, R.; Yasuzaki, Y.; Harashima, H. Dual function MITO-Porter, a nano carrier integrating both efficient cytoplasmic delivery and mitochondrial macromolecule delivery. Mol. Ther. 2011, 19, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Ishikawa, T.; Harashima, H. Validation of the use of an artificial mitochondrial reporter DNA vector containing a Cytomegalovirus promoter for mitochondrial transgene expression. Biomaterials 2017, 136, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.C.; Wang, X.B.; Zhou, M.; Fei, H. A Mitochondria-Targeting Gold-Peptide Nanoassembly for Enhanced Cancer-Cell Killing. Adv. Healthc. Mater. 2013, 2, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Boddapati, S.V.; D’Souza, G.G.; Erdogan, S.; Torchilin, V.P.; Weissig, V. Organelle-targeted nanocarriers: Specific delivery of liposomal ceramide to mitochondria enhances its cytotoxicity in vitro and in vivo. Nano Lett. 2008, 8, 2559–2563. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Dodwadkar, N.S.; Deshpande, P.P.; Torchilin, V.P. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J. Control. Release 2012, 159, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Dodwadkar, N.S.; Piroyan, A.; Torchilin, V.P. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials 2012, 33, 4773–4782. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Taylor, R.W.; Diekert, K.; Lill, R.; Turnbull, D.M.; Lightowlers, R.N. Peptide nucleic acid delivery to human mitochondria. Gene Ther. 1999, 6, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Flierl, A.; Jackson, C.; Cottrell, B.; Murdock, D.; Seibel, P.; Wallace, D.C. Targeted delivery of DNA to the mitochondrial compartment via import sequence-conjugated peptide nucleic acid. Mol. Ther. 2003, 7, 550–557. [Google Scholar] [CrossRef]
- Chuah, J.A.; Matsugami, A.; Hayashi, F.; Numata, K. Self-Assembled Peptide-Based System for Mitochondrial-Targeted Gene Delivery: Functional and Structural Insights. Biomacromolecules 2016, 17, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Ozdemir, S.S.; Chiodo, V.; Boye, S.L.; Boye, S.E.; Hauswirth, W.W.; Lewin, A.S.; et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, E1238–E1247. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ozdemir, S.S.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Chiodo, V.; Boye, S.L.; Hauswirth, W.W.; Lewin, A.S.; Guy, J. Mutant NADH dehydrogenase subunit 4 gene delivery to mitochondria by targeting sequence- modified adeno-associated virus induces visual loss and optic atrophy in mice. Mol. Vis. 2012, 18, 1668–1683. [Google Scholar] [PubMed]
- Suda, T.; Liu, D. Hydrodynamic gene delivery: Its principles and applications. Mol. Ther. 2007, 15, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; He, Y.F.; Shang, Y.Z.; Liu, H.L. Interaction between the Gemini Surfactant (12-6-12) and DNA. Acta Phys. Chim. Sin. 2011, 27, 156–162. [Google Scholar]
- Cardoso, A.M.; Faneca, H.; Almeida, J.A.; Pais, A.A.; Marques, E.F.; de Lima, M.C.; Jurado, A.S. Gemini surfactant dimethylene-1,2-bis(tetradecyldimethylammonium bromide)-based gene vectors: A biophysical approach to transfection efficiency. Biochim. Biophys. Acta 2011, 1808, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Del Gaizo, V.; Payne, R.M. A novel TAT-mitochondrial signal sequence fusion protein is processed, stays in mitochondria, and crosses the placenta. Mol. Ther. 2003, 7, 720–730. [Google Scholar] [CrossRef]
- Futaki, S.; Ohashi, W.; Suzuki, T.; Niwa, M.; Tanaka, S.; Ueda, K.; Harashima, H.; Sugiura, Y. Stearylated arginine-rich peptides: A new class of transfection systems. Bioconjug. Chem. 2001, 12, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Myc, A.; Silpe, J.E.; Sumit, M.; Wong, P.T.; McCarthy, K.; Desai, A.M.; Thomas, T.P.; Kotlyar, A.; Holl, M.M.B.; et al. Dendrimer-Based Multivalent Vancomycin Nanoplatform for Targeting the Drug-Resistant Bacterial Surface. ACS Nano 2013, 7, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Pandita, D.; Rodrigues, J.; Pego, A.P.; Granja, P.L.; Balian, G.; Tomas, H. Receptor-Mediated Gene Delivery Using PAMAM Dendrimers Conjugated with Peptides Recognized by Mesenchymal Stem Cells. Mol. Pharm. 2010, 7, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, M.A.; Orr, B.G.; Banaszak Holl, M.M. Diffusion NMR study of generation-five PAMAM dendrimer materials. J. Phys. Chem. B 2014, 118, 7195–7202. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Shao, N.M.; Zhang, Q.; Cheng, Y.Y. Mitochondrial targeting dendrimer allows efficient and safe gene delivery. J. Mater. Chem. B 2014, 2, 2546–2553. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Meijer, M. The Tom and Tim machine. Curr. Biol. 1997, 7, R100–R103. [Google Scholar] [CrossRef]
- Van der Laan, M.; Meinecke, M.; Dudek, J.; Hutu, D.P.; Lind, M.; Perschil, I.; Guiard, B.; Wagner, R.; Pfanner, N.; Rehling, P. Motor-free mitochondrial presequence translocase drives membrane integration of preproteins. Nat. Cell Biol. 2007, 9, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Esaki, M.; Kanamori, T.; Nishikawa, S.; Endo, T. Two distinct mechanisms drive protein translocation across the mitochondrial outer membrane in the late step of the cytochrome b(2) import pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 11770–11775. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Pichon, C.; Yaouanc, J.J.; Jaffres, P.A. Chemical vectors for gene delivery: A current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br. J. Pharmacol. 2009, 157, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.; Berns, K.I. Gene Therapy Using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.; Liu, F.; Chen, S.; Wang, M.; Cheng, A. Role of capsid proteins in parvoviruses infection. Virol. J. 2015, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Warrington, K.H.; Gorbatyuk, O.S.; Harrison, J.K.; Opie, S.R.; Zolotukhin, S.; Muzyczka, N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J. Virol. 2004, 78, 6595–6609. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Mehta, A.; Wang, G.F.; Hauswirth, W.W.; Chiodo, V.; Boye, S.L.; Guy, J. Next-generation sequencing of mitochondrial targeted AAV transfer of human ND4 in mice. Mol. Vis. 2013, 19, 1482–1491. [Google Scholar] [PubMed]
- Coutinho, E.; Batista, C.; Sousa, F.; Queiroz, J.; Costa, D. Mitochondrial Gene Therapy: Advances in Mitochondrial Gene Cloning, Plasmid Production, and Nanosystems Targeted to Mitochondria. Mol. Pharm. 2017, 14, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Harashima, H. Enhancement in selective mitochondrial association by direct modification of a mitochondrial targeting signal peptide on a liposomal based nanocarrier. Mitochondrion 2013, 13, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Mortensen, L.J.; Ravichandran, S.; Bentley, K.; DeLouise, L.A. Effect of Nanoparticle Surface Coating on Cell Toxicity and Mitochondria Uptake. J. Biomed. Nanotechnol. 2017, 13, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Bibb, M.J.; Van Etten, R.A.; Wright, C.T.; Walberg, M.W.; Clayton, D.A. Sequence and gene organization of mouse mitochondrial DNA. Cell 1981, 26 Pt 2, 167–180. [Google Scholar] [CrossRef]
- Bonawitz, N.D.; Clayton, D.A.; Shadel, G.S. Initiation and beyond: Multiple functions of the human mitochondril transcription machinery. Mol. Cell 2006, 24, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Walberg, M.W.; Clayton, D.A. Sequence and properties of the human KB cell and mouse L cell D-loop regions of mitochondrial DNA. Nucleic Acids Res. 1981, 9, 5411–5421. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Reyes, A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Zollo, O.; Tiranti, V.; Sondheimer, N. Transcriptional requirements of the distal heavy-strand promoter of mtDNA. Proc. Natl. Acad. Sci. USA 2012, 109, 6508–6512. [Google Scholar] [CrossRef] [PubMed]
- Boshart, M.; Weber, F.; Jahn, G.; Dorsch-Hasler, K.; Fleckenstein, B.; Schaffner, W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 1985, 41, 521–530. [Google Scholar] [CrossRef]
- Gammage, P.A.; Gaude, E.; Van Haute, L.; Rebelo-Guiomar, P.; Jackson, C.B.; Rorbach, J.; Pekalski, M.L.; Robinson, A.J.; Charpentier, M.; Concordet, J.P.; et al. Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs. Nucleic Acids Res. 2016, 44, 7804–7816. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Bacman, S.R.; Peralta, S.; Falk, M.J.; Chomyn, A.; Chan, D.C.; Williams, S.L.; Moraes, C.T. MitoTALEN: A General Approach to Reduce Mutant mtDNA Loads and Restore Oxidative Phosphorylation Function in Mitochondrial Diseases. Mol. Ther. 2015, 23, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.V.; Bacman, S.R.; Arguello, T.; Zekonyte, U.; Williams, S.L.; Edgell, D.R.; Moraes, C.T. mitoTev-TALE: A monomeric DNA editing enzyme to reduce mutant mitochondrial DNA levels. EMBO Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Borgeld, H.J.; Zhang, J.; Muramatsu, S.; Gong, J.S.; Yoneda, M.; Maruyama, W.; Naoi, M.; Ibi, T.; Sahashi, K.; et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J. Biomed. Sci. 2002, 9 Pt 1, 534–541. [Google Scholar]
- Yang, Y.; Wu, H.; Kang, X.; Liang, Y.; Lan, T.; Li, T.; Tan, T.; Peng, J.; Zhang, Q.; An, G.; et al. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell 2018, 9, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.P.; Ouvrier, R.A. Peripheral neuropathy associated with mitochondrial disease in children. Dev. Med. Child Neurol. 2012, 54, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Cwerman-Thibault, H.; Augustin, S.; Ellouze, S.; Sahel, J.A.; Corral-Debrinski, M. Gene therapy for mitochondrial diseases: Leber Hereditary Optic Neuropathy as the first candidate for a clinical trial. C. R. Biol. 2014, 337, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Man, P.Y.W.; Turnbull, D.M.; Chinnery, P.F. Leber hereditary optic neuropathy. J. Med. Genet. 2002, 39, 162–169. [Google Scholar] [CrossRef]
- Tzen, C.Y.; Thajeb, P.; Wu, T.Y.; Chen, S.C. Melas with point mutations involving tRNALeu (A3243G) and tRNAGlu(A14693g). Muscle Nerve 2003, 28, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Hajek, P.; Chomyn, A.; Chan, E.; Seo, B.B.; Matsuno-Yagi, A.; Yagi, T.; Attardi, G. Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. J. Biol. Chem. 2001, 276, 38808–38813. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.B.; Kitajima-Ihara, T.; Chan, E.K.; Scheffler, I.E.; Matsuno-Yagi, A.; Yagi, T. Molecular remedy of complex I defects: Rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9167–9171. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Classification | Key Acting Component | Delivery-Target Systems | Strategy | Advantages | Limitations | Ref. |
|---|---|---|---|---|---|---|
| Physical | Hydrodynamic injection | Rat, Mice | Cell penetration by hydrodynamic force | Simplicity | No mitochondria-targeting | [5,6,7] |
| Biolistics | Yeast | Cell penetration by bombardment | Cell type-independent | Potential cell damage, No mitochondria-targeting | [8] | |
| Chemical | Gemini surfactants | HeLa cell | Formation of cationic micelle-like structure with DNA | Works in small dose | Weak specificity to mitochondria | [9] |
| Rhodamine 123 | Normal human dermal fibroblast (NHDF) adult donor cell | Precipitation of DNA using lipophilic molecule with delocalized positive charge | Can be traced due to fluorescence | Expression from transferred DNA has not been confirmed | [10,11] | |
| DQAsomes | BT 20 cell | Transport of DNA by ampiphilic and cationic lipid-based vesicle | High specificity to mitochondria | Low transfection efficiency, Cytotoxicity | [12,13,14] | |
| DQA80s | Primary human dermal fibroblasts, HeLa cell | Lipid incorporation into DQAsomes | Improved transfection efficiency | Delivery of DNA around mitochondria not into the matrix | [15] | |
| MITO-Porter | HeLa cell, Rat | Membrane fusion by lipid-based nano carrier | Easy surface modification | [16,17,18,19] | ||
| R8-MITO-Porter | Rat | Enhancing cellular uptake by functionalization with cationic peptide | High fusogenic activity with mitochondrial outer membrane | Low fusogenic activity with mitochondrial inner membrane, Moderate cytotoxicity | [16,17] | |
| KALA-MITO-Porter | HeLa cell, Mice | Enhancing cellular uptake by functionalization with membrane destabilizing peptide | Improved transfection efficiency | High cytotoxicity | [19,20] | |
| STPP-liposome | 4T1 cell, Mice | Conjugation of stearyl residue to lipophilic and cationic TPP for ampiphilic property | Selective accumulation in mitochondria | Cytotoxicity | [21] | |
| TPP-PEG-PE liposome | HeLa, 4T1 cell, Mice | Substitution of stearyl moiety with biocompatible PEG-PE polymer | Decreased cytotoxicity | Transgene expression not confirmed | [22] | |
| TPP-PAMAM dendrimer | HeLa, MCF-7, 4T1, NIH 3T3 cell | DNA condensation by high positive surface charge, endosomal escape by free tertiary amine groups | Efficient endosomal escape and high serum resistance | Transgene expression not confirmed | [23] | |
| Biological | MTS-PNA | Myoblasts, Fibroblasts, NT 2, IMR 32, HeLa, HepG2, C2C12 cell | MTS-guided localization of DNA hybridized with PNA to mitochondria | High specificity to mitochondria via actions of translocase | Only can transfer short nucleic acids | [24,25] |
| MTS-KH peptide | HEK 293 cell | Mitochondrial localization of MTS-conjugated DNA-binding peptide and exogenous DNA complex | Can transfer large DNA with high specificity to mitochondria | [26] | ||
| MTS-AAV | Neuronal G11778A NT 2 cybrid, HEK 293T cell, Mice | Mitochondrial localization of DNA by inserting MTS into the AAV capsid | Proven effects of transgene expression | Inability to carry large DNA | [27,28] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, Y.-h.; Lim, K.-i. Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules 2018, 23, 2316. https://doi.org/10.3390/molecules23092316
Jang Y-h, Lim K-i. Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules. 2018; 23(9):2316. https://doi.org/10.3390/molecules23092316
Chicago/Turabian StyleJang, Yoon-ha, and Kwang-il Lim. 2018. "Recent Advances in Mitochondria-Targeted Gene Delivery" Molecules 23, no. 9: 2316. https://doi.org/10.3390/molecules23092316
APA StyleJang, Y.-h., & Lim, K.-i. (2018). Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules, 23(9), 2316. https://doi.org/10.3390/molecules23092316