
Oxadiazole/Pyridine-Based Ligands: A Structural Tuning for Enhancing G-Quadruplex Binding

 ,
,  ,
,
Abstract
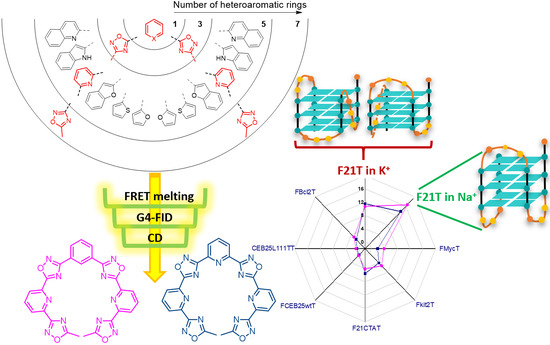
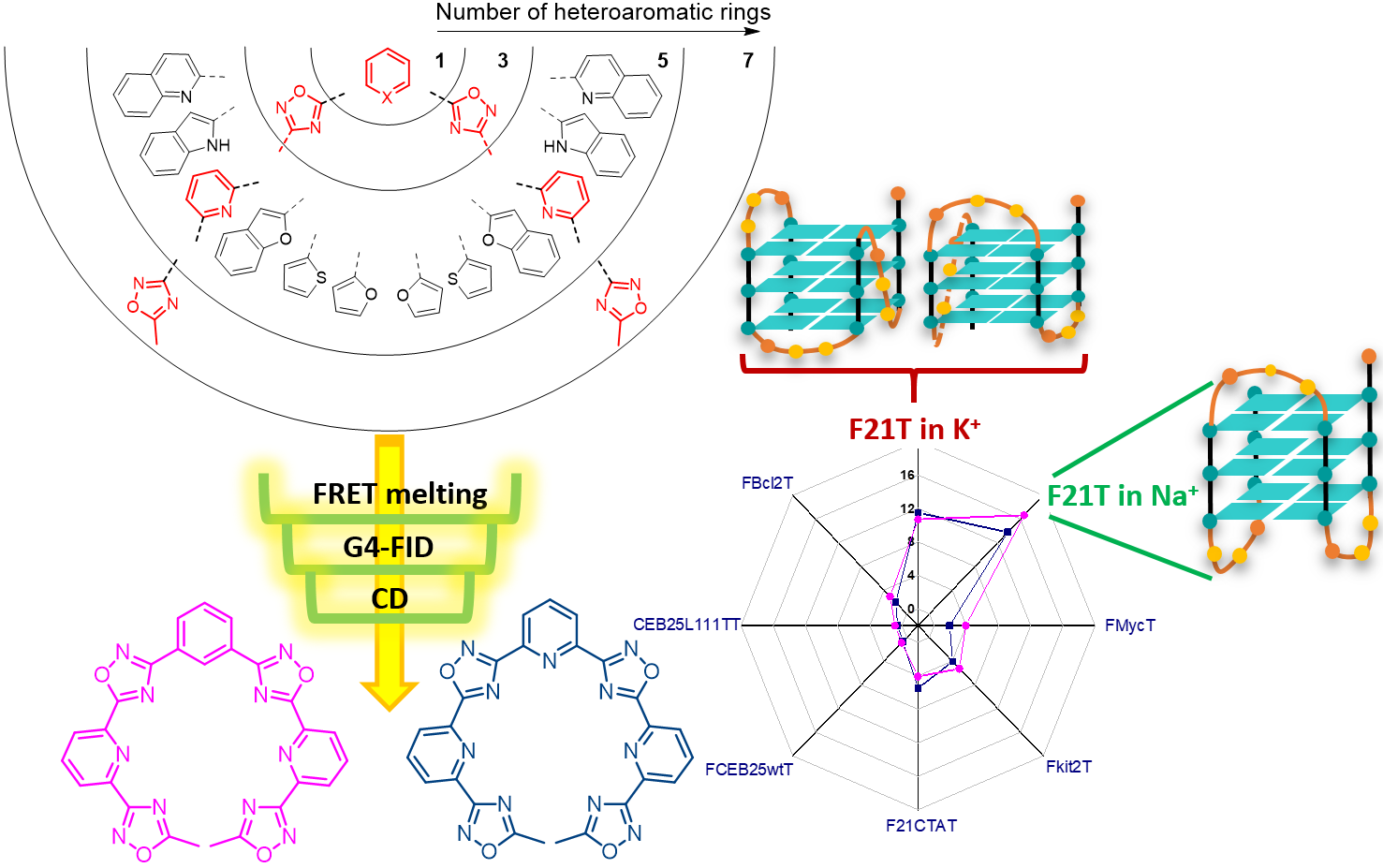
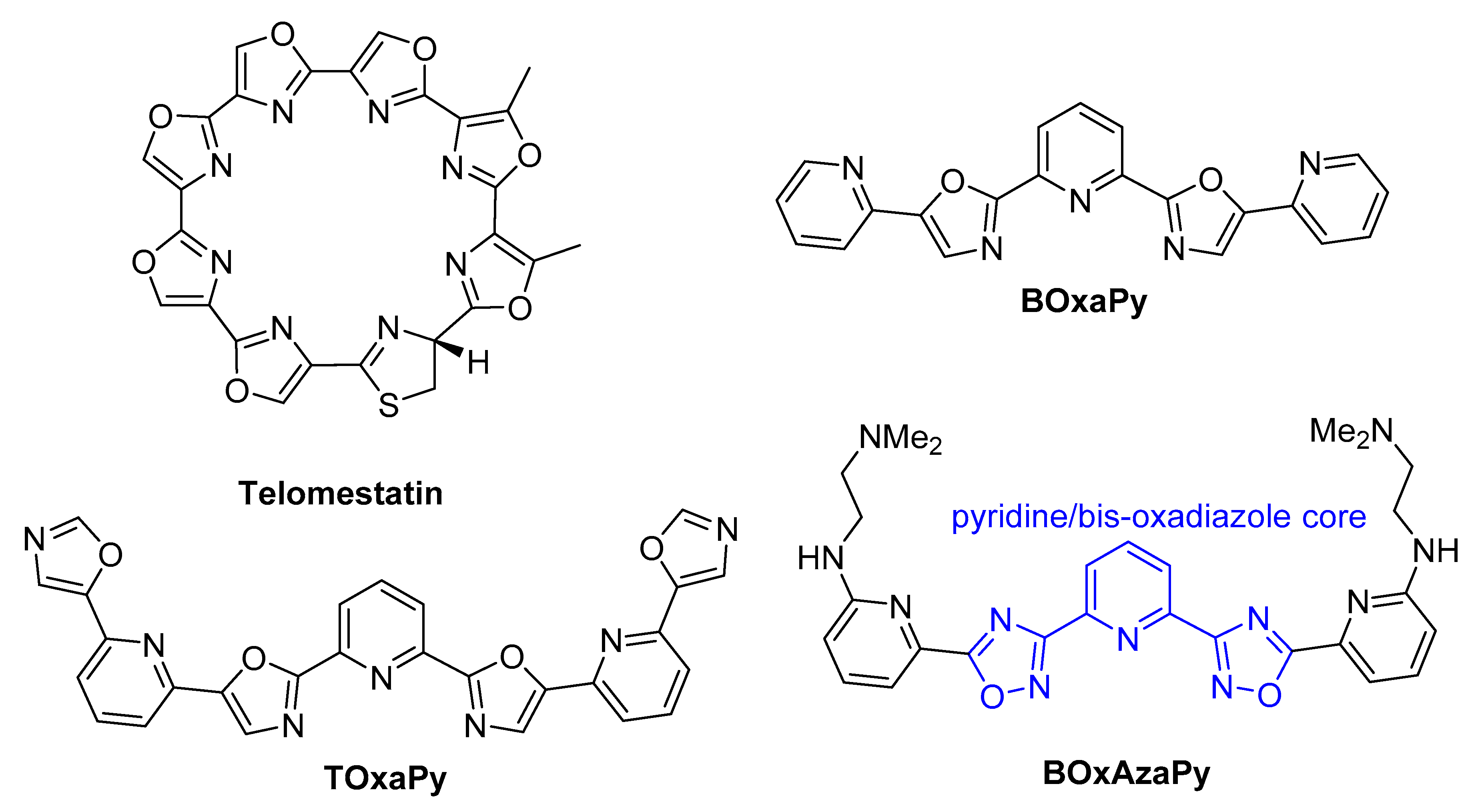
1. Introduction
2. Results and Discussion
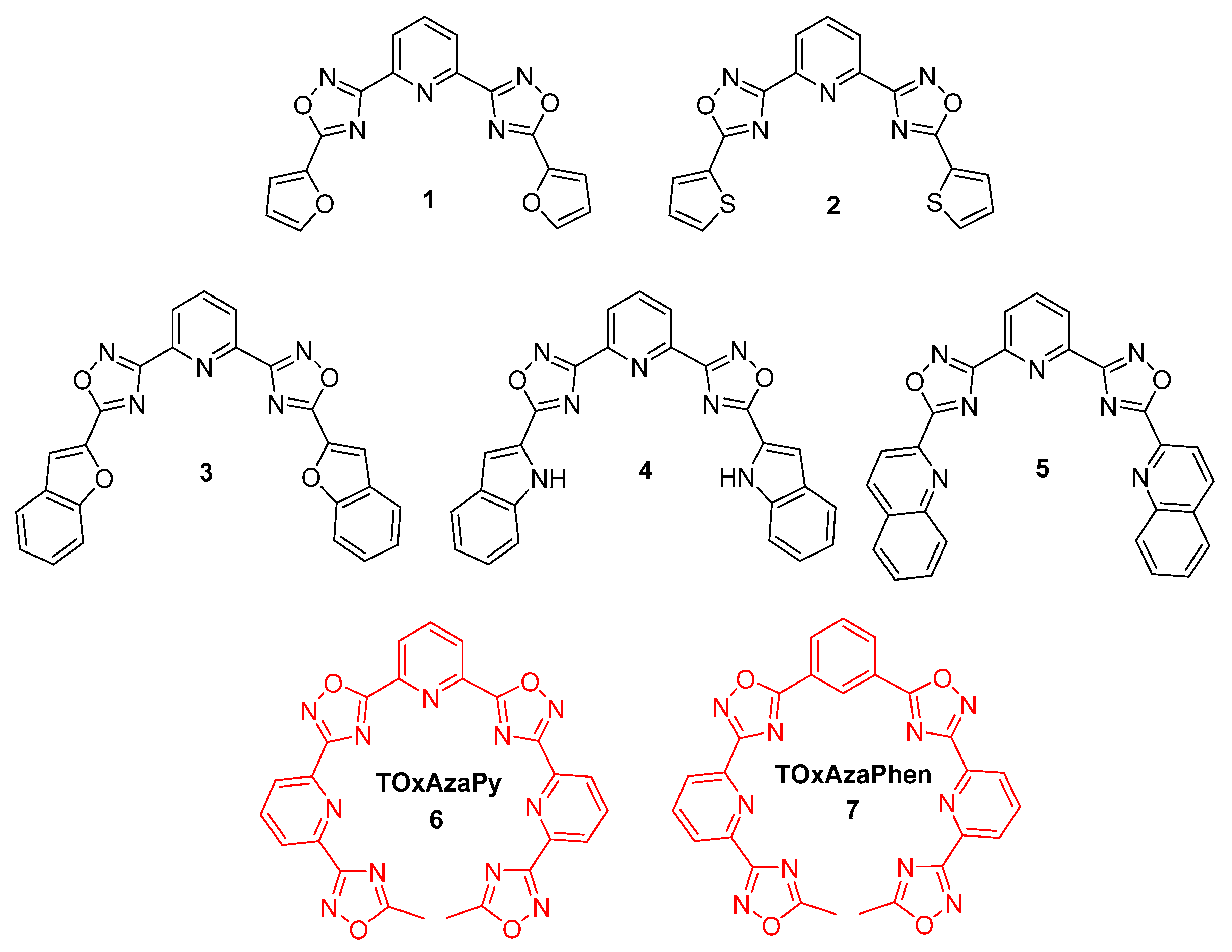
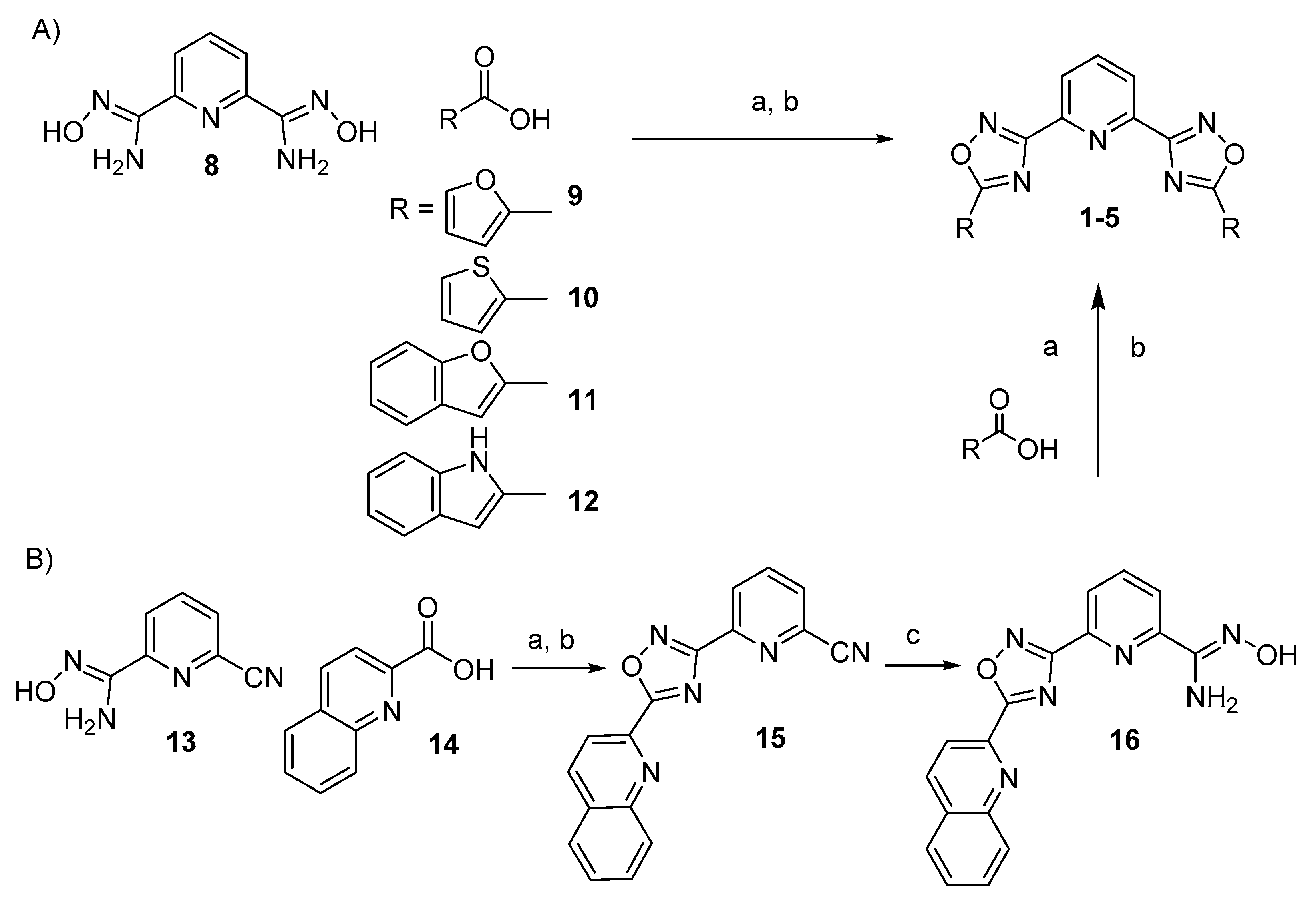
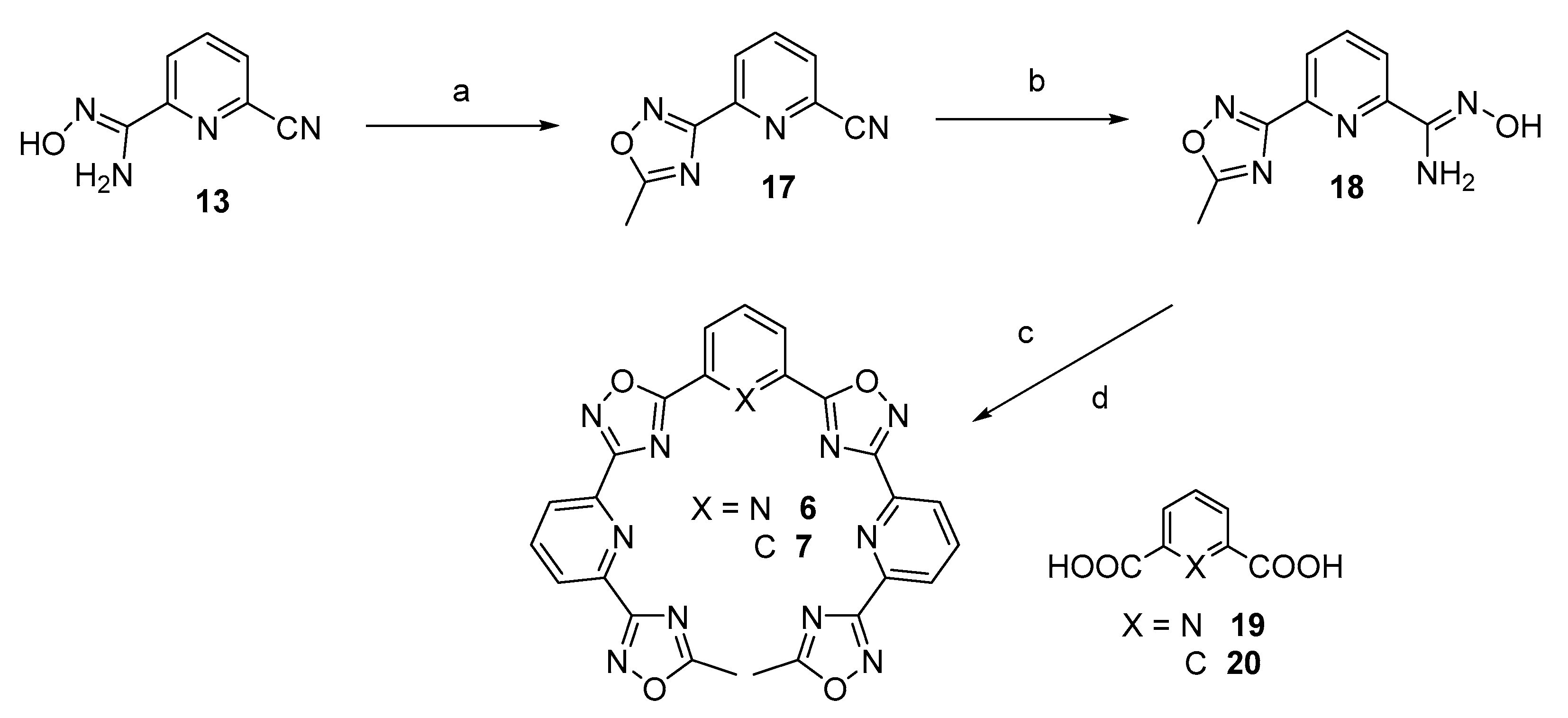
2.1. Design and Synthesis of the Polyheteroaryl Oxadiazole/Pyridine-Ligands
2.2. Screening of the Ligands
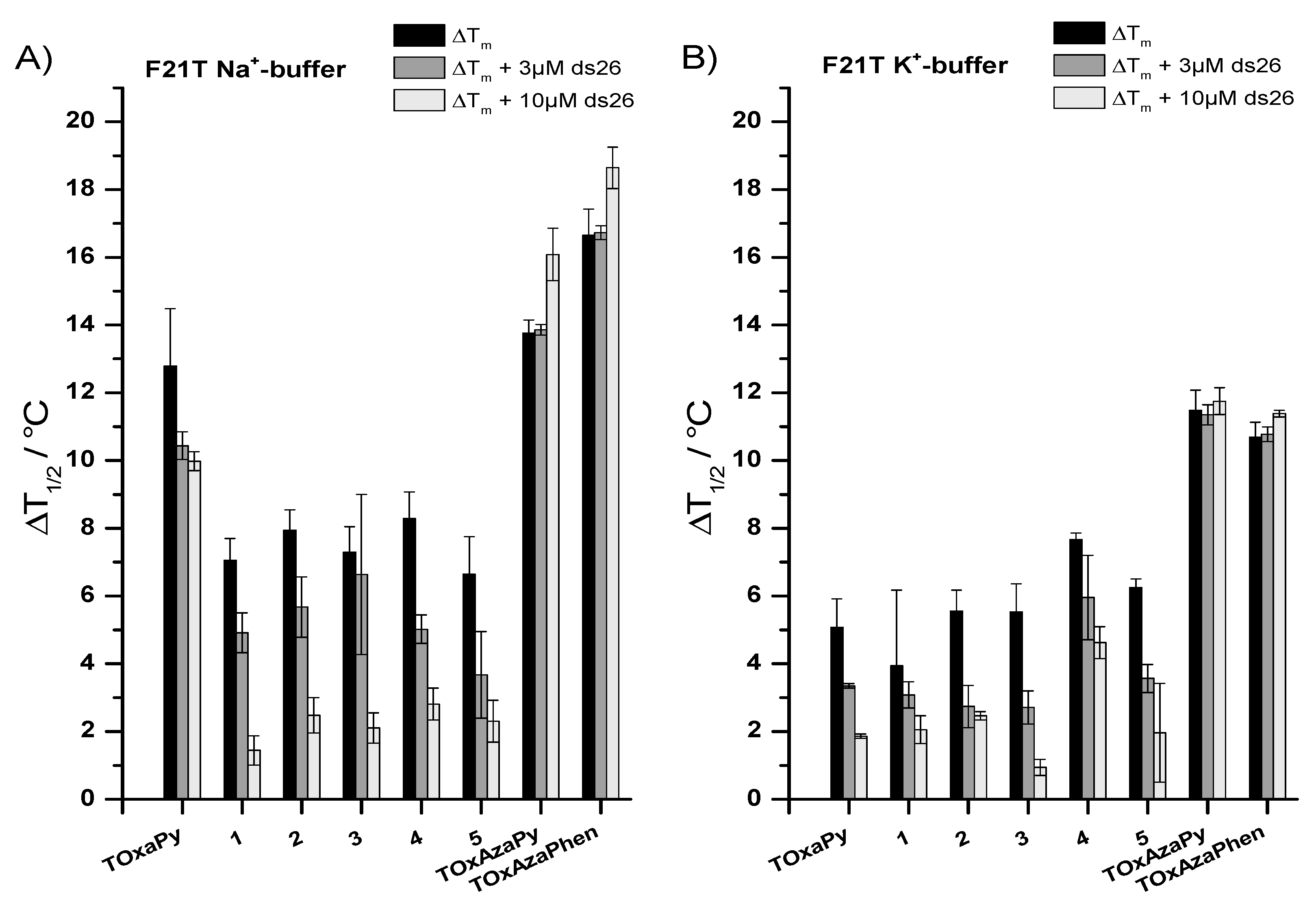
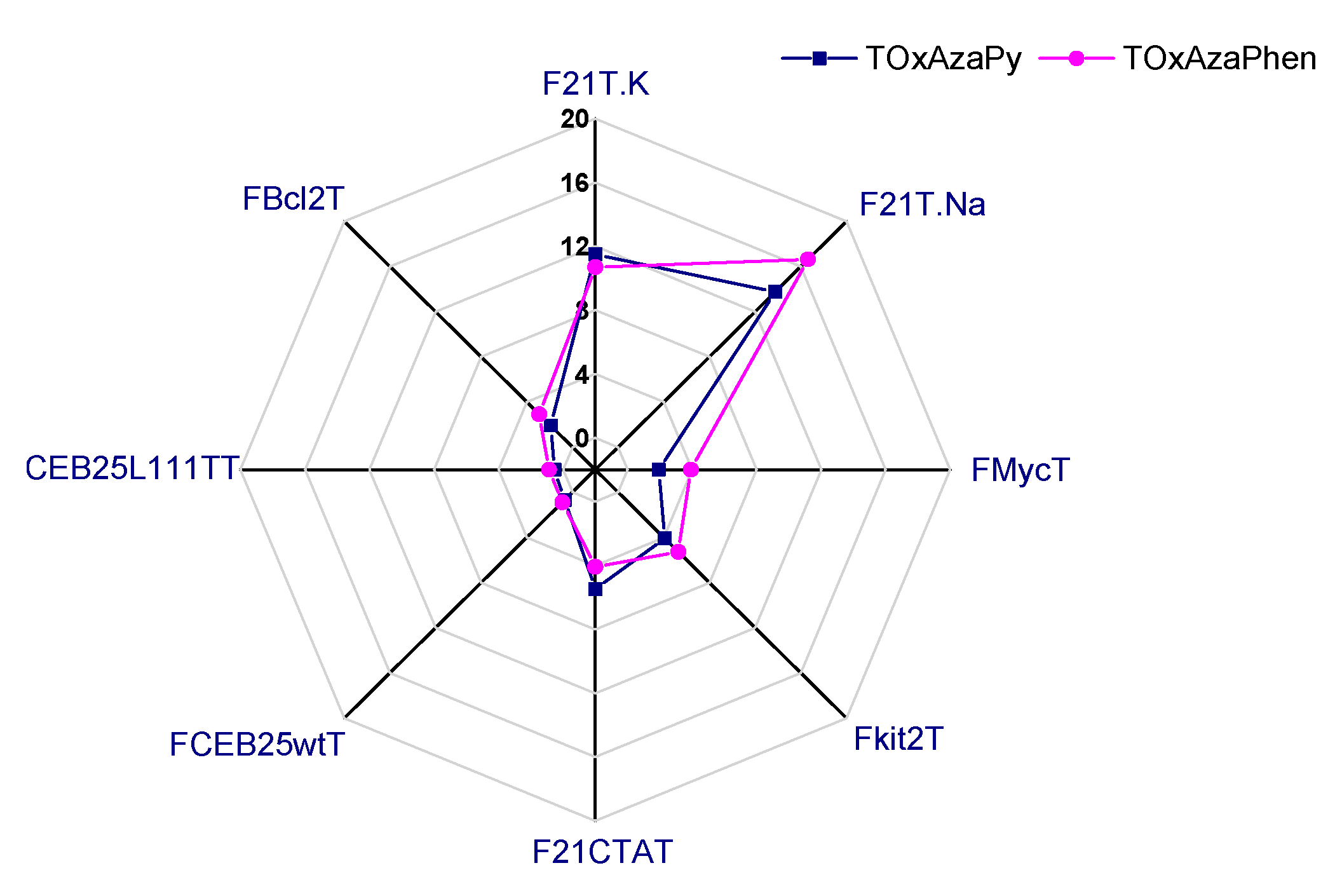
2.1.1. FRET-Melting
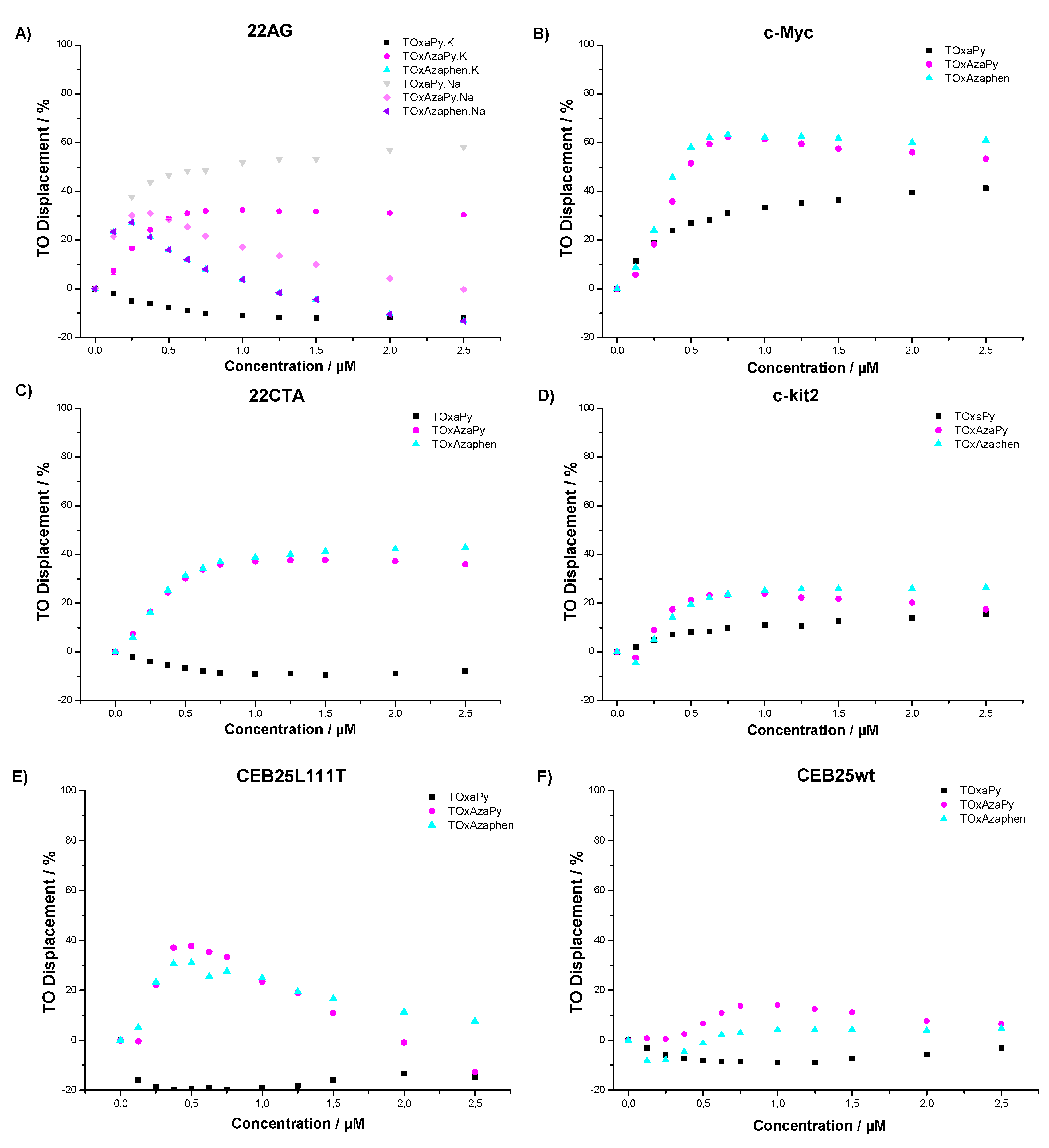
2.1.2. G4-FID
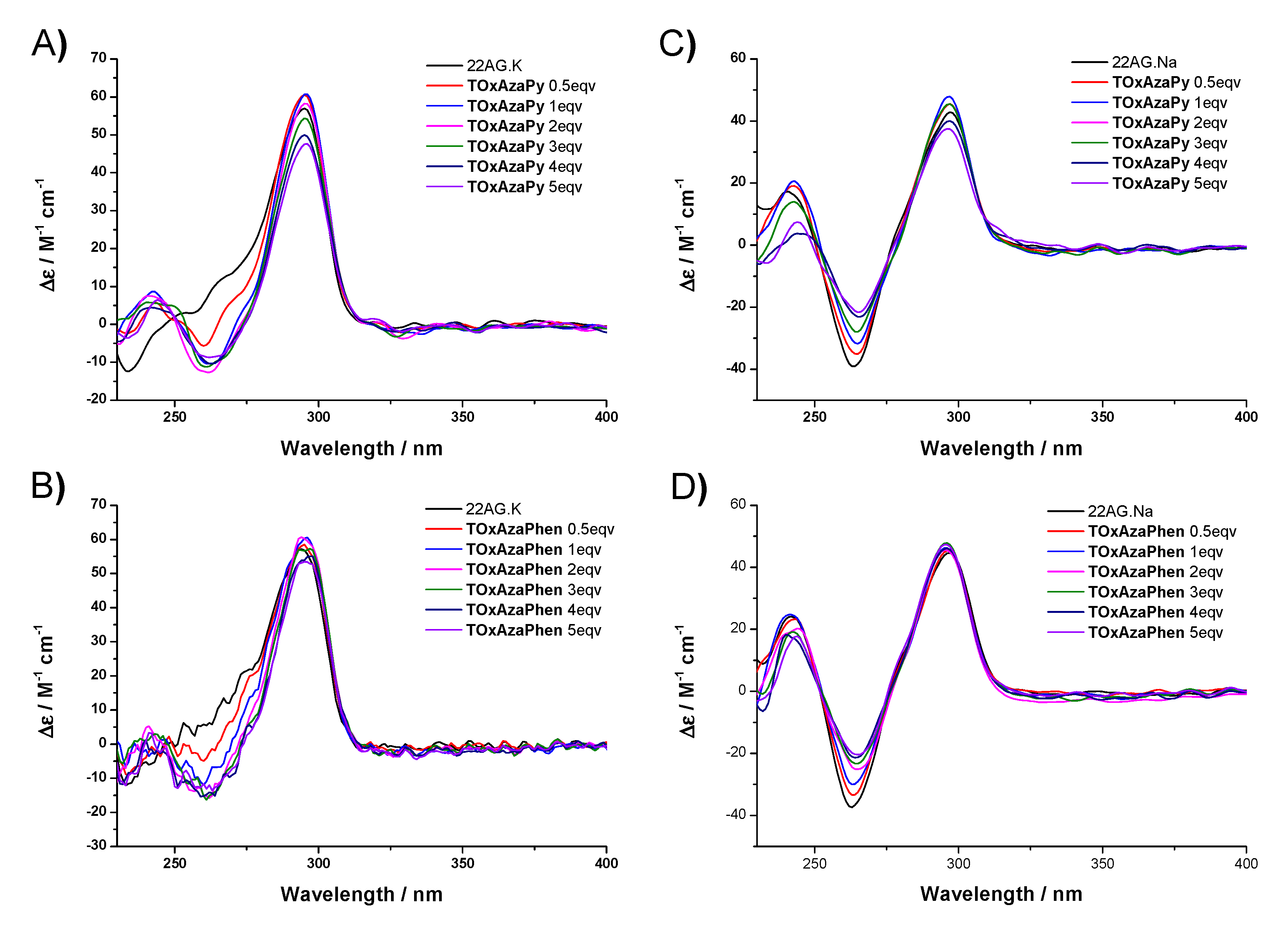
2.3. CD Titrations
3. Materials and Methods
3.1. General Information
3.2. Synthetic Methods
3.2.1. General Procedure for the Synthesis of 1–4
3.2.2. Stepwise Synthesis for (5)
3.2.3. Stepwise Synthesis of TOxAzaPy and TOxAzaPhen
3.2.4. General Procedure for the Synthesis of TOxAzaPy-Me (6) and TOxAzaPhen (7)
3.3. Biophysical Assays
3.3.1. FRET Melting Assay
3.3.2. G4-FID Assay
3.3.3. Circular Dichroism (CD) Titration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Neidle, S. The structures of quadruplex nucleic acids and their drug complexes. Curr. Opin. Struct. Biol. 2009, 19, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.D.; Sugiyama, H. First international meeting on quadruplex DNA. ACS Chem. Biol. 2007, 2, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.M.; Payet, L.; Huppert, J.L. Function and targeting of g-quadruplexes. Curr. Opin. Mol. Ther. 2009, 11, 146–155. [Google Scholar] [PubMed]
- Huppert, J.L. Structure, location and interactions of G-quadruplexes. FEBS J. 2010, 277, 3452–3458. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution structure of the biologically relevant g-quadruplex element in the human C-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Lipps, H.J.; Rhodes, D. G-quadruplex structures: In vivo evidence and function. Trends Cell Biol. 2009, 19, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Maizels, N. G4 motifs in human genes. Ann. N. Y. Acad. Sci. 2012, 1267, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Maizels, N.; Gray, L.T. The G4 genome. PLoS Genet. 2013, 9, e1003468. [Google Scholar] [CrossRef] [PubMed]
- Collie, G.W.; Parkinson, G.N. The application of DNA and RNA G-quadruplexes to therapeutic medicines. Chem. Soc. Rev. 2011, 40, 5867–5892. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- McLuckie, K.I.; Waller, Z.A.; Sanders, D.A.; Alves, D.; Rodriguez, R.; Dash, J.; McKenzie, G.J.; Venkitaraman, A.R.; Balasubramanian, S. G-quadruplex-binding benzo[a]phenoxazines down-regulate c-kit expression in human gastric carcinoma cells. J. Am. Chem. Soc. 2011, 133, 2658–2663. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.V.; Danford, F.L.; Gokhale, V.; Hurley, L.H.; Brooks, T.A. Demonstration that drug-targeted down-regulation of myc in non-hodgkins lymphoma is directly mediated through the promoter g-quadruplex. J. Biol. Chem. 2011, 286, 41018–41027. [Google Scholar] [CrossRef] [PubMed]
- Ohnmacht, S.A.; Micco, M.; Petrucci, V.; Todd, A.K.; Reszka, A.P.; Gunaratnam, M.; Carvalho, M.A.; Zloh, M.; Neidle, S. Sequences in the HSP90 promoter form g-quadruplex structures with selectivity for disubstituted phenyl bis-oxazole derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 5930–5935. [Google Scholar] [CrossRef] [PubMed]
- Arevalo-Ruiz, M.; Doria, F.; Belmonte-Reche, E.; De Rache, A.; Campos-Salinas, J.; Lucas, R.; Falomir, E.; Carda, M.; Perez-Victoria, J.M.; Mergny, J.L.; et al. Synthesis, binding properties, and differences in cell uptake of g-quadruplex ligands based on carbohydrate naphthalene diimide conjugates. Chem. Eur. J. 2017, 23, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Nadai, M.; Cimino-Reale, G.; Sattin, G.; Doria, F.; Butovskaya, E.; Zaffaroni, N.; Freccero, M.; Palumbo, M.; Richter, S.N.; Folini, M. Assessment of gene promoter gquadruplex binding and modulation by a naphthalene diimide derivative in tumor cells. Int. J. Oncol. 2015, 46, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Salvati, E.; Doria, F.; Manoli, F.; D’Angelo, C.; Biroccio, A.; Freccero, M.; Manet, I. A bimodal fluorescent and photocytotoxic naphthalene diimide for theranostic applications. Org. Biomol. Chem. 2016, 14, 7238–7249. [Google Scholar] [CrossRef] [PubMed]
- Monchaud, D.; Teulade-Fichou, M.P. A hitchhiker’s guide to g-quadruplex ligands. Org. Biomol. Chem. 2008, 6, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Temime-Smaali, N.; Guittat, L.; Sidibe, A.; Shin-ya, K.; Trentesaux, C.; Riou, J.F. The g-quadruplex ligand telomestatin impairs binding of topoisomerase iiialpha to g-quadruplex-forming oligonucleotides and uncaps telomeres in alt cells. PLoS ONE 2009, 4, e6919. [Google Scholar] [CrossRef] [PubMed]
- Tera, M.; Ishizuka, H.; Takagi, M.; Suganuma, M.; Shin-ya, K.; Nagasawa, K. Macrocyclic hexaoxazoles as sequence- and mode-selective g-quadruplex binders. Angew. Chem. Int. Ed. 2008, 47, 5557–5560. [Google Scholar] [CrossRef] [PubMed]
- Rzuczek, S.G.; Pilch, D.S.; Liu, A.; Liu, L.; LaVoie, E.J.; Rice, J.E. Macrocyclic pyridyl polyoxazoles: Selective rna and DNA g-quadruplex ligands as antitumor agents. J. Med. Chem. 2010, 53, 3632–3644. [Google Scholar] [CrossRef] [PubMed]
- Hamon, F.; Largy, E.; Guedin-Beaurepaire, A.; Rouchon-Dagois, M.; Sidibe, A.; Monchaud, D.; Mergny, J.L.; Riou, J.F.; Nguyen, C.H.; Teulade-Fichou, M.P. An acyclic oligoheteroaryle that discriminates strongly between diverse g-quadruplex topologies. Angew. Chem. Int. Ed. 2011, 50, 8745–8749. [Google Scholar] [CrossRef] [PubMed]
- Petenzi, M.; Verga, D.; Largy, E.; Hamon, F.; Doria, F.; Teulade-Fichou, M.P.; Guedin, A.; Mergny, J.L.; Mella, M.; Freccero, M. Cationic pentaheteroaryls as selective g-quadruplex ligands by solvent-free microwave-assisted synthesis. Chem. Eur. J. 2012, 18, 14487–14496. [Google Scholar] [CrossRef] [PubMed]
- Rizeq, N.; Georgiades, S.N. Investigation of ‘head-to-tail’-connected oligoaryl N,O-ligands as recognition motifs for cancer-relevant g-quadruplexes. Molecules 2017, 22, 2160. [Google Scholar] [CrossRef] [PubMed]
- Rizeq, N.; Georgiades, S.N. Linear and branched pyridyl–oxazole oligomers: Synthesis and circular dichroism detectable effect on c-myc g-quadruplex helicity. Eur. J. Org. Chem. 2016, 122–131. [Google Scholar] [CrossRef]
- Medeiros-Silva, J.; Guedin, A.; Salgado, G.F.; Mergny, J.L.; Queiroz, J.A.; Cabrita, E.J.; Cruz, C. Phenanthroline-bis-oxazole ligands for binding and stabilization of g-quadruplexes. Biochim. Biophys. Acta 2017, 1861, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- De Cian, A.; Guittat, L.; Kaiser, M.; Sacca, B.; Amrane, S.; Bourdoncle, A.; Alberti, P.; Teulade-Fichou, M.P.; Lacroix, L.; Mergny, J.L. Fluorescence-based melting assays for studying quadruplex ligands. Methods 2007, 42, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Kuryavyi, V.; Phan, A.T.; Patel, D.J. Solution structures of all parallel-stranded monomeric and dimeric g-quadruplex scaffolds of the human C-KIT2 promoter. Nucleic Acids Res. 2010, 38, 6757–6773. [Google Scholar] [CrossRef] [PubMed]
- Amrane, S.; Adrian, M.; Heddi, B.; Serero, A.; Nicolas, A.; Mergny, J.L.; Phan, A.T. Formation of pearl-necklace monomorphic g-quadruplexes in the human CEB25 minisatellite. J. Am. Chem. Soc. 2012, 134, 5807–5816. [Google Scholar] [CrossRef] [PubMed]
- Piazza, A.; Adrian, M.; Samazan, F.; Heddi, B.; Hamon, F.; Serero, A.; Lopes, J.; Teulade-Fichou, M.P.; Phan, A.T.; Nicolas, A. Short loop length and high thermal stability determine genomic instability induced by g-quadruplex-forming minisatellites. EMBO J. 2015, 34, 1718–1734. [Google Scholar] [CrossRef] [PubMed]
- Onyshchenko, M.I.; Gaynutdinov, T.I.; Englund, E.A.; Appella, D.H.; Neumann, R.D.; Panyutin, I.G. Stabilization of G-quadruplex in the BCL2 promoter region in double-stranded DNA by invading short pnas. Nucleic Acids Res. 2009, 37, 7570–7580. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.W.; Alberti, P.; Guedin, A.; Lacroix, L.; Riou, J.F.; Royle, N.J.; Mergny, J.L.; Phan, A.T. Sequence variant (CTAGGG)N in the human telomere favors a g-quadruplex structure containing a G.C.G.C tetrad. Nucleic Acids Res. 2009, 37, 6239–6248. [Google Scholar] [CrossRef] [PubMed]
- Viglasky, V.; Bauer, L.; Tluckova, K. Structural features of intra- and intermolecular G-quadruplexes derived from telomeric repeats. Biochemistry 2010, 49, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.S.; Brooks, S.C.; Graves, D.E. Interactions of actinomycin d with human telomeric g-quadruplex DNA. Biochemistry 2009, 48, 4440–4447. [Google Scholar] [CrossRef] [PubMed]
- Renciuk, D.; Kejnovska, I.; Skolakova, P.; Bednarova, K.; Motlova, J.; Vorlickova, M. Arrangements of human telomere DNA quadruplex in physiologically relevant K+ solutions. Nucleic Acids Res. 2009, 37, 6625–6634. [Google Scholar] [CrossRef] [PubMed]
- Gottarelli, G.; Lena, S.; Masiero, S.; Pieraccini, S.; Spada, G.P. The use of circular dichroism spectroscopy for studying the chiral molecular self-assembly: An overview. Chirality 2008, 20, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Marinelli, L.; Trotta, R.; Virno, A.; De Tito, S.; Romagnoli, R.; Pagano, B.; Limongelli, V.; Giancola, C.; Baraldi, P.G.; et al. Structural and conformational requisites in DNA quadruplex groove binding: Another piece to the puzzle. J. Am. Chem. Soc. 2010, 132, 6425–6433. [Google Scholar] [CrossRef] [PubMed]
- Balagurumoorthy, P.; Brahmachari, S.K. Structure and stability of human telomeric sequence. J. Biol. Chem. 1994, 269, 21858–21869. [Google Scholar] [PubMed]
- Largy, E.; Marchand, A.; Amrane, S.; Gabelica, V.; Mergny, J.L. Quadruplex turncoats: Cation-dependent folding and stability of quadruplex-DNA double switches. J. Am. Chem. Soc. 2016, 138, 2780–2792. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–7 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | Pathway | Compound | Yield (%) |
|---|---|---|---|
 | A | 1 | 11 |
 | A | 2 | 11 |
 | A | 3 | 28 |
 | A | 4 | 10 |
 | B | 5 | 53 |
| X | Compound | Yield (%) |
|---|---|---|
| N | 6 | 19 |
| C | 7 | 38 |
| G4 Sequences | Topology | TOxAzaPy ΔTm (°C) | TOxAzaPhen ΔTm (°C) |
|---|---|---|---|
| F21T.Na+ | Antiparallel [32] | 13.8 ± 0.4 | 16.6 ± 0.8 |
| F21T.K+ | Hybrid/mix of several topologies [32,33,34,35] | 11.5 ± 0.6 | 10.8 ± 0.4 |
| FMycT | parallel | 1.9 ± 0.1 | 3.9 ± 0.9 1 |
| Fkit2T | parallel | 4.1 ± 0.3 | 5.3 ± 0.4 2 |
| FCEB25wtT | parallel | 0.7 ± 0.9 | 0.9 ± 0.7 1 |
| FCEB25L111TT | parallel | 0.5 ± 0.3 | 0.9 ± 0.8 1 |
| FBcl2T | parallel | 1.9 ± 0.7 | 2.9 ± 0.7 2 |
| F21CTAT | antiparallel | 5.5 ± 0.2 | 4.1 ± 0.1 2 |
| Sequence Name | Sequence (5′-3′) |
|---|---|
| F21T | Fam-G3TTAG3TTAG3TTA G3-Tamra |
| FMycT | Fam-TTGAG3TG3TAG3TG3TAA-Tamra |
| F21CTAT | Fam-G3CTAG3CTAG3CTAG3-Tamra |
| FKit2T | Fam-G3CG3CGCGAG3AG4-Tamra |
| FCEB25wtT | Fam-AAG3TG3TGTAAGTGTG3TG3T-Tamra |
| FCEB25-L111TT | Fam-AAG3TG3TG3TG3T-Tamra |
| FCEB25-L121(AA)TT | Fam-AAG3TG3AAG3TG3T-Tamra |
| FCEB25-L121TT | Fam-AAG3TG3TTG3TG3T-Tamra |
| FBcl2T | Fam-AG4CG3CGCG3AG2AAG5CG3AGCG4CTG-Tamra |
| ds26 | CAATCGGATCGAATTCGATCCGATTG |
| Sequence Name | Sequence (5′-3′) |
|---|---|
| 22AG | AG3TTAG3TTAG3TTAG3 |
| Myc22 | TGAG3TG3TAG3TG3TAA |
| 22CTA | AG3CTAG3CTAG3CTAG3 |
| c-kit2 | G3CG3CGCGAG3AG4 |
| CEB25wt | AAG3TG3TGTAAGTGTG3TG3T |
| CEB25-L111T | AAG3TG3TG3TG3T |
| Bcl2 | AG4CG3CGCG3AG2AAG5CG3AGCG4CTG |
| ds26 | CAATCGGATCGAATTCGATCCGATTG |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doria, F.; Pirota, V.; Petenzi, M.; Teulade-Fichou, M.-P.; Verga, D.; Freccero, M. Oxadiazole/Pyridine-Based Ligands: A Structural Tuning for Enhancing G-Quadruplex Binding. Molecules 2018, 23, 2162. https://doi.org/10.3390/molecules23092162
Doria F, Pirota V, Petenzi M, Teulade-Fichou M-P, Verga D, Freccero M. Oxadiazole/Pyridine-Based Ligands: A Structural Tuning for Enhancing G-Quadruplex Binding. Molecules. 2018; 23(9):2162. https://doi.org/10.3390/molecules23092162
Chicago/Turabian StyleDoria, Filippo, Valentina Pirota, Michele Petenzi, Marie-Paule Teulade-Fichou, Daniela Verga, and Mauro Freccero. 2018. "Oxadiazole/Pyridine-Based Ligands: A Structural Tuning for Enhancing G-Quadruplex Binding" Molecules 23, no. 9: 2162. https://doi.org/10.3390/molecules23092162
APA StyleDoria, F., Pirota, V., Petenzi, M., Teulade-Fichou, M.-P., Verga, D., & Freccero, M. (2018). Oxadiazole/Pyridine-Based Ligands: A Structural Tuning for Enhancing G-Quadruplex Binding. Molecules, 23(9), 2162. https://doi.org/10.3390/molecules23092162