Ball Milling’s Effect on Pine Milled Wood Lignin’s Structure and Molar Mass

 ,
,  ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
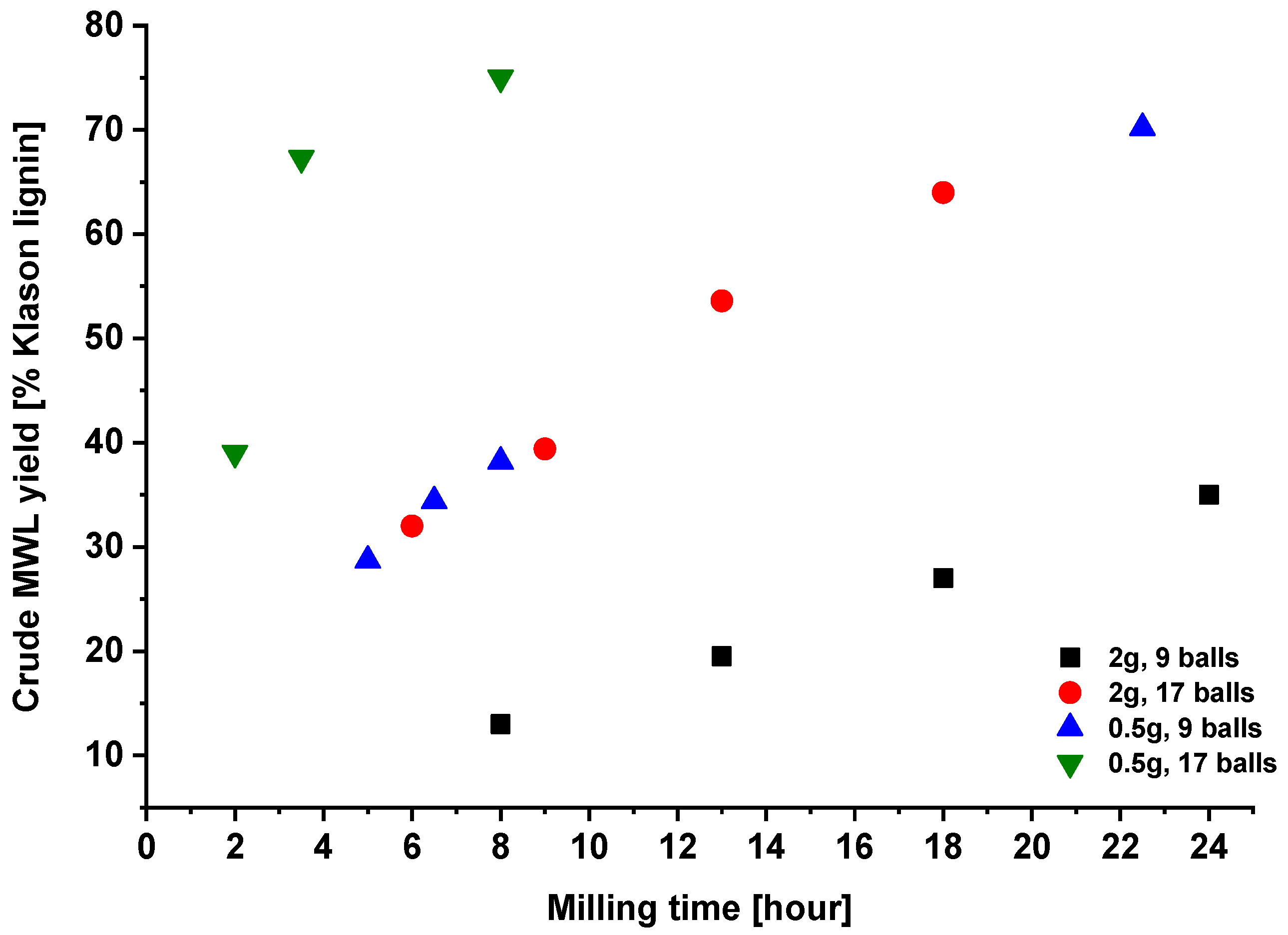
2.1. Effect of Milling Conditions on the Yield of Milled Wood Lignins
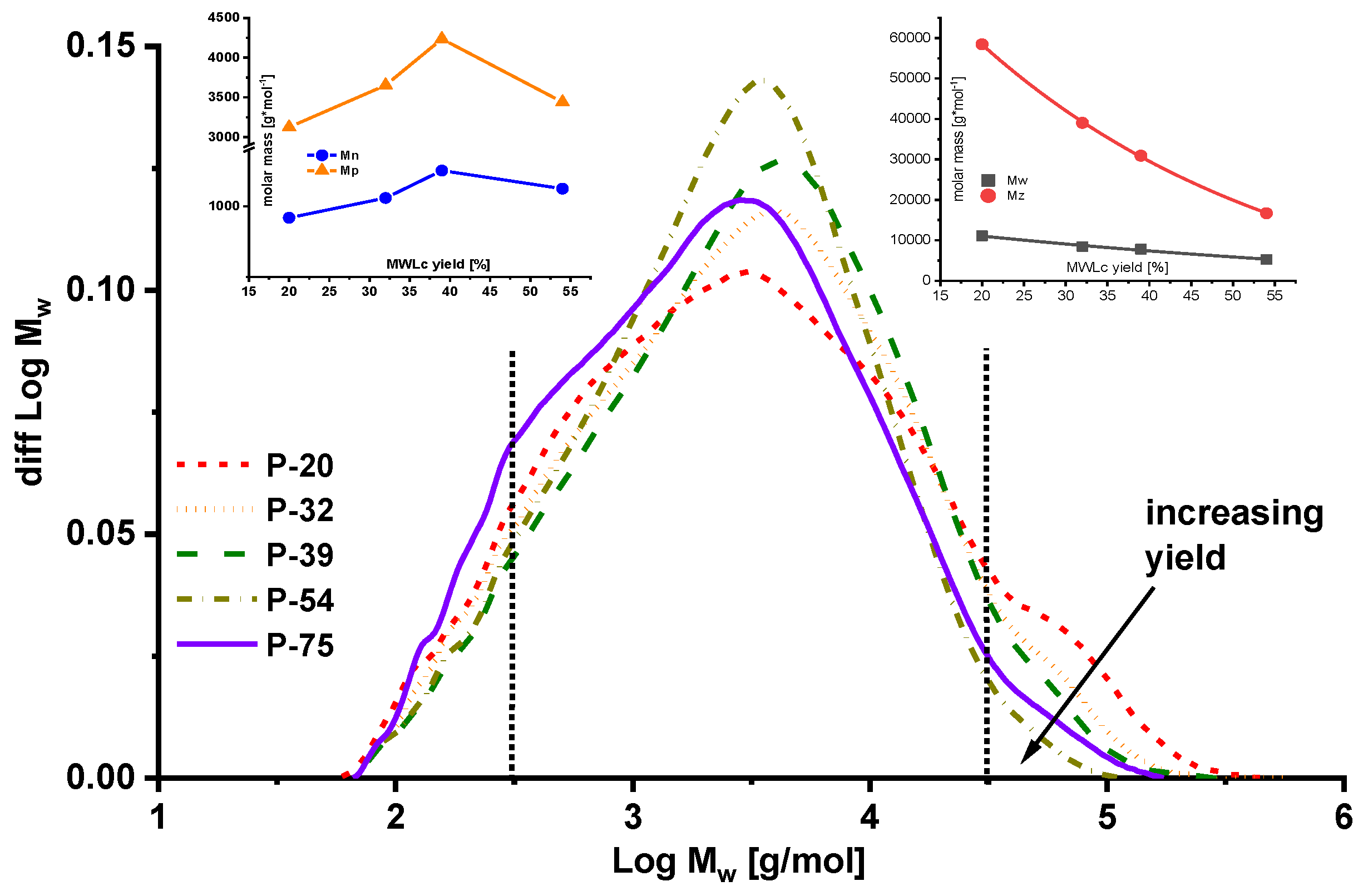
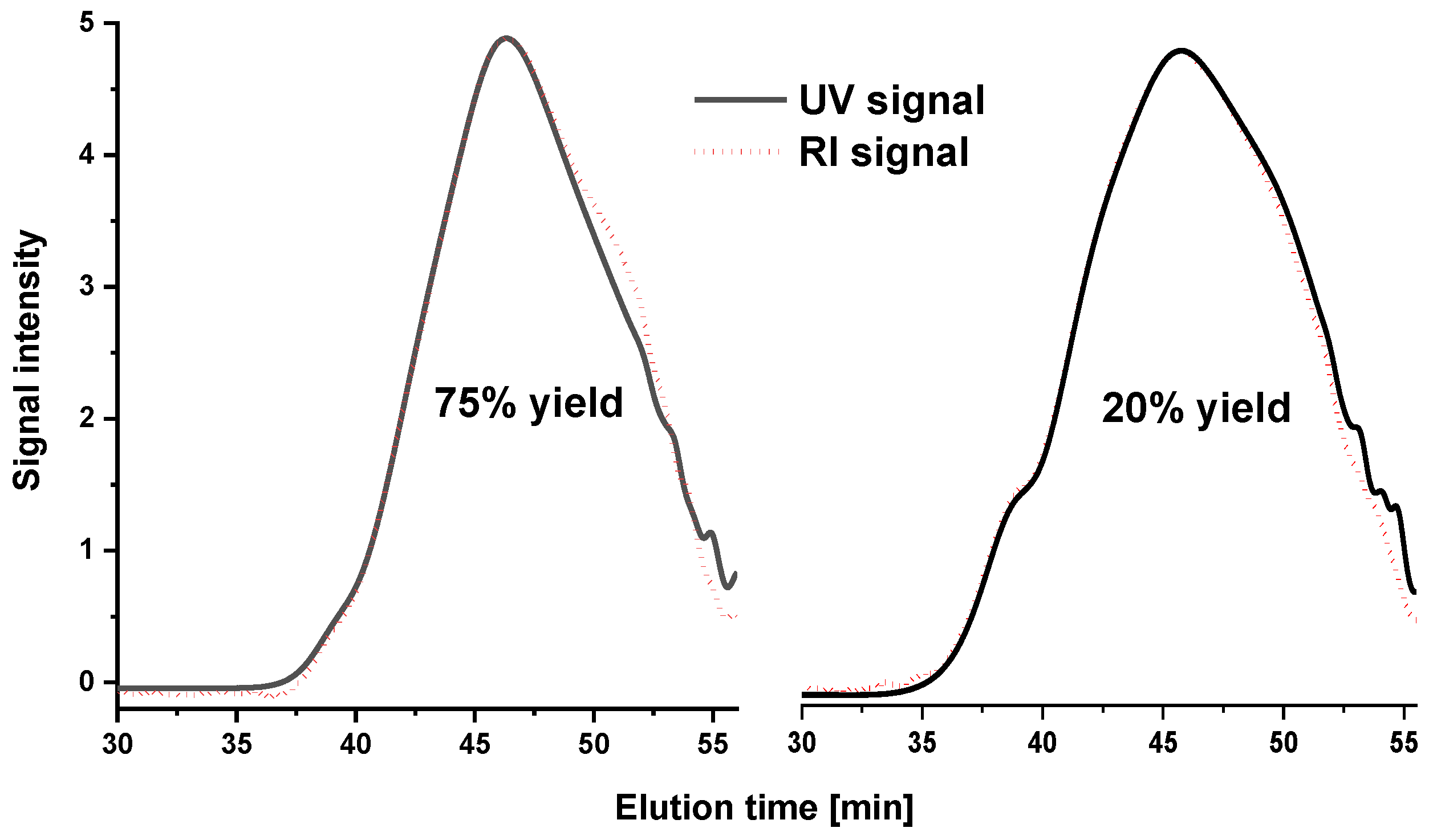
2.2. Molar Mass Distribution
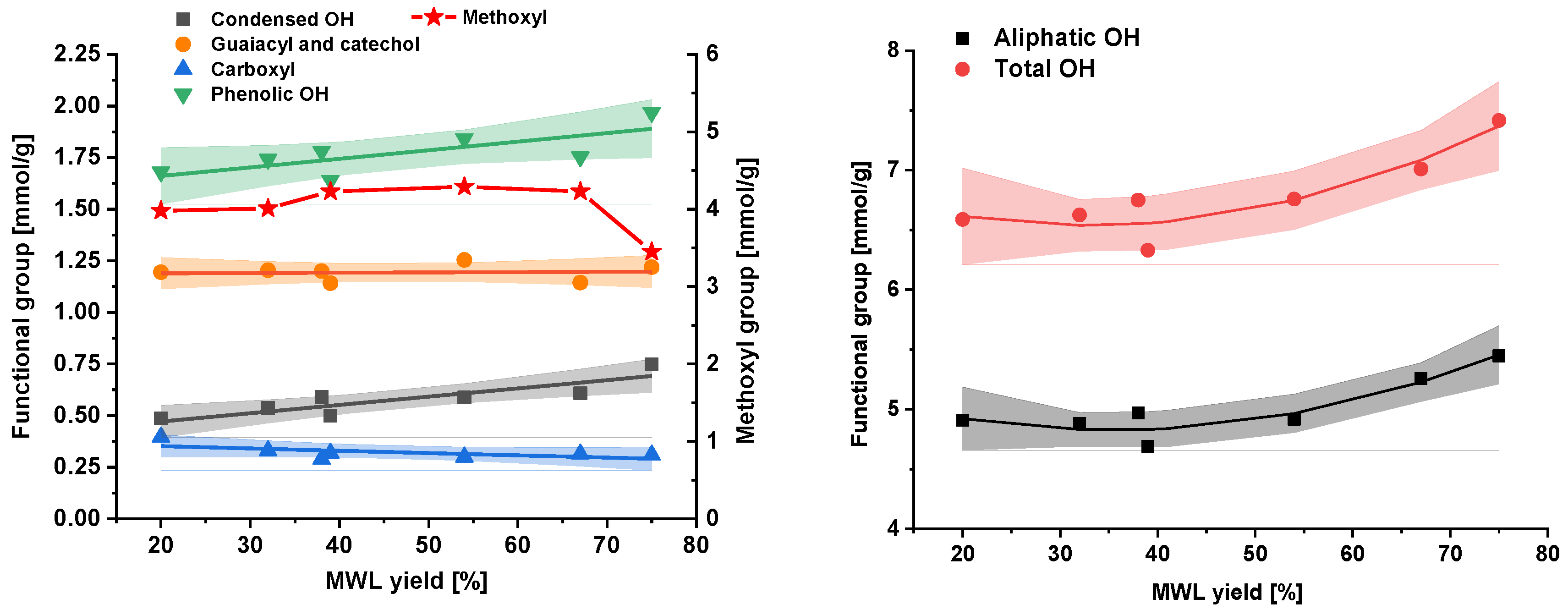
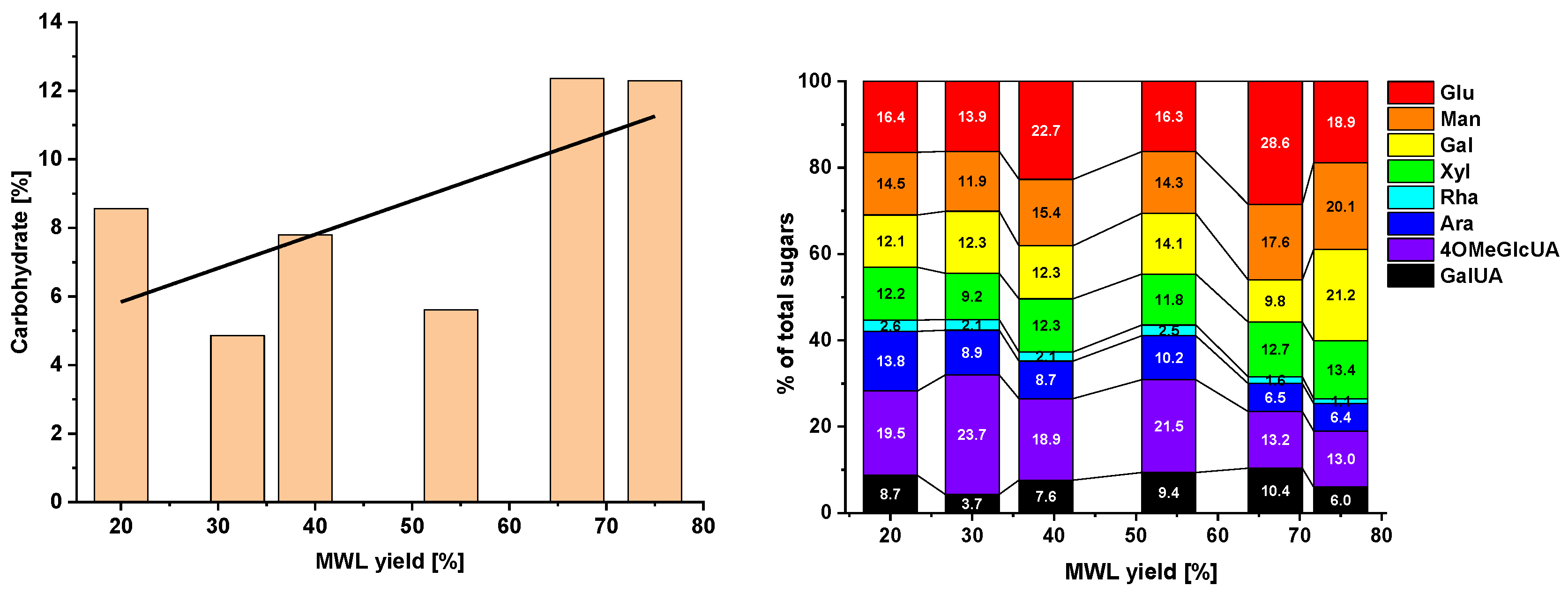
2.3. Functional Groups and Sugar Composition
3. Conclusions
4. Materials and Methods
4.1. Isolation of Milled Wood Lignin Preparations
4.2. Crude Milled Wood Lignin
4.3. Sugar Analysis
4.4. Molar Mass Analysis
4.5. Functional Groups Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Björkman, A. Isolation of Lignin from Finely Divided Wood with Neutral Solvents. Nature 1954, 174, 1057–1058. [Google Scholar] [CrossRef]
- Capanema, E.; Balakshin, M.; Katahira, R.; Chang, H.-M.; Jameel, H. How well do MWL and CEL preparations represent the whole hardwood lignin? J. Wood Chem. Technol. 2015, 35, 17–26. [Google Scholar] [CrossRef]
- Chang, H.-M.; Cowling, E.B.; Brown, W. Comparative Studies on Cellulolytic Enzyme Lignin and Milled Wood Lignin of Sweetgum and Spruce. Holzforsch. Int. J. Biol. Chem. Phys. Technol. Wood 1975, 29, 153–159. [Google Scholar] [CrossRef]
- Guerra, A.; Filpponen, I.; Lucia, L.A.; Saquing, C.; Baumberger, S.; Argyropoulos, D.S. Toward a better understanding of the lignin isolation process from wood. J. Agric. Food Chem. 2006, 54, 5939–5947. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Argyropoulos, D. An improved method for isolating lignin in high yield and purity. J. Pulp Pap. Sci. 2003, 29, 235–240. [Google Scholar]
- Lu, F.; Ralph, J. Non-degradative dissolution and acetylation of ball-milled plant cell walls: High-resolution solution-state NMR. Plant J. 2003, 35, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yokoyama, T.; Chang, H.-M.; Matsumoto, Y. Dissolution of Beech and Spruce Milled Woods in LiCl/DMSO. J. Agric. Food Chem. 2009, 57, 6167–6170. [Google Scholar] [CrossRef] [PubMed]
- Fasching, M.; Schroeder, P.; Wollboldt, R.P.; Weber, H.K.; Sixta, H. A new and facile method for isolation of lignin from wood based on complete wood dissolution. Holzforschung 2008, 62, 15–23. [Google Scholar] [CrossRef]
- Holtman, K.M.; Chang, H.-M.; Kadla, J.F. Solution-State nuclear magnetic resonance study of the similarities between milled wood lignin and cellulolytic enzyme lignin. J. Agric. Food Chem. 2004, 52, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yeh, T.-F.; Chang, H.-M.; Matsumoto, Y.; Kadla, J.F. Elucidation of the structure of cellulolytic enzyme lignin. Holzforschung 2006, 60, 389–397. [Google Scholar] [CrossRef]
- Rencoret, J.; Marques, G.; Gutiérrez, A.; Nieto, L.; Santos, J.; Jiménez-Barbero, J.; Martinez, A.T.; del Río, J.C. HSQC-NMR analysis of lignin in woody (Eucalyptus globulus and Picea abies) and non-woody (Agave sisalana) ball-milled plant materials at the gel state. Holzforschung 2008, 63, 691–698. [Google Scholar]
- Rencoret, J.; Gutiérrez, A.; Nieto, L.; Jiménez-Barbero, J.; Faulds, C.B.; Kim, H.; Ralph, J.; Martínez, Á.T.; del Río, J.C. Lignin composition and structure in young versus adult Eucalyptus globulus plants. Plant Physiol. 2011, 155, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Balakshin, M.; Capanema, E.; Gracz, H.; Chang, H.-M.; Jameel, H. Quantification of lignin-carbohydrate linkages with high-resolution NMR spectroscopy. Planta 2011, 233, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Matsumoto, Y.; Chang, H.-M.; Meshitsuka, G. Quantitative evaluation of milling effects on lignin structure during the isolation process of milled wood lignin. J. Wood Sci. 2005, 51, 89–91. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A.; Chang, H.-M. Recent advances in the isolation and analysis of lignins and lignin-carbohydrate complexes. In Characterization of Lignocellulosics; Hu, T., Ed.; Blackwell: Oxford, UK, 2008; pp. 148–166. [Google Scholar]
- Capanema, E.A.; Balakshin, M.Y.; Heerman, M.L.; Yeh, T.-F.; Yu, Q.; Nelson, C.; McKeand, S.E.; Li, B.; Jameel, H.; Chang, H.; et al. Chemical Properties in CAD-Deficient Pine and Their Effect on Pulping; International Symposium on Wood, Fibre and Pulping Chemistry (ISWFPC); Appita Inc: Carlton, Vic, Auckland, 2005; pp. 173–180. [Google Scholar]
- Fujimoto, A. Effect of Milling on the Structure of Lignin. Ph.D. Thesis, The University of Tokyo, Tokyo, Japan, 2005. [Google Scholar]
- Björkman, A. Studies on finely divided wood. Part 1. Extraction of lignin with neutral solvents. Sven Papperstidn 1956, 59, 477–485. [Google Scholar]
- Sundberg, A.; Sundberg, K.; Lillandt, C.; Holmbom, B. Determination of hemicelluloses and pectins in wood and pulp fibres by acid methanolysis and gas chromatography. NPPRJ 1996, 11, 216–219. [Google Scholar] [CrossRef]
- Sulaeva, I.; Zinovyev, G.; Plankeele, J.-M.; Sumerskii, I.; Rosenau, T.; Potthast, A. Fast track to molar mass distributions of technical lignins. ChemSusChem 2017, 10, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Granata, A.; Argyropoulos, D. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a reagent for the accurate determination of the uncondensed and condensed phenolic moieties in lignins. J. Agric. Food Chem. 1995, 43, 1538–1544. [Google Scholar] [CrossRef]
- Korntner, P.; Sumerskii, I.; Bacher, M.; Rosenau, T.; Potthast, A. Characterization of technical lignins by NMR spectroscopy: Optimization of functional group analysis by 31P NMR spectroscopy. Holzforschung 2015, 69, 807–814. [Google Scholar] [CrossRef]
- Sumerskii, I.; Zweckmair, T.; Hettegger, H.; Zinovyev, G.; Bacher, M.; Rosenau, T.; Potthast, A. A fast track for the accurate determination of methoxyl and ethoxyl groups in lignin. RSC Adv. 2017, 7, 22974–22982. [Google Scholar] [CrossRef]
Sample Availability: Samples of some lignin preparations are available from the authors. |





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zinovyev, G.; Sumerskii, I.; Rosenau, T.; Balakshin, M.; Potthast, A. Ball Milling’s Effect on Pine Milled Wood Lignin’s Structure and Molar Mass. Molecules 2018, 23, 2223. https://doi.org/10.3390/molecules23092223
Zinovyev G, Sumerskii I, Rosenau T, Balakshin M, Potthast A. Ball Milling’s Effect on Pine Milled Wood Lignin’s Structure and Molar Mass. Molecules. 2018; 23(9):2223. https://doi.org/10.3390/molecules23092223
Chicago/Turabian StyleZinovyev, Grigory, Ivan Sumerskii, Thomas Rosenau, Mikhail Balakshin, and Antje Potthast. 2018. "Ball Milling’s Effect on Pine Milled Wood Lignin’s Structure and Molar Mass" Molecules 23, no. 9: 2223. https://doi.org/10.3390/molecules23092223
APA StyleZinovyev, G., Sumerskii, I., Rosenau, T., Balakshin, M., & Potthast, A. (2018). Ball Milling’s Effect on Pine Milled Wood Lignin’s Structure and Molar Mass. Molecules, 23(9), 2223. https://doi.org/10.3390/molecules23092223