Comparative Analyses of Chloroplast Genomes of Cucurbitaceae Species: Lights into Selective Pressures and Phylogenetic Relationships

 , ,
, ,
Abstract
1. Introduction
2. Results
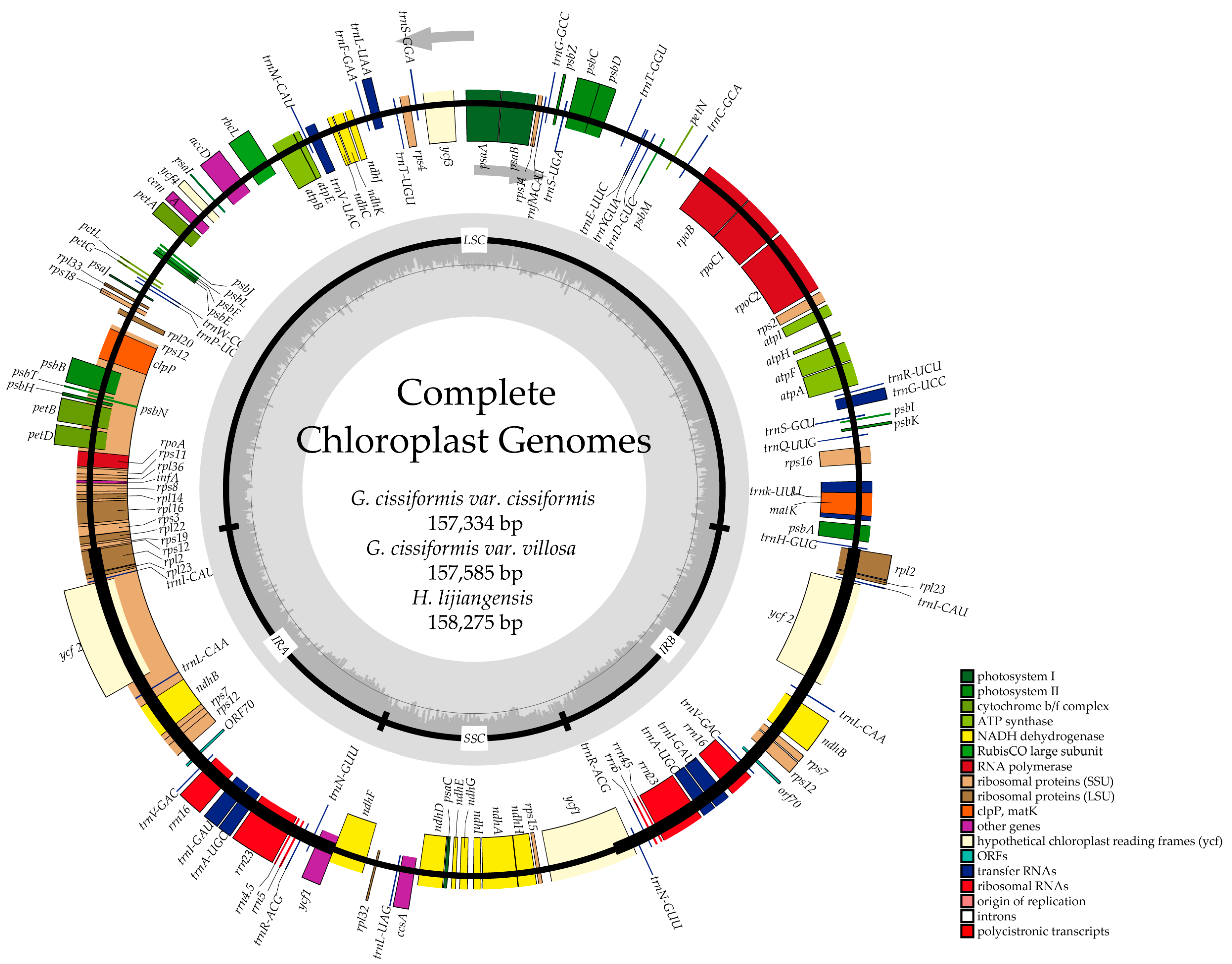
2.1. Genome Features
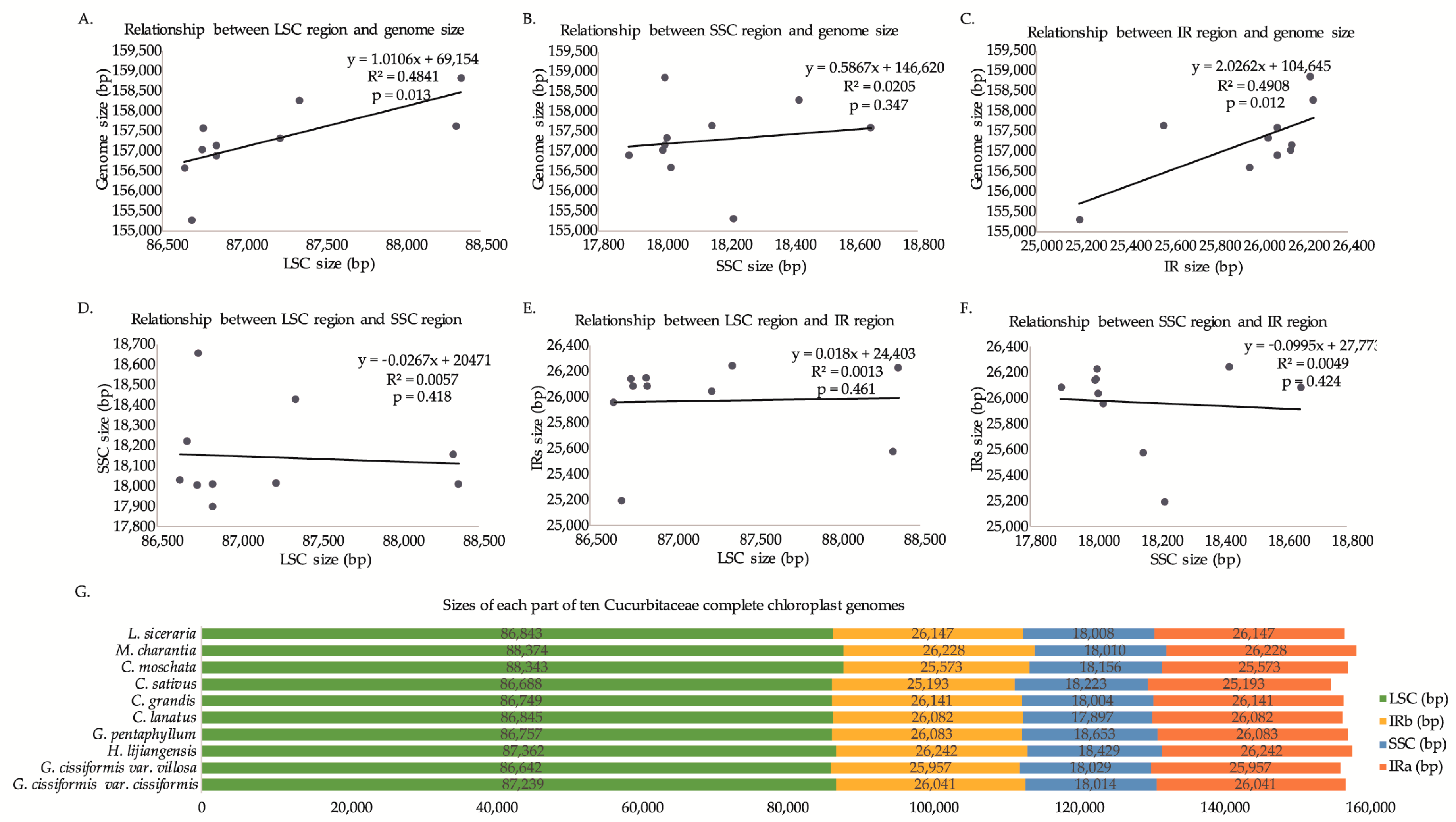
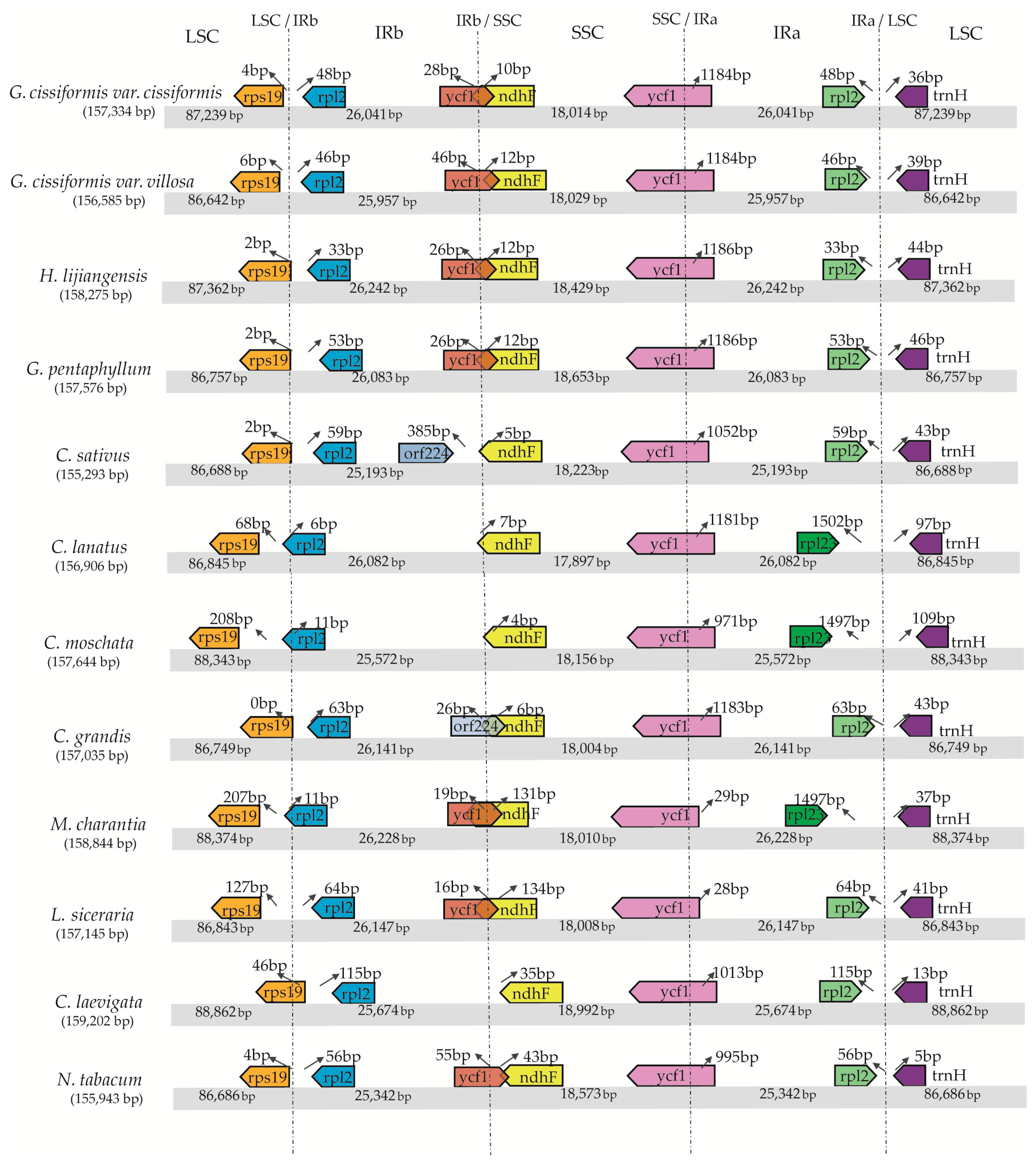
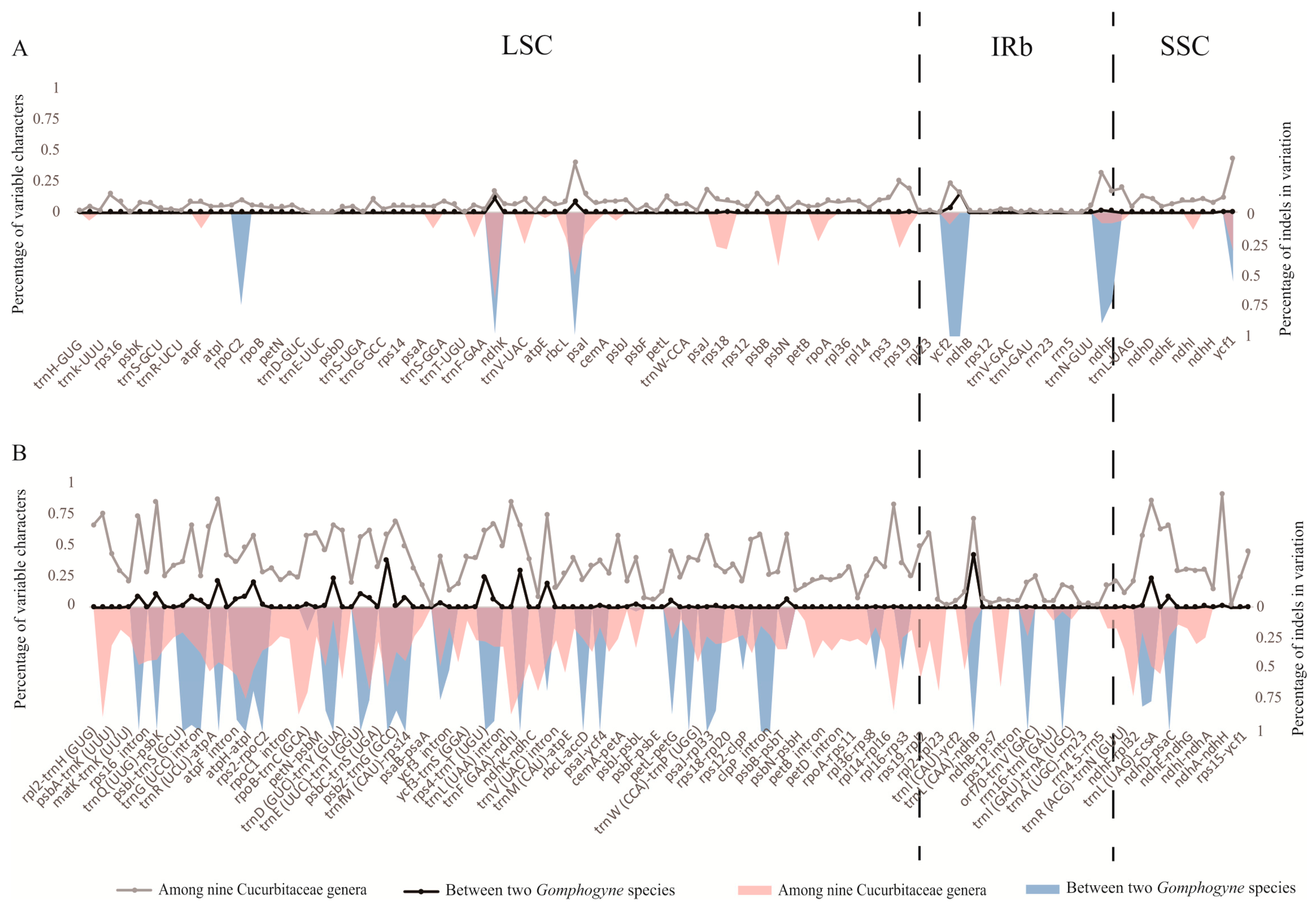
2.2. IR/SC Boundary, Genome Rearrangement and Sequence Divergence
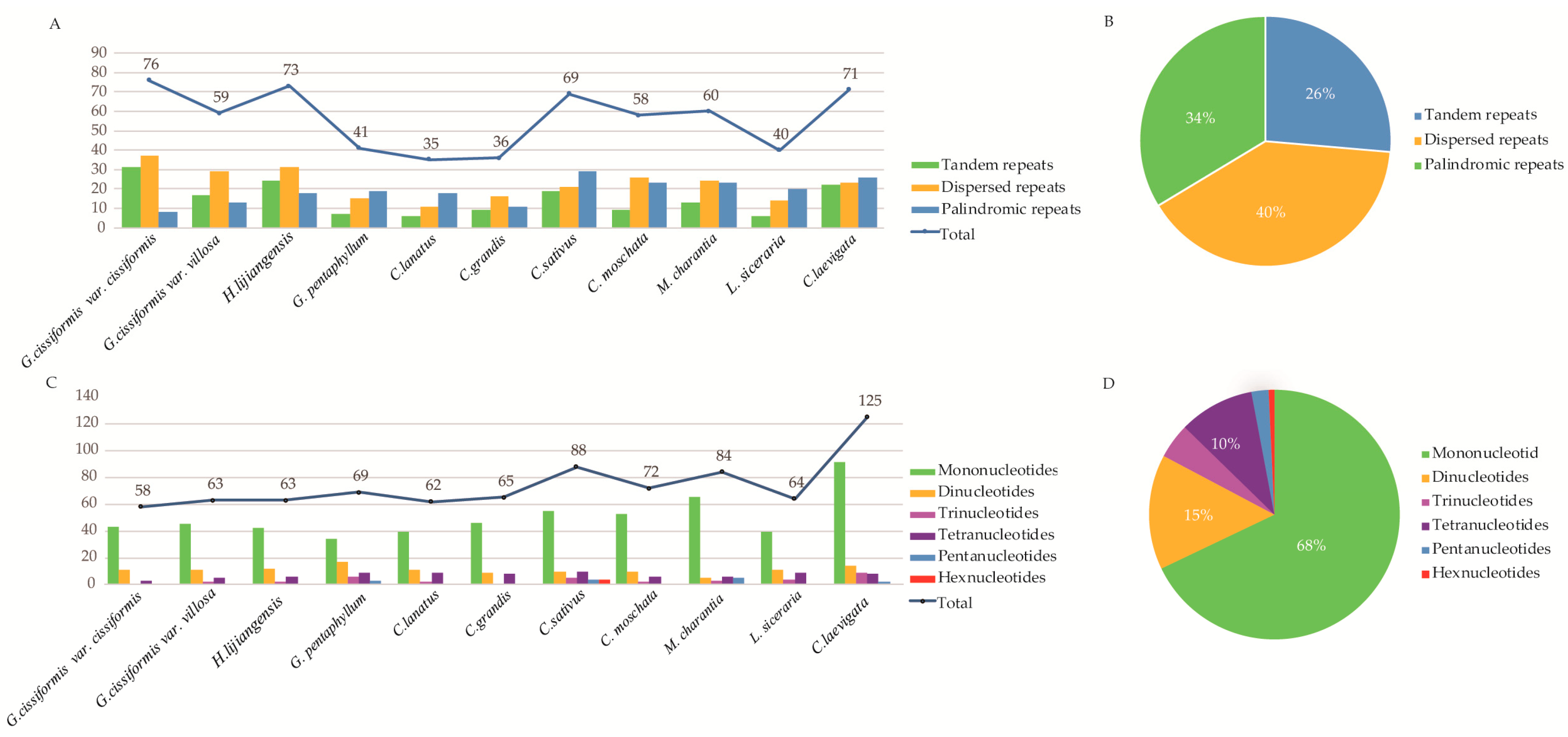
2.3. Repeat Analysis and Microsatellites (SSR)
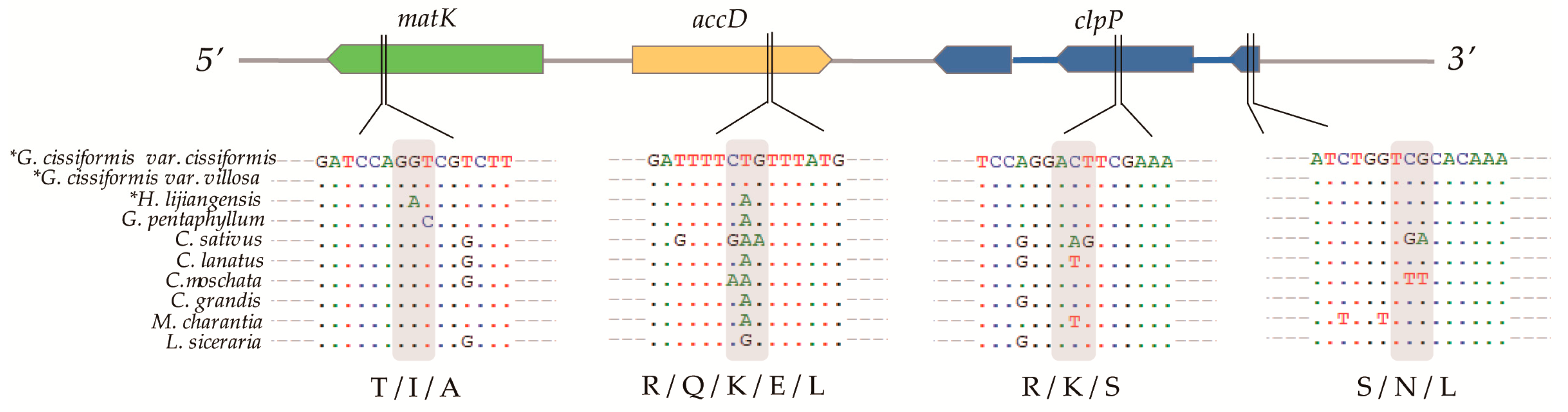
2.4. Selective Pressures Events
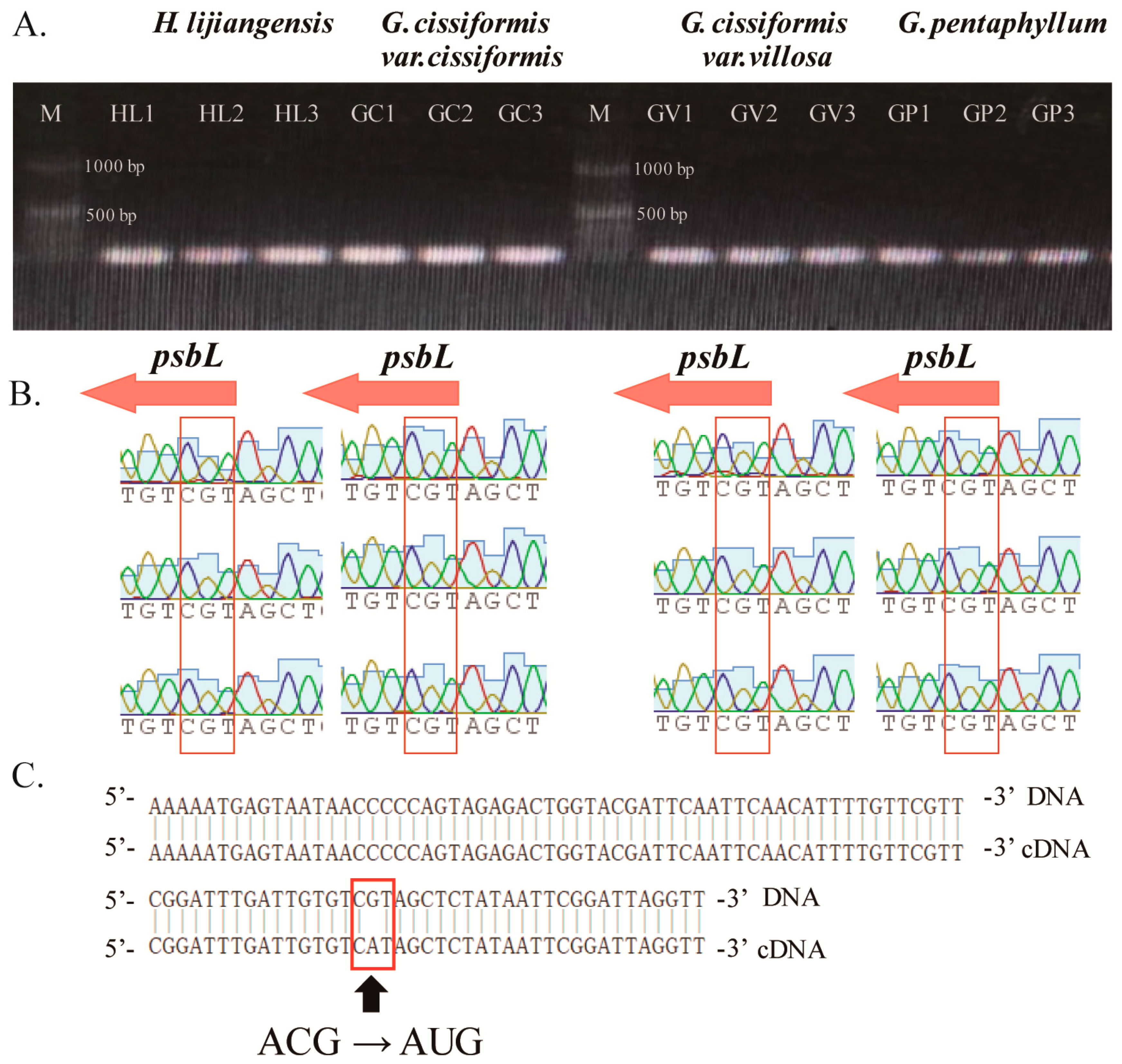
2.5. Codon Usage Bias and Unconventional Initiation Codon
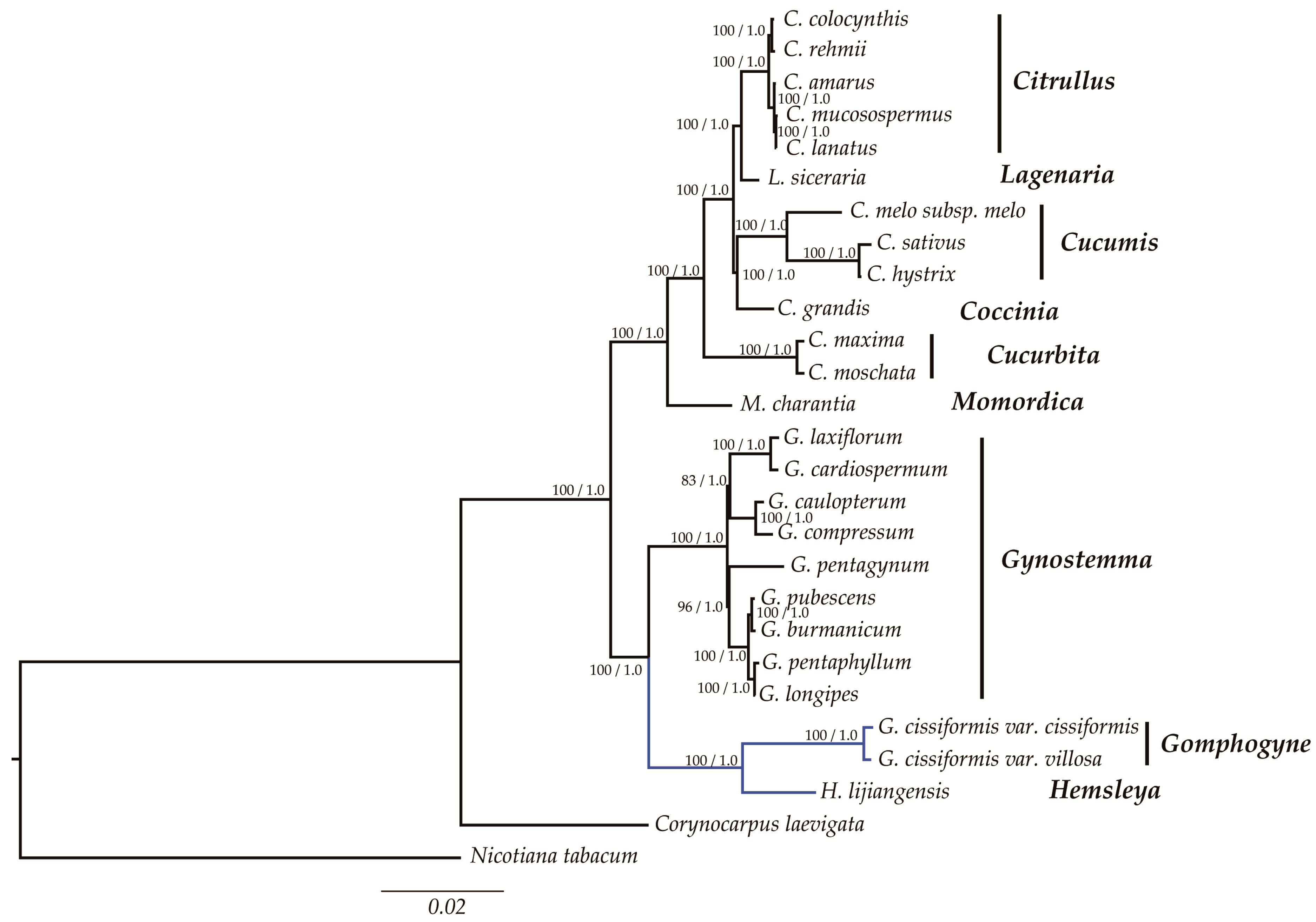
2.6. Phylogenetic Analysis
3. Discussion
3.1. Evolution and Variation of Chloroplast Sequences
3.2. Selective Genes and RNA Editing
3.3. Phylogeny of Cucurbitaceae
4. Materials and Methods
4.1. Plant Materials and DNA Extraction
4.2. Illumina Sequencing, Assembly, and Annotation
4.3. Comparison of Complete Chloroplast Genomes
4.4. Sequence Divergence
4.5. Microsatellites and Repeated Sequences
4.6. Selective Pressure Analysis
4.7. Codon Usage Bias and Unconventional Initiation Codon
4.8. Phylogenetic Relationships
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaefer, H.; Heibl, C.; Renner, S.S. Gourds afloat: A dated phylogeny reveals an asian origin of the gourd family (cucurbitaceae) and numerous oversea dispersal events. Proc. Roy. Soc. B-Biol. Sci. 2009, 276, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Huang, L.; Chen, S.; Charles, J. Flora of China; Missouri Botanical Garden Press: Beijing, China, 2011; Volume 19. [Google Scholar]
- Kocyan, A.; Zhang, L.-B.; Schaefer, H.; Renner, S.S. A multi-locus chloroplast phylogeny for the cucurbitaceae and its implications for character evolution and classification. Mol. Phylogenet. Evol. 2007, 44, 553–577. [Google Scholar] [CrossRef] [PubMed]
- Heiser, C.B. The Gourd Book; University of Oklahoma Press: Norman, OK, USA, 2016. [Google Scholar]
- Tsai, Y.C.; Lin, C.L.; Chen, B.H. Preparative chromatography of flavonoids and saponins in gynostemma pentaphyllum and their antiproliferation effect on hepatoma cell. Phytomedicine 2010, 18, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Liu, W.; Huang, H.; Slavin, M.; Zhao, Y.; Whent, M.; Blackford, J.; Lutterodt, H.; Zhou, H.; Chen, P. Chemical composition of five commercial gynostemma pentaphyllum samples and their radical scavenging, antiproliferative, and anti-inflammatory properties. J. Agric. Food Chem. 2010, 58, 11243–11249. [Google Scholar] [CrossRef] [PubMed]
- Nie, R.; Chen, Z. The research history and present status on the chemical components of genus hemsleya (cucurbitaceae). Acta Bot. Yunnanica 1986, 8, 115–124. [Google Scholar]
- Dinan, L.; Whiting, P.; Sarker, S.D.; Kasai, R.; Yamasaki, K. Cucurbitane-type compounds from hemsleya carnosiflora antagonize ecdysteroid action in the drosophila melanogaster bii cell line. Cell Mol. Life Sci. 1997, 53, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; Gao, J. Three new cucurbitacins from hemsleya lijiangensis. Chin. Chem. Lett. 2003, 14, 389–392. [Google Scholar]
- Wu, J.; Wu, Y.; Yang, B.B. Anticancer activity of hemsleya amabilis extract. Life Sci. 2002, 71, 2161–2170. [Google Scholar] [CrossRef]
- Jeffrey, C. A new system of cucurbitaceae. Bot. Zhum. 2005, 90, 332–335. [Google Scholar]
- Piperno, D.R.; Stothert, K.E. Phytolith Evidence for Early Holocene Cucurbita Domestication in Southwest Ecuador. Science 2003, 299, 1054–1057. [Google Scholar] [CrossRef] [PubMed]
- Kistler, L.; Montenegro, Á.; Smith, B.D.; Gifford, J.A.; Green, R.E.; Newsom, L.A.; Shapiro, B. Transoceanic drift and the domestication of african bottle gourds in the americas. Proc. Natl. Acad. Sci. USA 2014, 111, 2937. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, C.; Renner, S.S. Watermelon origin solved with molecular phylogenetics including linnaean material: Another example of museomics. New Phytol. 2015, 205, 526–532. [Google Scholar]
- Kistler, L.; Newsom, L.A.; Ryan, T.M.; Clarke, A.C.; Smith, B.D.; Perry, G.H. Gourds and squashes (Cucurbita spp.) adapted to megafaunal extinction and ecological anachronism through domestication. Proc. Natl. Acad. Sci. USA 2015, 112, 15107–15112. [Google Scholar] [CrossRef] [PubMed]
- Sanjur, O.I.; Piperno, D.R.; Andres, T.C.; Wessel-Beaver, L. Phylogenetic relationships among domesticated and wild species of Cucurbita (Cucurbitaceae) inferred from a mitochondrial gene: Implications for crop plant evolution and areas of origin. Proc. Natl. Acad. Sci. USA 2002, 99, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.C.; Burtenshaw, M.K.; McLenachan, P.A.; Erickson, D.L.; Penny, D. Reconstructing the origins and dispersal of the polynesian bottle gourd (lagenaria siceraria). Mol. Biol. Evol. 2006, 23, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, H.; Renner, S.S. Phylogenetic relationships in the order cucurbitales and a new classification of the gourd family (cucurbitaceae). Taxon 2011, 60, 122–138. [Google Scholar]
- Li, H.; Li, D. Systematic position of gomphogyne (cucurbitaceae) inferred from its, rpl16 and trns-trnr DNA sequences. J. Syst. Evol. 2008, 46, 595–599. [Google Scholar]
- Li, H.-T.; Yang, J.-B.; Li, D.-Z.; Möller, M.; Shah, A. A molecular phylogenetic study of hemsleya (cucurbitaceae) based on its, rpl16, trnh-psba, and trnl DNA sequences. Plant Syst. Evol. 2010, 285, 23–32. [Google Scholar] [CrossRef]
- Barrett, C.F.; Baker, W.J.; Comer, J.R.; Conran, J.G.; Lahmeyer, S.C.; Leebens-Mack, J.H.; Li, J.; Lim, G.S.; Mayfield-Jones, D.R.; Perez, L.; et al. Plastid genomes reveal support for deep phylogenetic relationships and extensive rate variation among palms and other commelinid monocots. New Phytol. 2016, 209, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Yang, Y.; Zhou, T.; Liu, Z.-L. Comparative plastid genomes of primula species: Sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 2018, 19, 1050. [Google Scholar] [CrossRef] [PubMed]
- Edger, P.P.; Hall, J.C.; Harkess, A.; Tang, M.; Coombs, J.; Mohammadin, S.; Schranz, M.E.; Xiong, Z.; Leebens-Mack, J.; Meyers, B.C.; et al. Brassicales phylogeny inferred from 72 plastid genes: A reanalysis of the phylogenetic localization of two paleopolyploid events and origin of novel chemical defenses. Am. J. Bot. 2018, 105, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; de Pamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, T.; Kanwal, N.; Zhao, Y.; Bai, G.; Zhao, G. Completion of eight gynostemma bl. (cucurbitaceae) chloroplast genomes: Characterization, comparative analysis, and phylogenetic relationships. Front. Plant Sci. 2017, 8, 1583. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-J.; Cheng, C.-L.; Chang, C.-C.; Wu, C.-L.; Su, T.-M.; Chaw, S.-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yonezawa, T.; Zhong, Y.; Hasegawa, M. Episodic evolution and adaptation of chloroplast genomes in ancestral grasses. PLoS ONE 2009, 4, e5297. [Google Scholar] [CrossRef] [PubMed]
- Rabah, S.O.; Shrestha, B.; Hajrah, N.H.; Sabir, M.J.; Alharby, H.F.; Sabir, M.J.; Alhebshi, A.M.; Sabir, J.S.M.; Gilbert, L.E.; Ruhlman, T.A.; et al. Passiflora plastome sequencing reveals widespread genomic rearrangements. J. Syst. Evol. 2018. [CrossRef]
- Diekmann, K.; Hodkinson, T.R.; Wolfe, K.H.; van den Bekerom, R.; Dix, P.J.; Barth, S. Complete chloroplast genome sequence of a major allogamous forage species, perennial ryegrass (lolium perenne L.). DNA Res. 2009, 16, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Davis, J.I.; Soreng, R.J.; Garvin, D.; Anderson, M.J. Chloroplast DNA inversions and the origin of the grass family (poaceae). Proc. Natl. Acad. Sci. USA 1992, 89, 7722–7726. [Google Scholar] [CrossRef] [PubMed]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats and nucleotide substitution rates. Mol. Biol. Evol. 2013, 31, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Vanin, E.F. Processed pseudogenes: Characteristics and evolution. Annu. Rev. Genet. 1985, 19, 253–272. [Google Scholar] [CrossRef] [PubMed]
- Balakirev, E.S.; Ayala, F.J. Pseudogenes: Are they “junk” or functional DNA? Annu. Rev. Genet. 2003, 37, 123–151. [Google Scholar] [CrossRef] [PubMed]
- Asif, H.; Khan, A.; Iqbal, A.; Khan, I.A.; Heinze, B.; Azim, M.K. The chloroplast genome sequence of syzygium cumini (L.) and its relationship with other angiosperms. Tree Genet. Genomes 2013, 9, 867–877. [Google Scholar] [CrossRef]
- Nashima, K.; Terakami, S.; Nishitani, C.; Kunihisa, M.; Shoda, M.; Takeuchi, M.; Urasaki, N.; Tarora, K.; Yamamoto, T.; Katayama, H. Complete chloroplast genome sequence of pineapple (ananas comosus). Tree Genet. Genomes 2015, 11, 60. [Google Scholar] [CrossRef]
- Do, H.D.K.; Kim, J.S.; Kim, J.-H. Comparative genomics of four liliales families inferred from the complete chloroplast genome sequence of veratrum patulum o. Loes. (melanthiaceae). Gene 2013, 530, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Tabata, S. Complete structure of the chloroplast genome of arabidopsis thaliana. DNA Res. 1999, 6, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Lee, H.-L. Complete chloroplast genome sequences from korean ginseng (panax schinseng nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.R.; Dastidar, S.G.; Cai, Z.; Penaflor, C.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (buxaceae), chloranthus (chloranthaceae), dioscorea (dioscoreaceae), and illicium (schisandraceae). Mol. Phylogenet. Evol. 2007, 45, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Naydenov, K.D.; Naydenov, M.K.; Alexandrov, A.; Vasilevski, K.; Gyuleva, V.; Matevski, V.; Nikolic, B.; Goudiaby, V.; Bogunic, F.; Paitaridou, D. Ancient split of major genetic lineages of european black pine: Evidence from chloroplast DNA. Tree Genet. Genomes 2016, 12, 68. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Yin, K.; Zhang, Y.; Li, Y.; Du, F. Different natural selection pressures on the atpf gene in evergreen sclerophyllous and deciduous oak species: Evidence from comparative analysis of the complete chloroplast genome of quercus aquifolioides with other oak species. Int. J. Mol. Sci. 2018, 19, 1042. [Google Scholar] [CrossRef] [PubMed]
- Tomoko, O. Synonymous and nonsynonymous substitutions in mammalian genes and the nearly neutral theory. J. Mol. Evol. 1995, 40, 56–63. [Google Scholar] [CrossRef]
- Kimura, M. The neutral theory of molecular evolution and the world view of the neutralists. Genome 1989, 31, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Nagano, Y. Plant acetyl-coa carboxylase: Structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci. Biotech. Bioch. 2004, 68, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accd gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Tsudzuki, T.; Sugiura, M. The genomics of land plant chloroplasts: Gene content and alteration of genomic information by rna editing. Photosynth. Res. 2001, 70, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-C.; Sung, T.-Y.; Li, Y.-C.; Hsu, S.-J.; Lin, C.-L.; Hsieh, M.-H. Editing of accd and ndhf chloroplast transcripts is partially affected in the arabidopsis vanilla cream1 mutant. Plant Mol. Biol. 2010, 73, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Madoka, Y.; Tomizawa, K.-I.; Mizoi, J.; Nishida, I.; Nagano, Y.; Sasaki, Y. Chloroplast transformation with modified accd operon increases acetyl-coa carboxylase and causes extension of leaf longevity and increase in seed yield in tobacco. Plant Cell Physiol. 2002, 43, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Ogihara, Y. Phylogenetic affinities of the grasses to other monocots as revealed by molecular analysis of chloroplast DNA. Curr. Genet. 1996, 29, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Shikanai, T.; Shimizu, K.; Ueda, K.; Nishimura, Y.; Kuroiwa, T.; Hashimoto, T. The chloroplast clpp gene, encoding a proteolytic subunit of atp-dependent protease, is indispensable for chloroplast development in tobacco. Plant Cell Physiol. 2001, 42, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.K.; Gustafsson, P.; Lidholm, J.Å. Identification and expression of the chloroplast clpp gene in the conifer pinus contorta. Plant Mol. Biol. 1994, 26, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, H.; Maliga, P. The plastid clpp1 protease gene is essential for plant development. Nature 2003, 425, 86. [Google Scholar] [CrossRef] [PubMed]
- Maliga, P. Plastid transformation in higher plants. Annu. Rev. Plant Biol. 2004, 55, 289–313. [Google Scholar] [CrossRef] [PubMed]
- Zoschke, R.; Nakamura, M.; Liere, K.; Sugiura, M.; Börner, T.; Schmitz-Linneweber, C. An organellar maturase associates with multiple group ii introns. Proc. Natl. Acad. Sci. USA 2010, 107, 3245–3250. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, H.; Link, G. The chloroplast trnalys(uuu) gene from mustard (sinapis alba) contains a class ii intron potentially coding for a maturase-related polypeptide. Curr. Genet. 1987, 11, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Mohr, G.; Perlman, P.S.; Lambowitz, A.M. Evolutionary relationships among group ii intron-encoded proteins and identification of a conserved domain that may be related to maturase function. Nucleic Acids Res. 1993, 21, 4991–4997. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Soltis, D.E. Phylogenetic inference in saxifragaceae sensu stricto and gilia (polemoniaceae) using matk sequences. Ann. Mo. Bot. Gard. 1995, 82, 149–175. [Google Scholar] [CrossRef]
- Gadek, P.A.; Wilson, P.G.; Quinn, C.J. Phylogenetic reconstruction in myrtaceae using mat k, with particular reference to the position of psiloxylon and heteropyxis. Aust. Syst. Bot. 1996, 9, 283–290. [Google Scholar] [CrossRef]
- Hilu, K.; Liang, H. The matk gene: Sequence variation and application in plant systematics. Am. J. Bot. 1997, 84, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N.; Kawanishi, Y. DNA G + C content of the third codon position and codon usage biases of human genes. Gene 2000, 261, 53–62. [Google Scholar] [CrossRef]
- Bellgard, M.; Schibeci, D.; Trifonov, E.; Gojobori, T. Early detection of G + C differences in bacterial species inferred from the comparative analysis of the two completely sequenced helicobacter pylori strains. J. Mol. Evol. 2001, 53, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Shimda, H.; Sugiuro, M. Fine structural features of the chloroplast genome: Comparison of the sequenced chloroplast genomes. Nucleic Acids Res. 1991, 19, 983–995. [Google Scholar] [CrossRef]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H., Jr.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [PubMed]
- Tangphatsornruang, S.; Sangsrakru, D.; Chanprasert, J.; Uthaipaisanwong, P.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. The chloroplast genome sequence of mungbean (vigna radiata) determined by high-throughput pyrosequencing: Structural organization and phylogenetic relationships. DNA Res. 2009, 17, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Delannoy, E.; Fujii, S.; Colas, d.F.-S.C.; Brundrett, M.; Small, I. Rampant gene loss in the underground orchid rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol. Biol. Evol. 2011, 28, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, J.; Feng, L.; Zhou, T.; Bai, G.; Yang, J.; Zhao, G. Plastid genome comparative and phylogenetic analyses of the key genera in fagaceae: Highlighting the effect of codon composition bias in phylogenetic inference. Front. Plant Sci. 2018, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Tsudzuki, T.; Wakasugi, T.; Sugiura, M. Comparative analysis of rna editing sites in higher plant chloroplasts. J. Mol. Evol. 2001, 53, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Maier, R.M.; Zeltz, P.; Kössel, H.; Bonnard, G.; Gualberto, J.M.; Grienenberger, J.M. Rna editing in plant mitochondria and chloroplasts. Plant Mol. Biol. 1996, 32, 343–365. [Google Scholar] [CrossRef] [PubMed]
- Hoch, B.; Maier, R.M.; Appel, K.; Igloi, G.L.; Kössel, H. Editing of a chloroplast mrna by creation of an initiation codon. Nature 1991, 353, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Freyer, R.; Kiefer-Meyer, M.C.; Kössel, H. Occurrence of plastid rna editing in all major lineages of land plants. Proc. Natl. Acad. Sci. USA 1997, 94, 6285–6290. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Funk, H.L.C.; Poltnigg, P.; Sabater, B.; Martin, M.; Maier, R. Editing of plastid rna in arabidopsis thaliana ecotypes. Plant J. 2010, 43, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Maier, R.M.; Neckermann, K.; Igloi, G.L.; Kössel, H. Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 1995, 251, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Corneille, S.; Lutz, K.; Maliga, P. Conservation of rna editing between rice and maize plastids: Are most editing events dispensable? Mol. Gen. Genet. 2000, 264, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Guzowska-Nowowiejska, M.; Fiedorowicz, E.; Pląder, W. Cucumber, melon, pumpkin, and squash: Are rules of editing in flowering plants chloroplast genes so well known indeed? Gene 2009, 434, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Hirose, T.; Horihata, M.; Tsudzuki, T.; Kössel, H.; Sugiura, M. Creation of a novel protein-coding region at the RNA level in black pine chloroplasts: The pattern of RNA editing in the gymnosperm chloroplast is different from that in angiosperms. Proc. Natl. Acad. Sci. USA 1996, 93, 8766–8770. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, K.; Kakehi, T.; Shima, Y.; Iinuma, H.; Masuzawa, T.; Ueno, M. Extensive rna editing and possible double-stranded structures determining editing sites in the atpb transcripts of hornwort chloroplasts. Nucleic Acids Res. 1997, 25, 4830–4834. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, S.; Kobayashi, T.; Sugiyama, R.; Hoshida, H.; Shiina, T.; Toyoshima, Y. Role of psii-l protein (psbl gene product) on the electron transfer in photosystem ii complex. 1. Over-production of wild-type and mutant versions of psii-l protein and reconstitution into the psii core complex. Plant Mol. Biol. 1997, 34, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, M.; Camara, B.; Weil, J.H.; Schantz, R. The psbl gene from bell pepper (capsicum annuum): Plastid rna editing also occurs in non-photosynthetic chromoplasts. Plant Mol. Biol. 1992, 20, 1185–1188. [Google Scholar] [CrossRef] [PubMed]
- Kudla, J.; Igloi, G.L.; Metzlaff, M.; Hagemann, R.; Kössel, H. Rna editing in tobacco chloroplasts leads to the formation of a translatable psbl mrna by a c to u substitution within the initiation codon. Embo J. 1992, 11, 1099–1103. [Google Scholar] [PubMed]
- Bock, R.; Hagemann, R.; Kössel, H.; Kudla, J. Tissue- and stage-specific modulation of rna editing of the psbf and psbl transcript from spinach plastids—A new regulatory mechanism? Mol. Gen. Genet. 1993, 240, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Raman, G.; Park, S. The complete chloroplast genome sequence of ampelopsis: Gene organization, comparative analysis, and phylogenetic relationships to other angiosperms. Front. Plant Sci. 2016, 7, 341. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Sugiura, M. Both RNA editing and RNA cleavage are required for translation of tobacco chloroplast ndhDmRNA: A possible regulatory mechanism for the expression of a chloroplast operon consisting of functionally unrelated genes. Embo J. 1997, 16, 6804–6811. [Google Scholar] [CrossRef] [PubMed]
- Bock, D.G.; Kane, N.C.; Ebert, D.P.; Rieseberg, L.H. Genome skimming reveals the origin of the jerusalem artichoke tuber crop species: Neither from jerusalem nor an artichoke. New Phytol. 2014, 201, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genuscitrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Gao, L.M.; Soltis, D.E.; Soltis, P.S.; Yang, J.B.; Fang, L.; Yang, S.X.; Li, D.Z. Insights into the historical assembly of east asian subtropical evergreen broadleaved forests revealed by the temporal history of the tea family. New Phytol. 2017, 215, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Z. Phylogeny and Evolution of the Genus Hemsleya; Yunnan Science and Technology Press: Kunming, China, 1993. [Google Scholar]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Zhu, Q.; Cui, H.; Zhao, Y.; Gao, P.; Liu, S.; Wang, P.; Luan, F. The complete chloroplast genome sequence of the Citrullus lanatus L. Subsp. Vulgaris (cucurbitaceae). Mitochondrial DNA B 2016, 1, 943–944. [Google Scholar] [CrossRef]
- Sousa, A.; Bellot, S.; Fuchs, J.; Houben, A.; Renner, S.S. Analysis of transposable elements and organellar DNA in male and female genomes of a species with a huge y chromosome reveals distinct y centromeres. Plant J. 2016, 88, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Pląder, W.; Yukawa, Y.; Sugiura, M.; Malepszy, S. The complete structure of the cucumber (Cucumis sativus L.) chloroplast genome: Its composition and comparative analysis. Cell. Mol. Biol. Lett. 2007, 12, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.G.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mrna transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer 3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with dogma. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. Vista: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. Dnasp v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. Mega5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Ma, P.-F.; Li, D.-Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting est databases for the development and characterization of gene-derived ssr-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Paml 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. Kaks_calculator: Calculating ka and ks through model selection and model averaging. Genom. Proteom. Bioinf. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 1999. [Google Scholar]
- Zhao, Y.-M.; Zhou, T.; Li, Z.-H.; Zhao, G.-F. Characterization of global transcriptome using illumina paired-end sequencing and development of est-ssr markers in two species of Gynostemma (Cucurbitaceae). Molecules 2015, 20, 21214–21231. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml-vi-hpc: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | * G. cissiformis var. cissiformis | * G. cissiformis var. villosa | * H. lijiangensis | G. pentaphyllum | C. lanatus |
| Locations | 24.20° N, 99.50° E | 24.20° N, 99.50° E | 27.17° N, 100.06° E | / | / |
| Assembly reads | 1,274,004 | 1,505,442 | 1,483,091 | / | / |
| Mean coverage | 1112.4 | 1358.4 | 1392.7 | / | / |
| Size (bp) | 157,334 | 156,585 | 158,275 | 157,576 | 156,906 |
| LSC (bp) | 87,239 | 86,642 | 87,362 | 86,757 | 86,845 |
| SSC (bp) | 18,014 | 18,029 | 18,429 | 18,653 | 17,897 |
| IRs (bp) | 26,041 | 25,957 | 26,242 | 26,083 | 26,082 |
| Number of total genes | 133 | 133 | 133 | 133 | 122 |
| Number of protein-coding genes | 87 | 87 | 87 | 87 | 85 |
| Number of tRNA genes | 37 | 37 | 37 | 37 | 29 |
| Number of rRNA genes | 8 | 8 | 8 | 8 | 8 |
| Pseudogene | infA | infA | infA | infA | / |
| Overall GC content (%) | 37 | 37 | 37 | 37 | 37.2 |
| GC content in LSC (%) | 34.8 | 34.8 | 34.8 | 34.8 | 34.9 |
| GC content in SSC (%) | 31.1 | 31 | 31 | 30.6 | 31.5 |
| GC content in IR (%) | 42.8 | 42.7 | 42.8 | 42.8 | 42.8 |
| GenBank number | MH256801 | MF784515 | MG733988 | KX852298 | KY014105 |
| Species | C. grandis | C. sativus | C. moschata | M. charantia | L. siceraria |
| Locations | / | / | / | / | / |
| Assembly reads | / | / | / | / | / |
| Mean coverage | / | / | / | / | / |
| Size (bp) | 157,035 | 155,293 | 157,644 | 158,844 | 157,145 |
| LSC (bp) | 86,749 | 86,688 | 88,343 | 88,374 | 86,843 |
| SSC (bp) | 18,004 | 18,223 | 18,156 | 18,010 | 18,008 |
| IRs (bp) | 26,141 | 25,193 | 25,573 | 26,228 | 26,147 |
| Number of total genes | 132 | 132 | 135 | 130 | 130 |
| Number of protein-coding genes | 85 | 85 | 85 | 85 | 86 |
| Number of tRNA genes | 39 | 38 | 42 | 38 | 37 |
| Number of rRNA genes | 8 | 8 | 8 | 8 | 8 |
| Pseudogene | / | rps16 | / | ycf1 | ycf1 |
| Overall GC content (%) | 37.1 | 37.1 | 37.1 | 36.7 | 37.1 |
| GC content in LSC (%) | 34.8 | 34.8 | 34.9 | 34.3 | 34.9 |
| GC content in SSC (%) | 31.3 | 31.8 | 31.5 | 30.7 | 31.4 |
| GC content in IR (%) | 42.8 | 42.8 | 43.1 | 42.8 | 42.8 |
| GenBank number | KX147312 | AJ970307 | MF991116 | MG022622 | MG022623 |
| Category | Gene Group | Gene Name |
|---|---|---|
| Self-replication | Ribosomal protein (small subunit) (14) | rps2 rps3 rps4 rps7 (×2) rps8 rps11 * rps12 (×2) rps14 rps15 * rps16 rps18 rps19 |
| Ribosomal protein (large subunit) (11) | * rpl2 (×2) rpl14 * rpl16 rpl20 rpl22 rpl23 (×2) rpl32 rpl33 rpl36 | |
| RNA polymerase (4) | rpoA rpoB * rpoC1 rpoC2 | |
| Transfer RNAs (37) | * trnA-UGC (×2) trnC-GCA trnD-GUC trnE-UUC trnF-GAA trnfM-CAU * trnG-UCC trnG-GCC trnH-GUG trnI-CAU(×2) * trnI-GAU (×2) * trnK-UUU trnL-CAA(×2) trnL-UAG * trnL-UAA trnM-CAU trnN-GUU(×2) trnP-UGG trnQ-UUG trnR-ACG(×2) trnR-UCU trnS-GCU trnS-GGA trnS-UGA trnT-GGU trnT-UGU trnV-GAC(×2) * trnV-UAC trnW-CCA trnY-GUA | |
| Ribosomal RNAs (8) | rrn4.5(×2) rrn5(×2) rrn16(×2) rrn23(×2) | |
| Photosynthesis | Photosystem I (5) | psaA psaB psaC psaI psaJ |
| Photosystem II (15) | psbA psbB psbC psbD psbE psbF psbH psbI psbJ psbK psbL psbM psbN psbT psbZ | |
| Cytochrome b/f complex (6) | petA * petB * petD petG petL petN | |
| ATP synthase (6) | atpA atpB atpE * atpF atpH atpI | |
| NADH dehydrogenase (12) | * ndhA * ndhB (×2) ndhC ndhD ndhE ndhF ndhG ndhH ndhI ndhJ ndhK | |
| Rubisco large subunit (1) | rbcL | |
| Other genes | Maturase (1) | matK |
| membrane protein (1) | cemA | |
| Acetyl-CoA carboxylase gene (1) | accD | |
| ATP-dependent protease subunit (1) | clpP | |
| c-type Cytochrome biogenesis (1) | ccsA | |
| Assembly/stability of photosystem I (2) | ycf3 ycf4 | |
| Conserved reading frames (ycfs) (4) | ycf1(×2) ycf2(×2) | |
| hypothetical chloroplast protein (2) | orf70(×2) | |
| Pseudogene | Translation-related gene (1) | infA |
| Genes | Model | df | lnL/ω Value | LRTs | No. of Sites (BEB) | Consistent Sites |
|---|---|---|---|---|---|---|
| clpP | M0 (one ratio) | 19 | ω = 0.96975 | |||
| M1 (neutral) | 20 | −1396.7593 | M1 vs. M2: | 2 | 12 S/N/L | |
| M2 (selection) | 22 | −1389.6667 | 14.1852 ** | AGT/AAT/CTT | ||
| M7 (beta) | 20 | −1396.7638 | M7 vs. M8: | 4 | 87 R/K/S | |
| M8 (beta&ω) | 22 | −1389.6672 | 14.1933 ** | CGA/AAA/TCA | ||
| atpE | M0 (one ratio) | 19 | ω = 0.70941 | |||
| M1 (neutral) | 20 | −787.914712 | M1 vs. M2: | 0 | / | |
| M2 (selection) | 22 | −785.703588 | 4.42225 | |||
| M7 (beta) | 20 | −787.942937 | M7 vs. M8: | 0 | ||
| M8 (beta&ω) | 22 | −785.704169 | 4.47754 | |||
| psbL | M0 (one ratio) | 19 | ω = 0.61775 | |||
| M1 (neutral) | 20 | −158.807141 | M1 vs. M2: | 0 | / | |
| M2 (selection) | 22 | −158.807091 | 0.00010 | |||
| M7 (beta) | 20 | −158.807137 | M7 vs. M8: | 0 | ||
| M8 (beta&ω) | 22 | −158.80712 | 0.00003 | |||
| accD | M0 (one ratio) | 19 | ω = 0.53161 | |||
| M1 (neutral) | 20 | −3221.0459 | M1 vs. M2: | 1 | 308 R/Q/K/E/L | |
| M2 (selection) | 22 | −3214.0011 | 14.0896 ** | CGG/CAG/CTG/AAG/GAA | ||
| M7 (beta) | 20 | −3221.5205 | M7 vs. M8: | 4 | ||
| M8 (beta&ω) | 22 | −3214.0768 | 14.8875 ** | |||
| matK | M0 (one ratio) | 19 | ω = 0.52255 | |||
| M1 (neutral) | 20 | −3688.5152 | M1 vs. M2: | 1 | 337 T/I/A | |
| M2 (selection) | 22 | −3683.8284 | 9.3735 ** | TCA/GGA/GCA | ||
| M7 (beta) | 20 | −3689.0422 | M7 vs. M8: | 8 | ||
| M8 (beta&ω) | 22 | −3683.9212 | 10.2421 ** |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Zhou, T.; Yang, J.; Sun, J.; Ju, M.; Zhao, Y.; Zhao, G. Comparative Analyses of Chloroplast Genomes of Cucurbitaceae Species: Lights into Selective Pressures and Phylogenetic Relationships. Molecules 2018, 23, 2165. https://doi.org/10.3390/molecules23092165
Zhang X, Zhou T, Yang J, Sun J, Ju M, Zhao Y, Zhao G. Comparative Analyses of Chloroplast Genomes of Cucurbitaceae Species: Lights into Selective Pressures and Phylogenetic Relationships. Molecules. 2018; 23(9):2165. https://doi.org/10.3390/molecules23092165
Chicago/Turabian StyleZhang, Xiao, Tao Zhou, Jia Yang, Jingjing Sun, Miaomiao Ju, Yuemei Zhao, and Guifang Zhao. 2018. "Comparative Analyses of Chloroplast Genomes of Cucurbitaceae Species: Lights into Selective Pressures and Phylogenetic Relationships" Molecules 23, no. 9: 2165. https://doi.org/10.3390/molecules23092165
APA StyleZhang, X., Zhou, T., Yang, J., Sun, J., Ju, M., Zhao, Y., & Zhao, G. (2018). Comparative Analyses of Chloroplast Genomes of Cucurbitaceae Species: Lights into Selective Pressures and Phylogenetic Relationships. Molecules, 23(9), 2165. https://doi.org/10.3390/molecules23092165