
Diverse Derivatives of Selenoureas: A Synthetic and Single Crystal Structural Study




Abstract
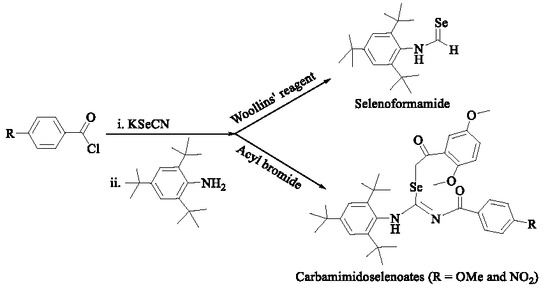
1. Introduction
2. Results and Discussion
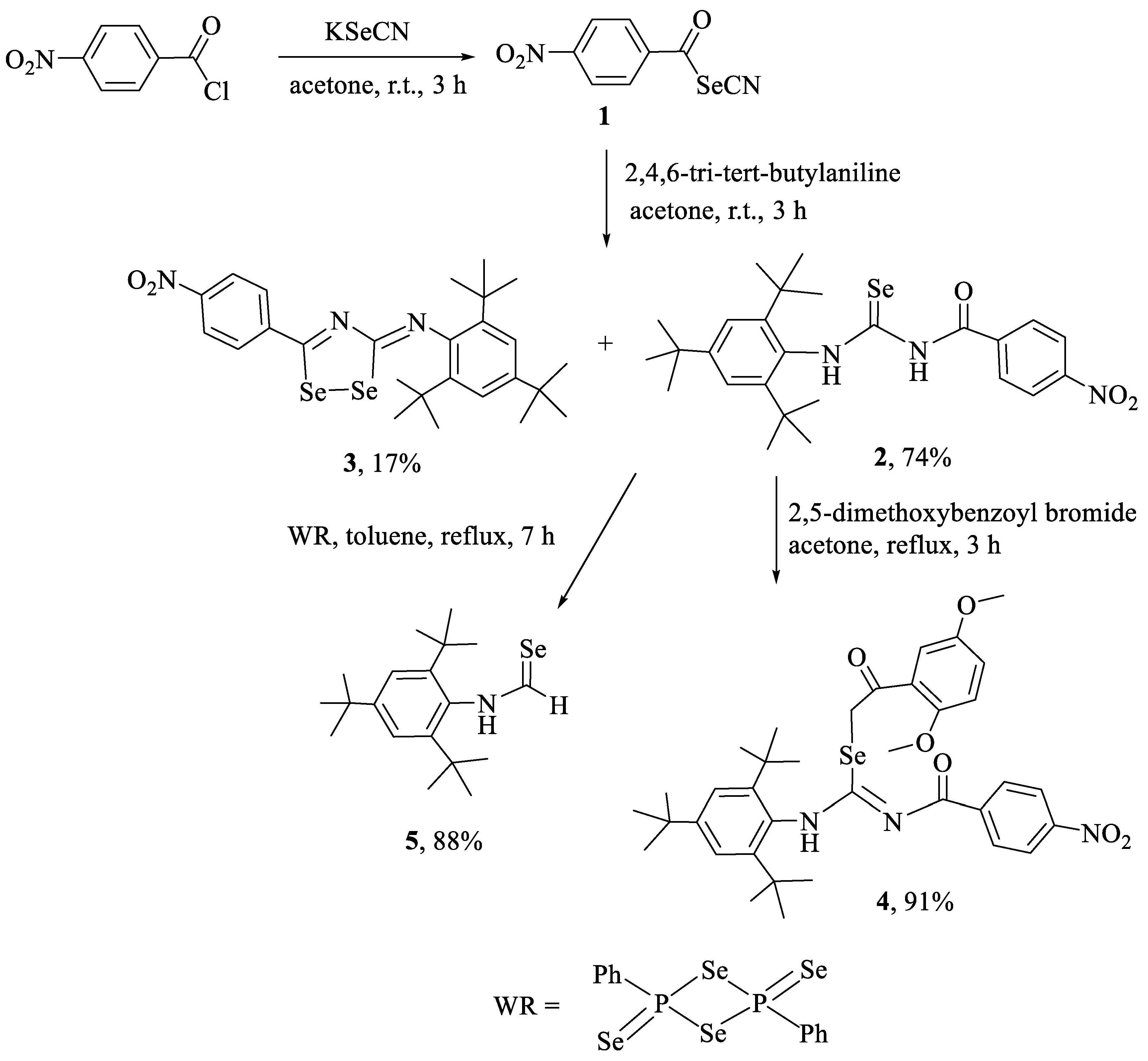
2.1. Synthesis and Characterization
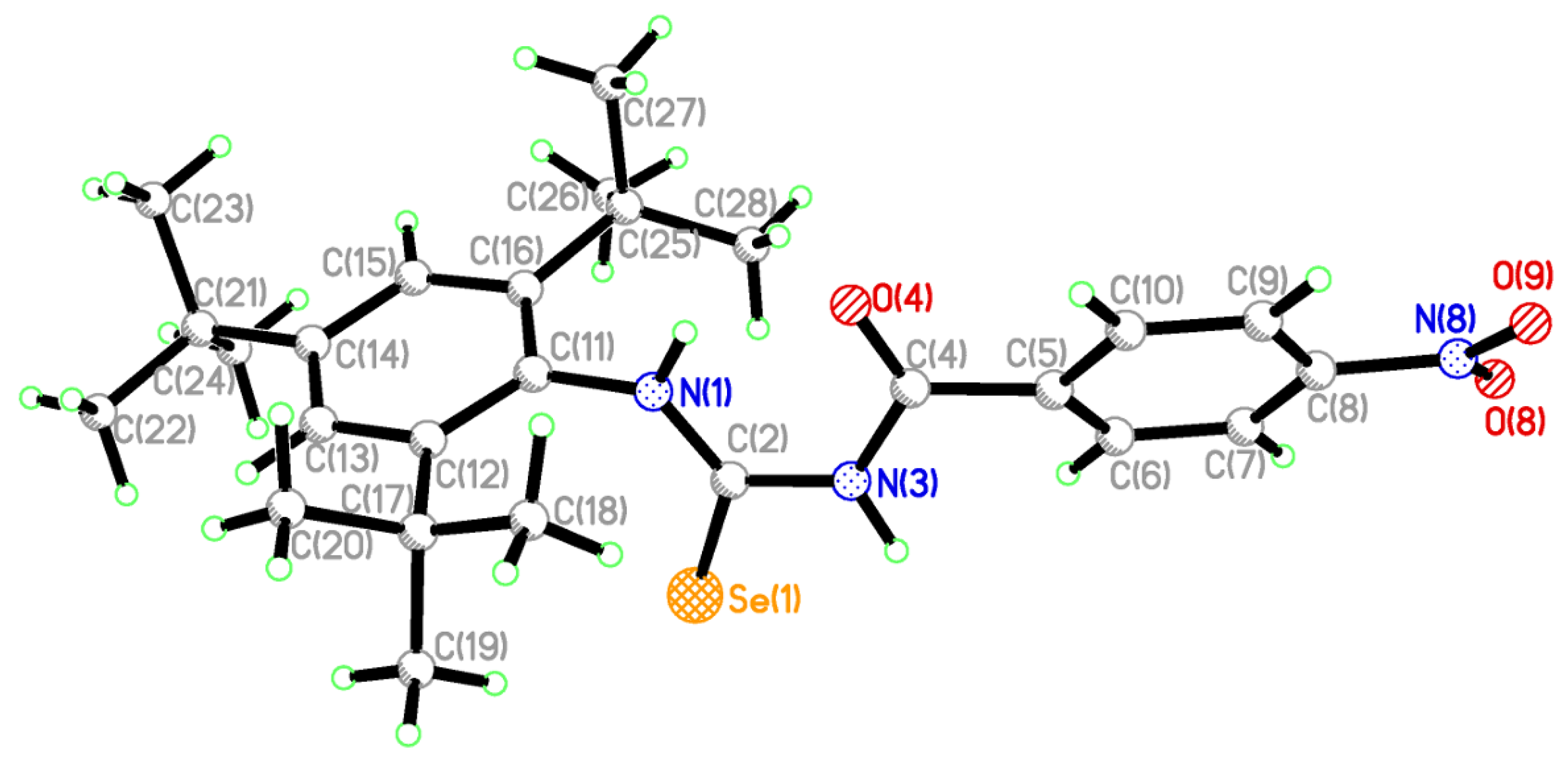
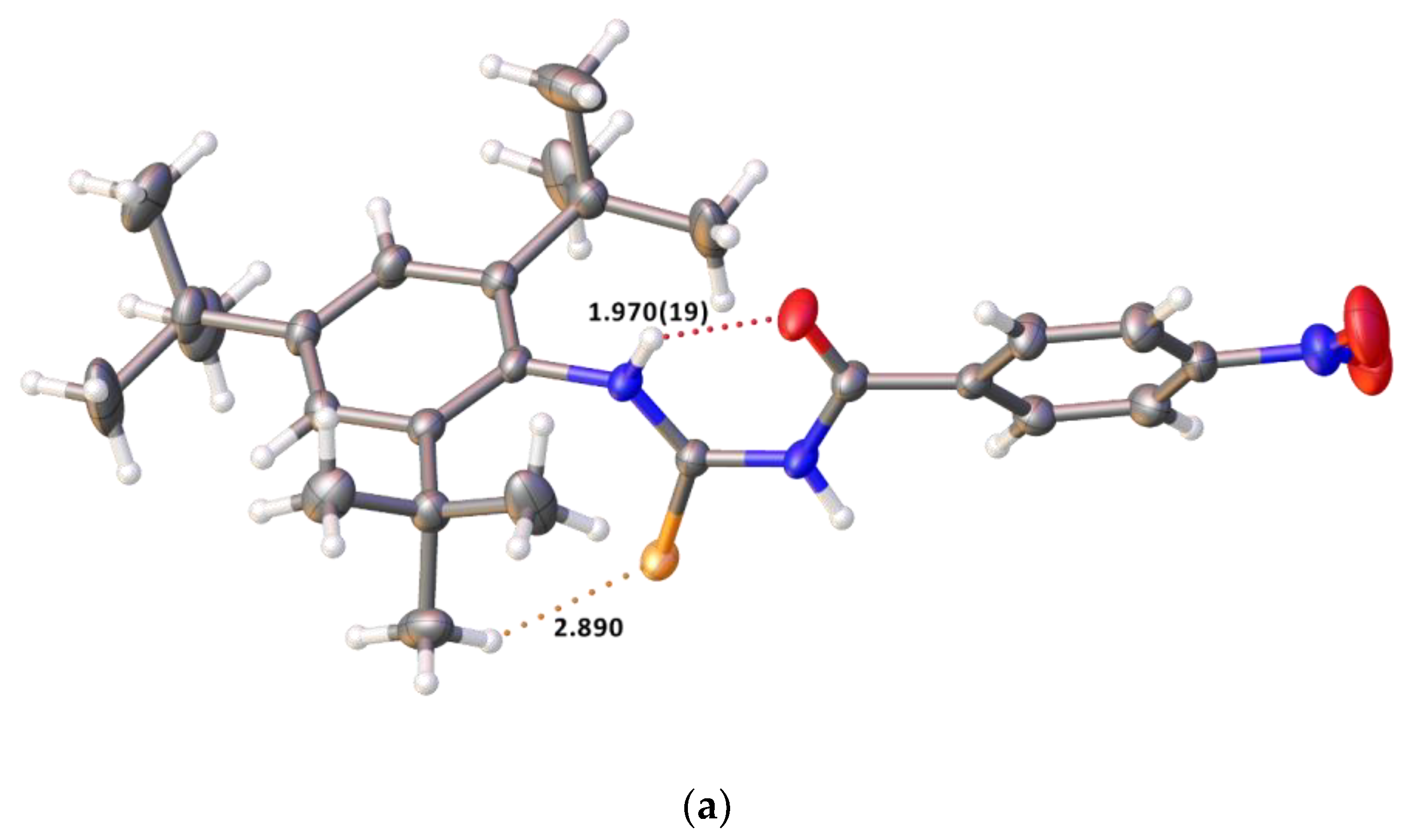
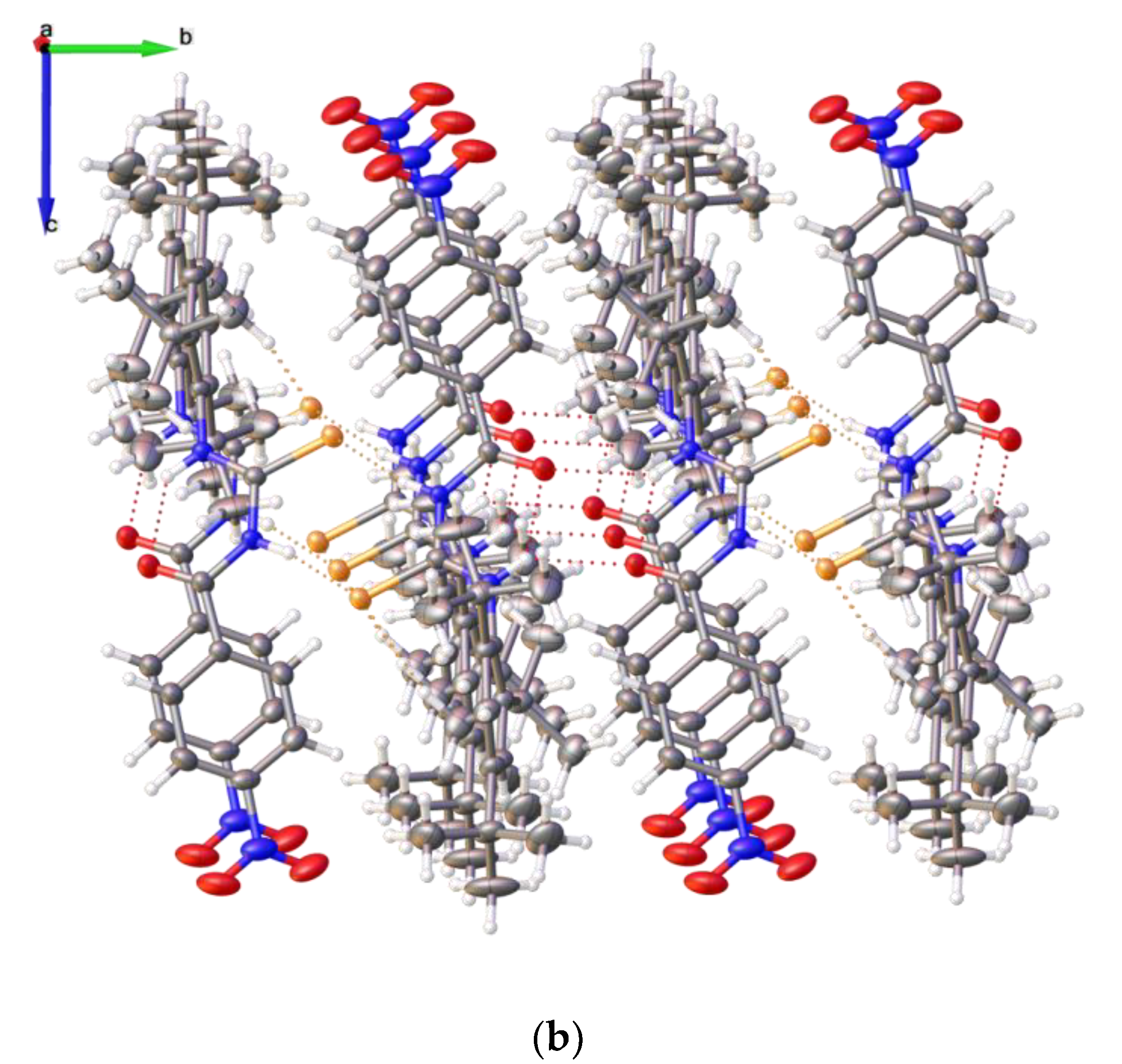
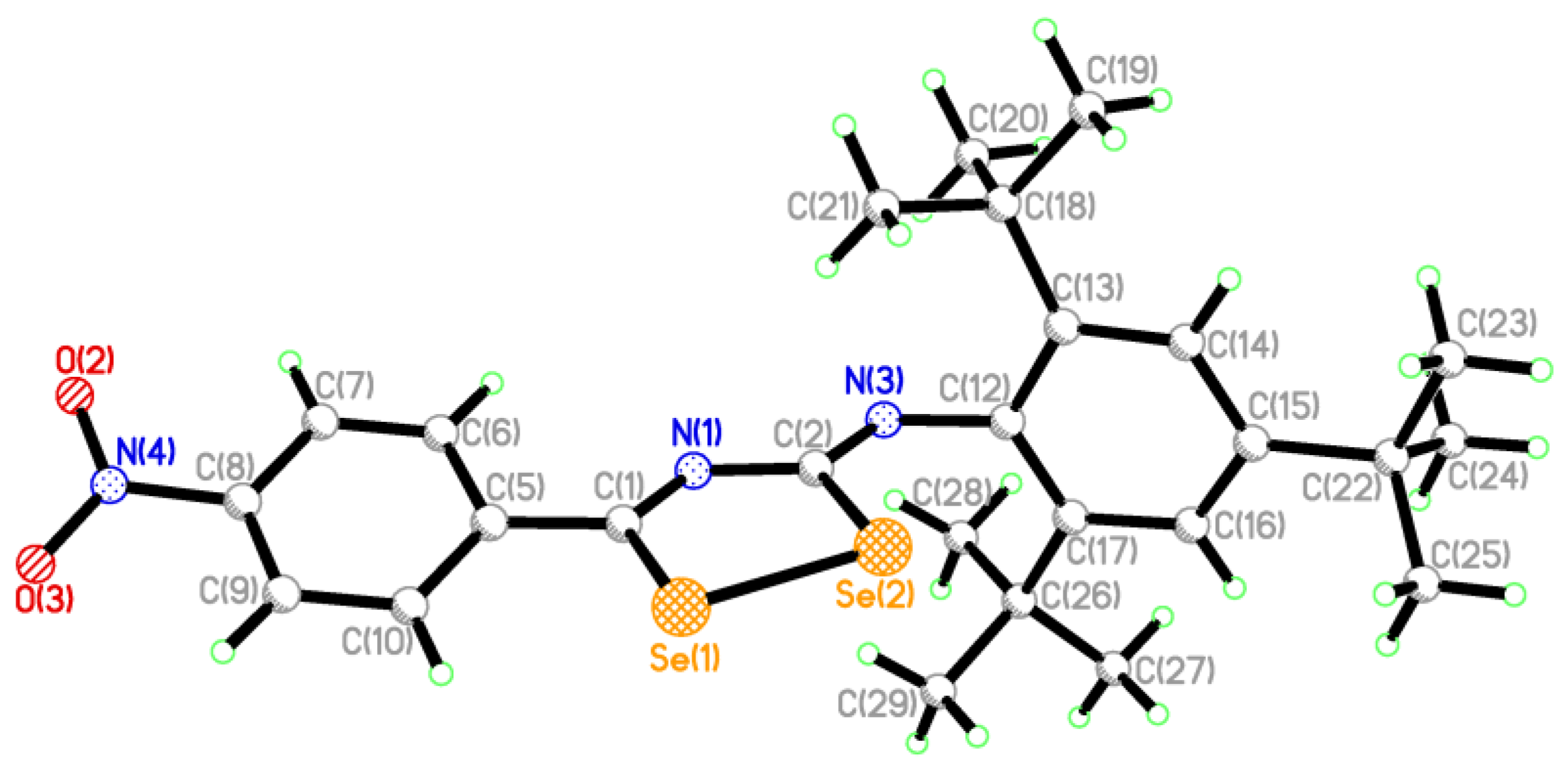
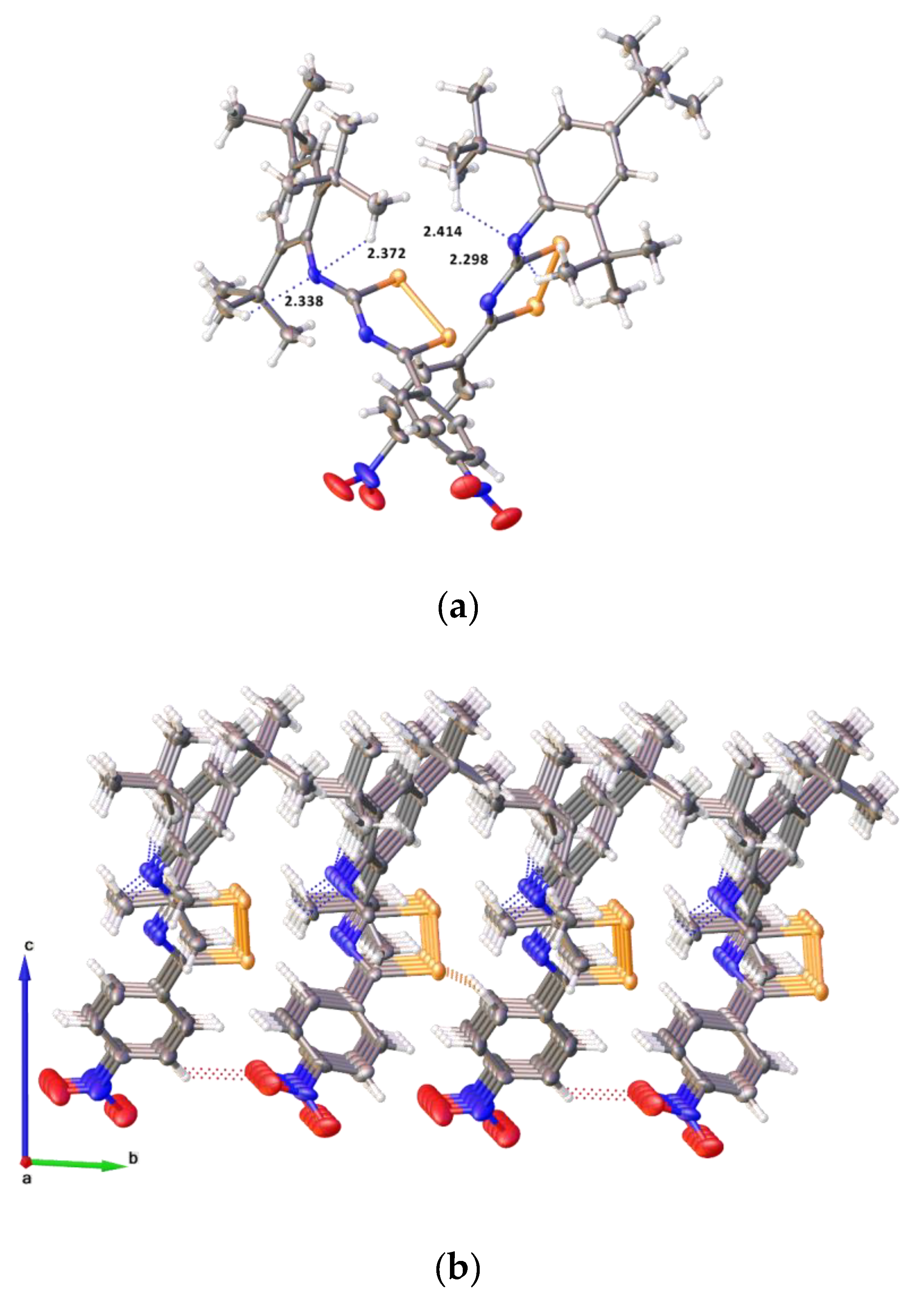
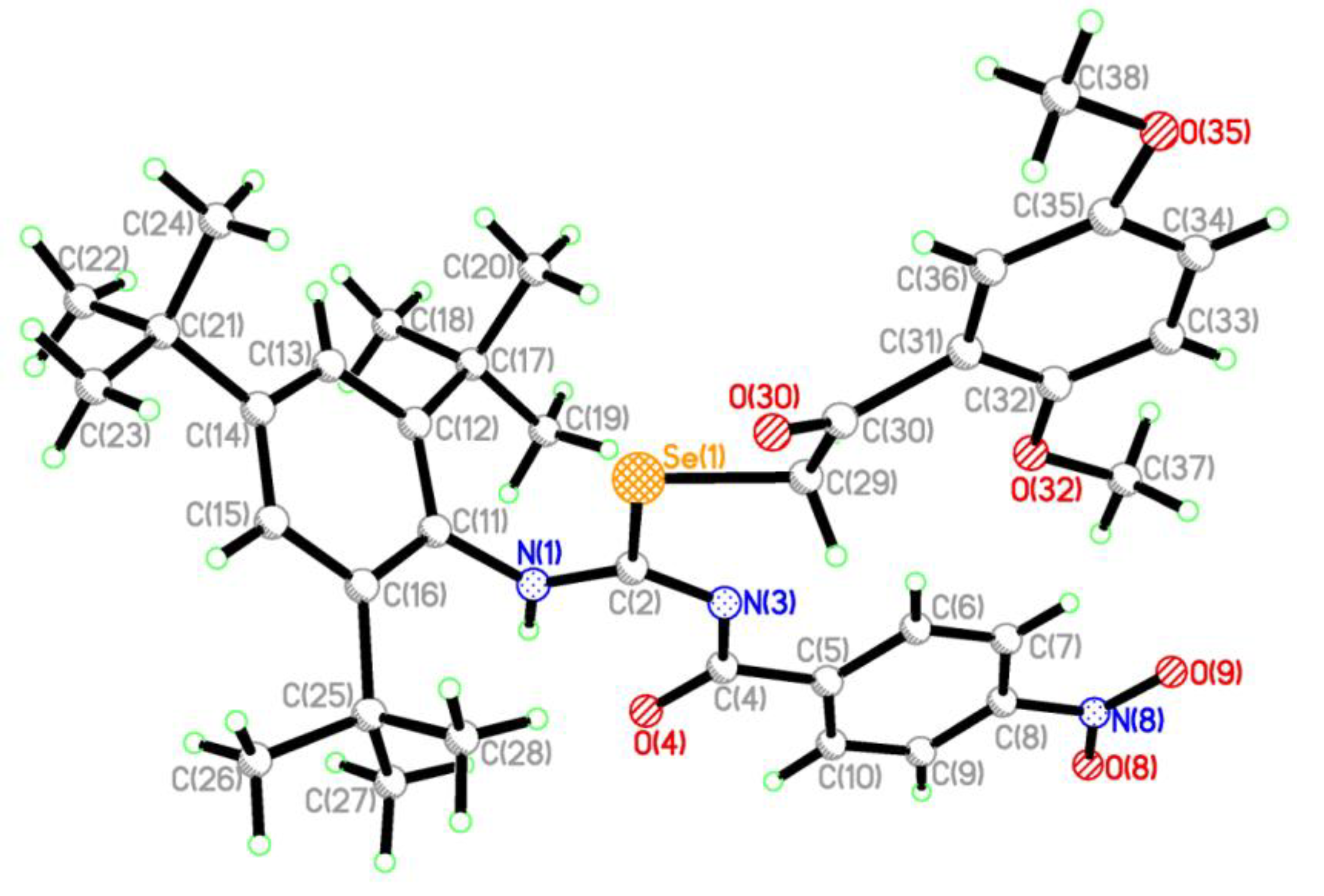
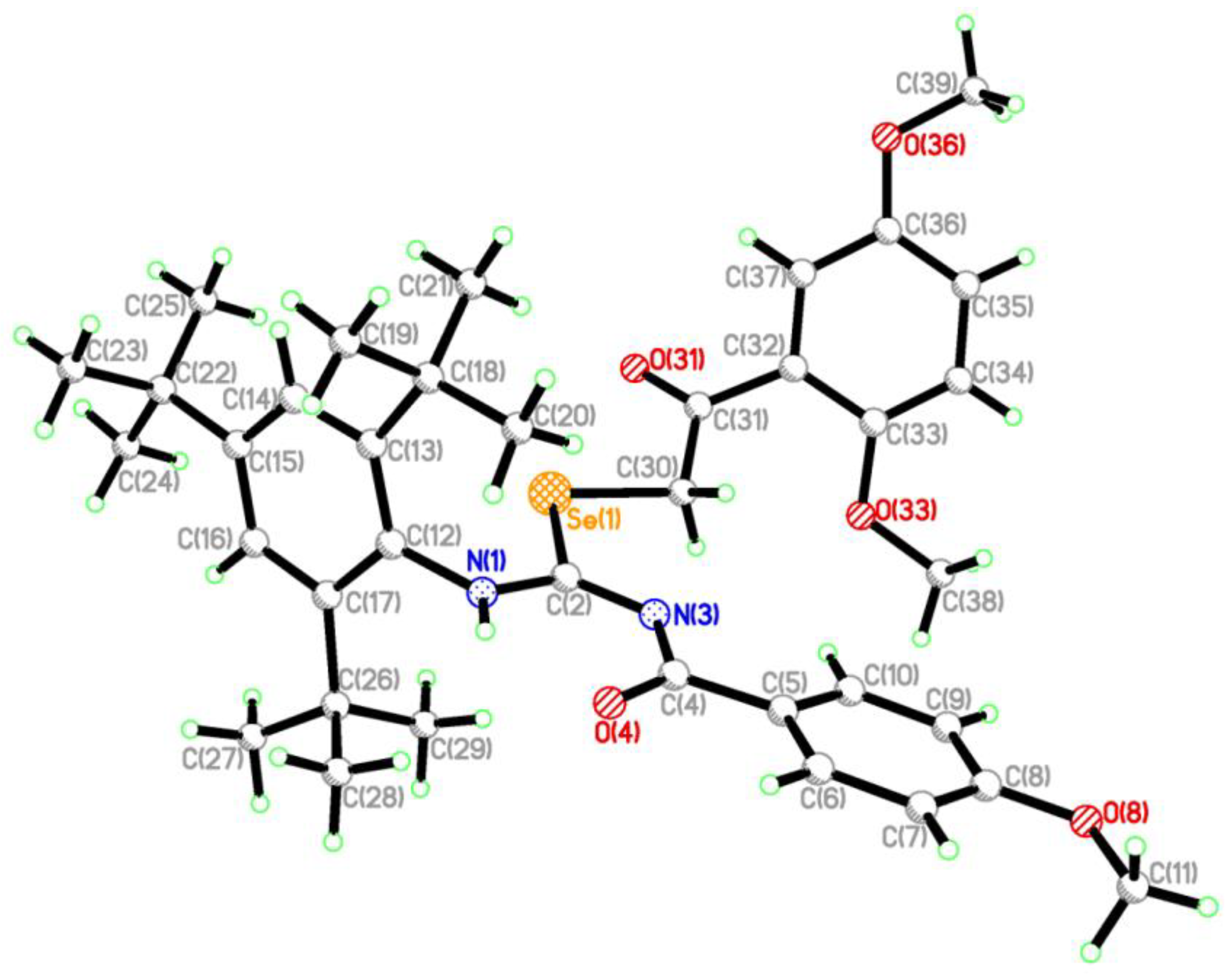
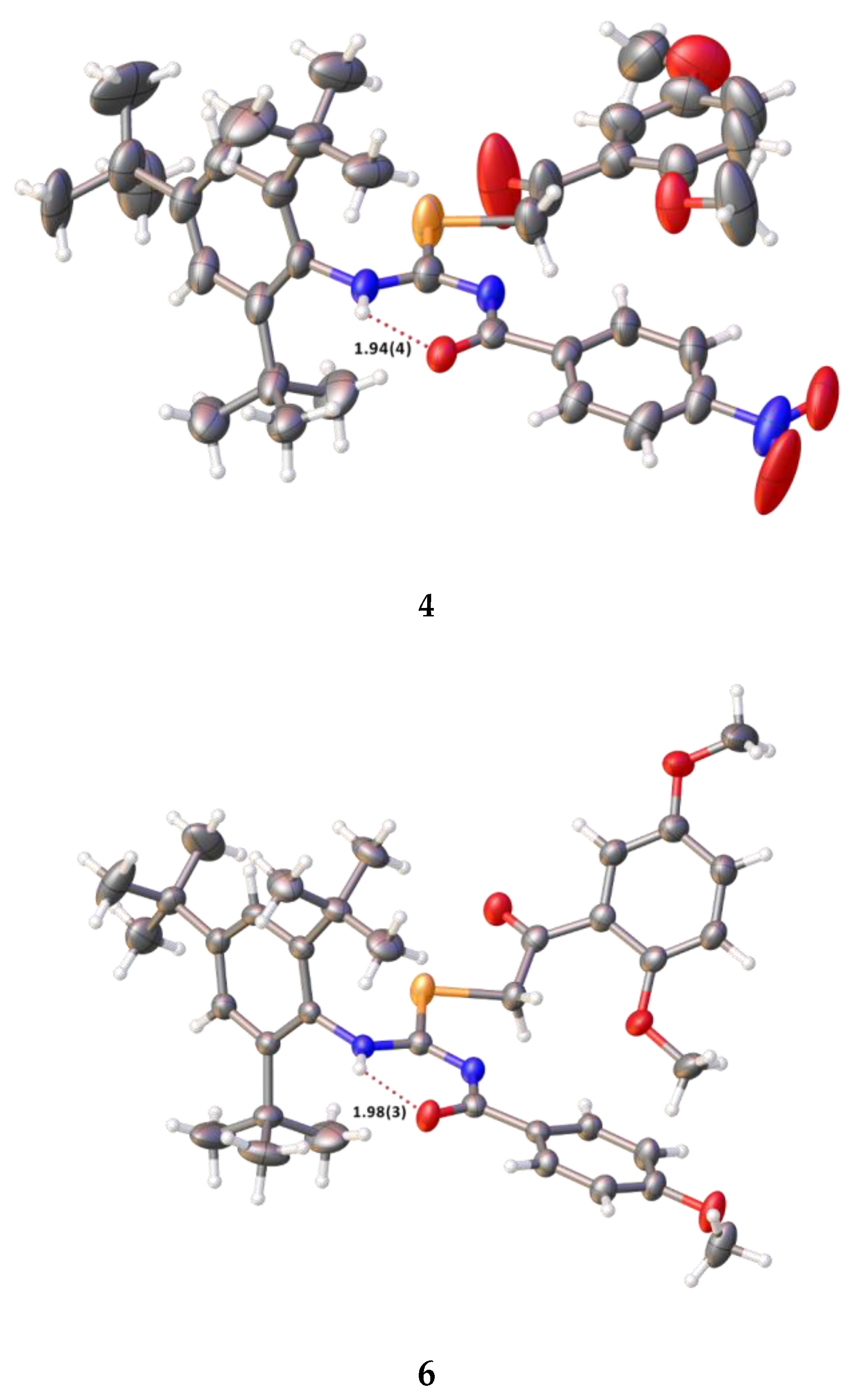
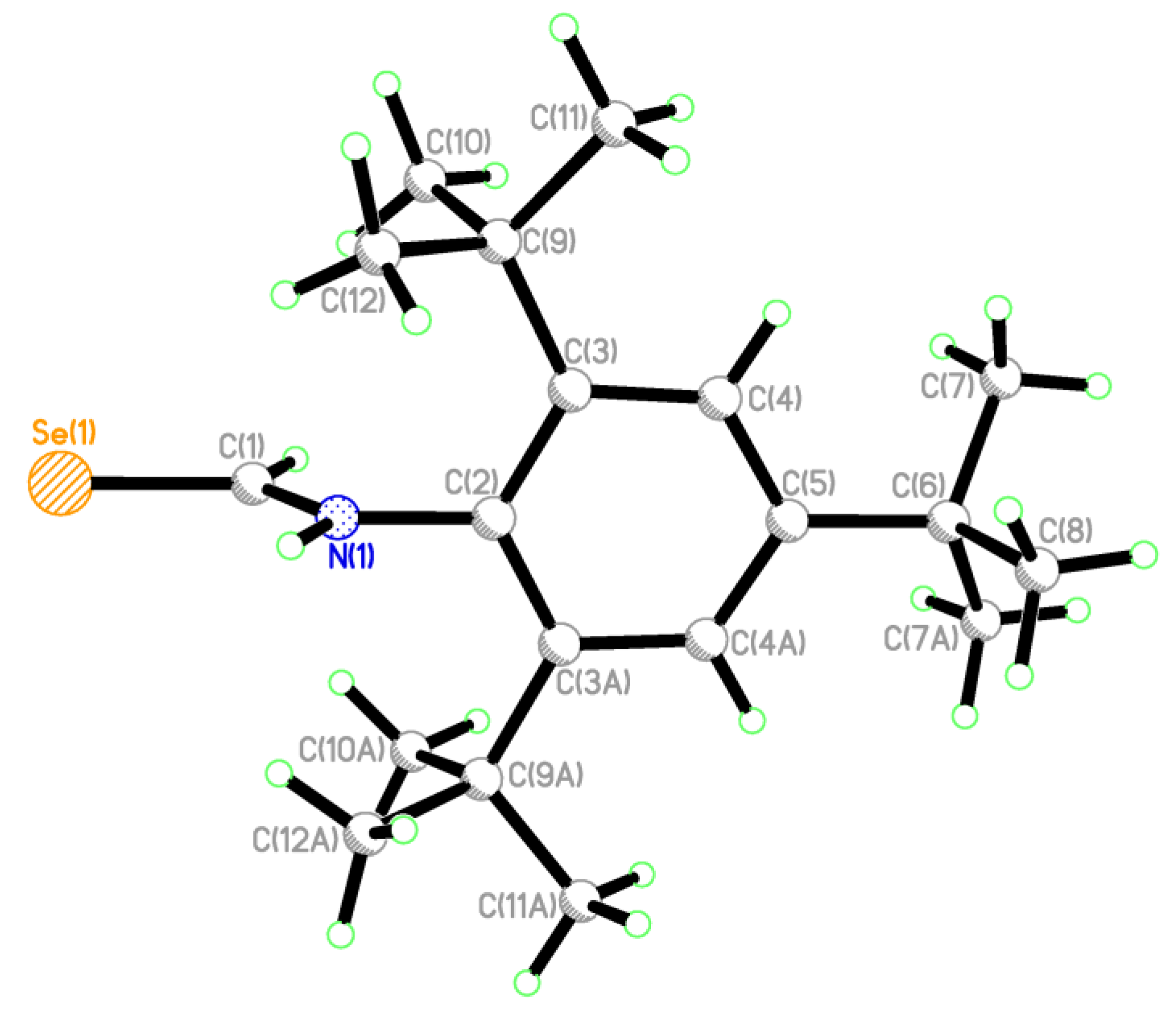
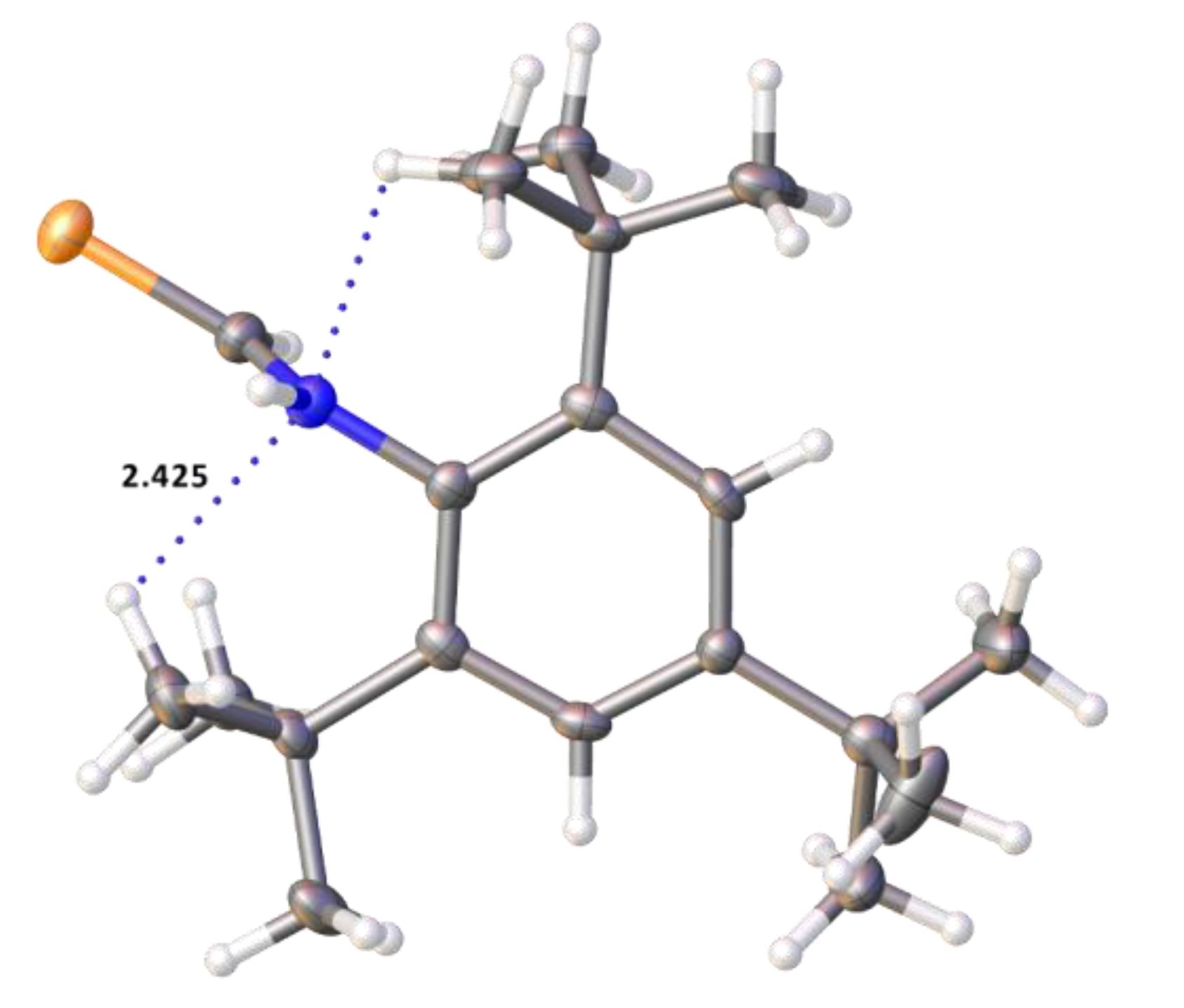
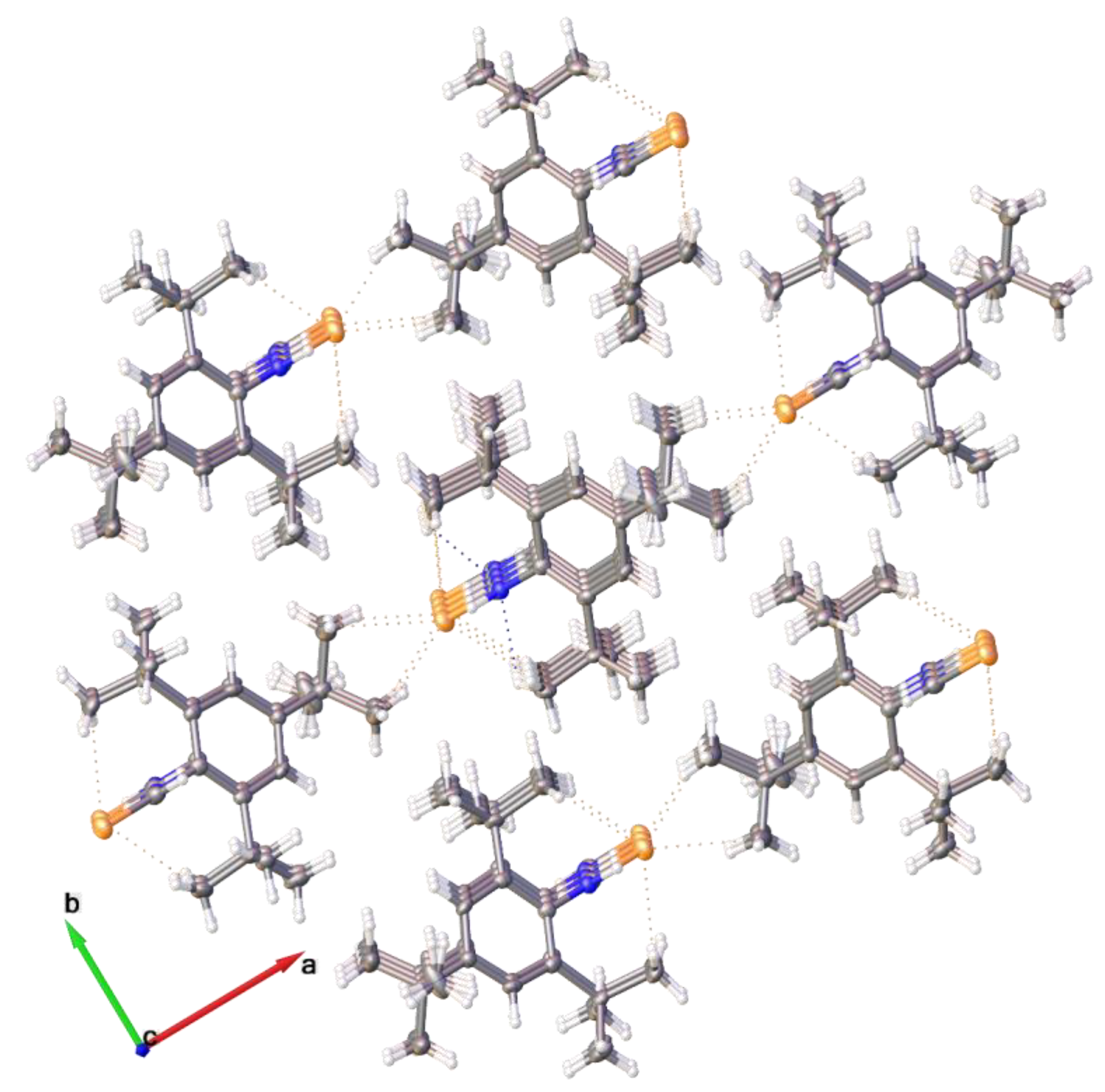
2.2. Single Crystal Structure Analysis
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. Synthesis of Compounds 2 and 3
3.2.2. Synthesis of 2-(2,5-dimethoxyphenyl)-2-oxoethyl-N′-(4-nitrobenzoyl)-N-(2,4,6-tri-tert-butylphenyl)-carbamimidoselenoate (4)
3.2.3. Synthesis of 2-(2,5-dimethoxyphenyl)-2-oxoethyl-N′-(4-methoxybenzoyl)-N-(2,4,6-tri-tert-butylphenyl)-carbamimidoselenoate (6)
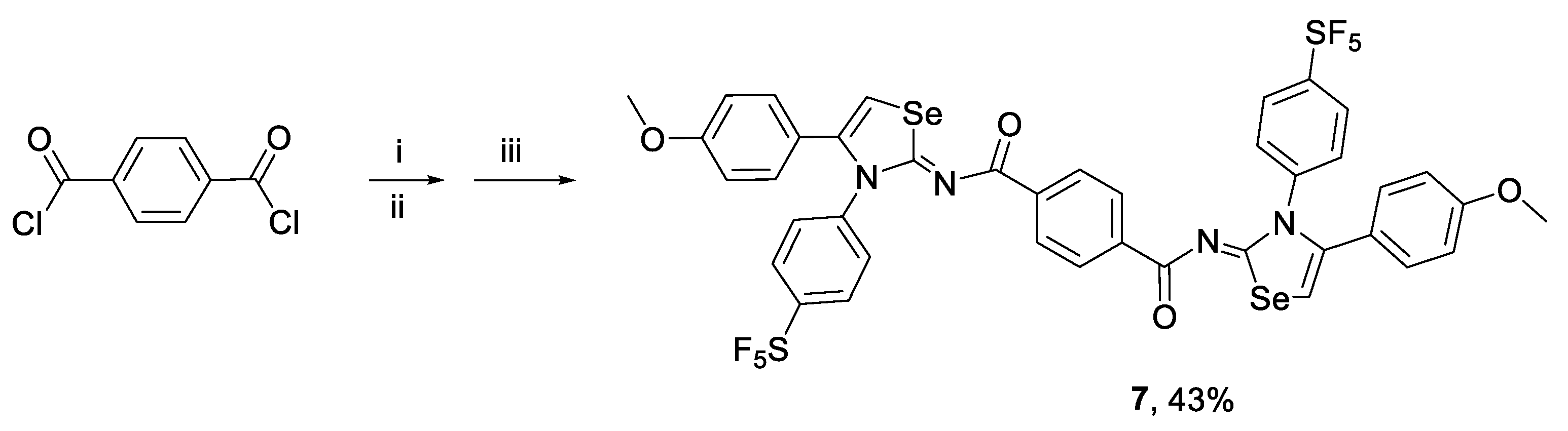
3.2.4. Synthesis of 1,4-bis[N-(4-(4-methoxyphenyl)-3-(4-(pentafluoro-l6-sulfanyl)phenyl)-1,3-selenazol-2(3H)-ylidene)]terephthalamide (7)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garud, D.R.; Koketsu, M.; Ishihara, H. Isoselenocyanates: A power tool for the synthesis of selenium-containing heterocycles. Molecules 2007, 12, 504–535. [Google Scholar] [CrossRef] [PubMed]
- Heimgartner, H.; Zhou, Y.; Atanassov, P.K.; Sommen, G.L. Isoselenocyantates as building blocks for selenium-containing heterocycles. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 840–855. [Google Scholar] [CrossRef]
- Ninomiya, M.; Garud, D.R.; Koketsu, M. Selenium-containing heterocycles using selenoamides, selenoureas, selenazadienes, and isoselenocyanates. Heterocycles 2010, 81, 2027–2055. [Google Scholar]
- Barton, D.H.R.; Parekh, S.I.; Tajbakhsh, M.; Theodorakis, E.A.; Tse, C.L. A convenient and high yielding procedure for the preparation of isoselenocyanates. Synthesis and reactivity of O-alkylselenocarbamates. Tetrahedron 1994, 50, 639–654. [Google Scholar] [CrossRef]
- Atanassov, P.K.; Zhou, Y.; Linden, A. Heimgartner, H. Synthesis of bis(2,4-diarylimidazol-5-yl) diselennides from N-benzylbenzidoyl isoselenocyanates. Helv. Chim. Acta 2002, 85, 1102–1117. [Google Scholar] [CrossRef]
- Zakrzewski, J.; Krawczyk, M. Synthesis and pesticidal properties of thio and seleno analogs of some urea herbicides. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 1880–1903. [Google Scholar] [CrossRef]
- Koketsu, M.; Takahashi, A.; Ishihara, H. A facile preparation of selenohydantoins using isoselenocyanate. J. Heterocycl. Chem. 2007, 44, 79–81. [Google Scholar] [CrossRef]
- Köhler, R.; Beyer, L.; Moll, M.; Hantschmann, A.; Richter, R.; Sieler, J.; Szargan, R.; Weber, L.; Hoyer, E. 2-Benzoylamino-5-dimethylamino-1,6-6aλ4-triselena-3,4-diazapentalene. Tetrahedron 1990, 46, 7735–7738. [Google Scholar]
- Zhou, Y.; Heimgartner, H. Selenium-containing heterocycles from isoselenocyanates: synthesis of 1,2,3-selenadiazole derivatives. Helv. Chim. Acta 2000, 83, 539–553. [Google Scholar] [CrossRef]
- L’abbé, G.; Dekerk, J.P.; Martens, C.; Toppet, S. Chemistry of N-sulfonyl-substituted thiiranimines. J. Org. Chem. 1980, 45, 4366–4371. [Google Scholar] [CrossRef]
- Koketsu, M.; Suzuki, N.; Ishihara, H. Preparation of isoselenocyanate and synthesis of carbodiimide by oxidation of selenourea. J. Org. Chem. 1999, 64, 6473–6475. [Google Scholar] [CrossRef]
- Fujiwara, S.I.; Kambe, N.; Sonoda, N. Organoselenium Chemistry: A Practical Approach; Back, T.G., Ed.; Oxford University Press: London, UK, 1999; pp. 223–240. [Google Scholar]
- Koketsu, M.; Ishihara, H. Thioureas and selenoureas and their applications. Curr. Org. Synth. 2006, 3, 439–455. [Google Scholar] [CrossRef]
- Koketsu, M.; Yamamura, Y.; Ishihara, H. Synthesis of selenosemicarbazides and 1,2,4-triazoles. Heterocycles 2006, 68, 1191–1200. [Google Scholar] [CrossRef]
- Koketsu, M.; Yamamura, Y.; Ando, H.; Ishihara, H. Synthesis of 1,3-selenazetidines and 4H-1,3,5-oxadiazines using acyl isoselenocyanates. Heterocycles 2006, 68, 1267–1273. [Google Scholar] [CrossRef]
- Koketsu, M.; Otsuka, T.; Ishihara, H. Steroids. Synthesis of 1,3-selenazetidine derivatives from imines and thiocarbamoyl isoselenocycanate. Heterocycles 2006, 68, 2107–2112. [Google Scholar] [CrossRef]
- Koketsu, M.; Sakai, T.; Kiyokuni, T.; Garud, D.R.; Ando, H.; Ishihara, H. One-pot synthesis of 2-imino-1,3-selenazolidines by reaction of isoselenocyanates with propargylamine. Heterocycles 2006, 68, 1607–1615. [Google Scholar] [CrossRef]
- Koketsu, M.; Yamamura, Y.; Ishihara, H. Synthesis of 2-selenoxoperhydro-1,3-selenazin-4-ones and 2-selenoxo-1,3-selenazolidin-4-ones via diselenocarbamate intermediates. Synthesis 2006, 2738–2742. [Google Scholar] [CrossRef]
- Koketsu, M.; Otsuka, T.; Swenson, D.; Ishihara, H. The synthesis of 1-thia-6-oxa-6aλ4-seleno-3-azapentalene and a 3H-1,2,4-dithiazole. Org. Biomol. Chem. 2007, 5, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Koketsu, M.; Yamamura, Y.; Aoki, H.; Ishihara, H. The preparation of acylselenourea and selenocarbamate using isoselenocyanate. Phosphorus Sulfur Silicon Relat. Elem. 2006, 181, 2699–2708. [Google Scholar] [CrossRef]
- Segi, M.; Kojima, A.; Nakajima, T.; Suga, S. A facile synthesis of seleno- and telluroformamides. Synlett 1991, 105–106. [Google Scholar] [CrossRef]
- Li, G.M.; Zingaro, R.A. Chalcogenoamides: Convenient preparations and reactions with metal compounds. Phosphorus Sulfur Silicon Relat. Elem. 1998, 136–138, 525–530. [Google Scholar] [CrossRef]
- Shibara, F.; Sugiura, R.; Murai, T. Direct thionation and selenation of amides using elemental sulfur and selenium and hydrochlorosilanes in the presence of amines. Org. Lett. 2009, 11, 3064–3067. [Google Scholar] [CrossRef] [PubMed]
- Linden, A.; Zhou, Y.; Hermgartner, H. Intra- and intermolecular Se···X (X = Se, O) interactions in selenium-containing heterocycles: 3-benzoylimino-5-(morpholin-4-yl)-1,2,4-diselenazole. Acta Crystallogr. 2014, C70, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Richter, R.; Sieler, J.; Hansen, L.K.; Kohler, R.; Beyer, L.; Hoyer, E. Structure of 2-benzoylamino-5-diethylamino-1,66aλ4-triselena-3,4-diazapentalene. Acta Chem. Scand. 1991, 45, 1–5. [Google Scholar] [CrossRef]
- Li, G.M.; Zingaro, R.A.; Segi, M.; Reibenspies, J.H.; Nakajima, T. Synthesis and structure of telluroamides and selenoamides. The first crystallographic study of telluroamides. Organometallics 1997, 16, 756–762. [Google Scholar] [CrossRef]
- Hummel, H.U.; Fischer, T.; Gruss, D.; Franke, A.; Dietzsch, W. Synthesis and molecular structures of some novel anionic diselenides. J. Chem. Soc. Dalton Trans. 1992, 2781–2785. [Google Scholar] [CrossRef]
- Landman, M.; van der Westhuizen, B.; Bezuidenhout, D.I.; Liles, D.C. N-(2,4,6-Trimethylphenyl)formamide. Acta Crystallogr. 2011, E67, o120. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Li, Y.; Slawin, A.M.Z.; Woollins, J.D. Synthesis of primary arylselenoamids by reaction of aryl nitriles with Woollins’ reagent. Org. Lett. 2006, 8, 5251–5254. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hua, G.; Slawin, A.M.Z.; Woollins, J.D. The X-ray crystal structures of primary aryl substituted selenoamides. Molecules 2009, 14, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Fuller, A.L.; Scott-Hayward, L.A.S.; Li, Y.; Bühl, M.; Slawin, A.M.Z.; Woollins, J.D. Automated Chemical Crystallography. J. Am. Chem. Soc. 2010, 132, 5799–5802. [Google Scholar] [CrossRef] [PubMed]
- Rigaku. CrystalClear-SM Expert (Version 3.1b27); Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2013. [Google Scholar]
- Rigaku. CrystalClear-SM Expert (Version 2.1); Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2014. [Google Scholar]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Crystallogr. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G.; Spagna, R. SIR2011: A new package for crystal structure determination and refinement. J. Appl. Crystallogr. 2012, 45, 357–361. [Google Scholar] [CrossRef]
- Beurskens, P.T.; Beurskens, G.; de Gelder, R.; Garcia-Granda, S.; Gould, R.O.; Israel, R.; Smits, J.M.M. The DIRDIF-99 Program System; Crystallography Laboratory, University of Nijmegen: Nijmegen, The Netherlands, 1999. [Google Scholar]
- Spek, A.L. Platon squeeze: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Rigaku. CrystalStructure (Version 4.3.0); Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2018. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Sample Availability: Samples of all compounds are not available from the authors. |















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
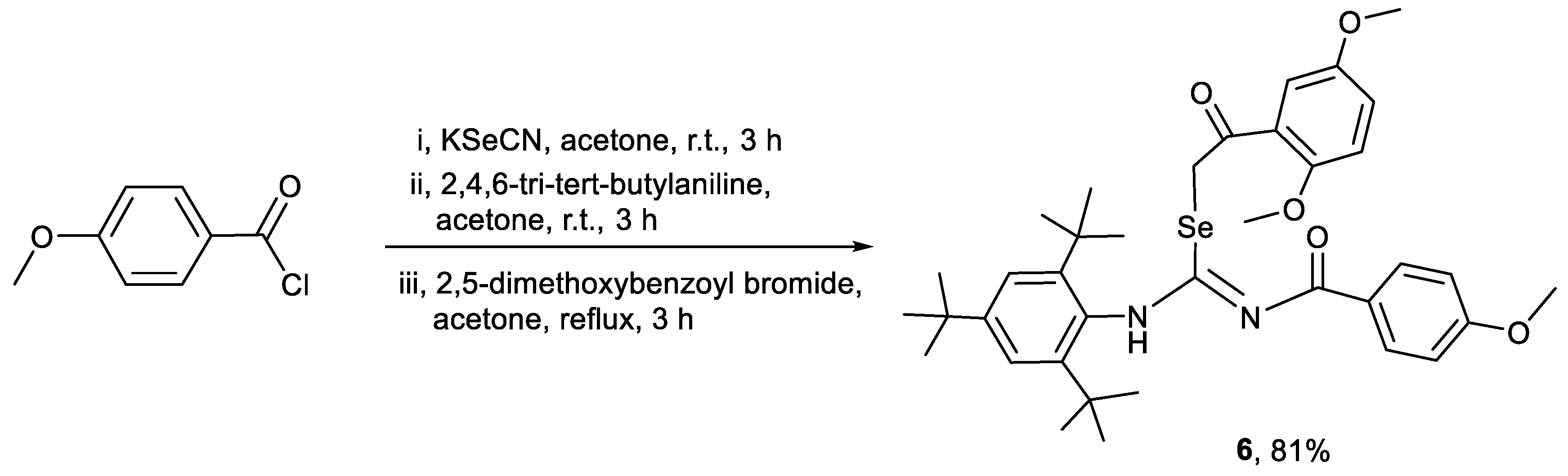{kind=link}
{kind=link}
| Compound | 2·CHCl3 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|
| Formula | C27H36Cl3N3O3Se | C26H33N3O2Se2 | C36H45N3O6Se | C19H31NSe | C37H48N2O5Se |
| M | 635.92 | 577.49 | 694.73 | 352.42 | 679.76 |
| T/K | 173 | 125 | 173 | 125 | 173 |
| Crystal system | triclinic | triclinic | triclinic | orthorhombic | monoclinic |
| Space group | P | P | P | Pnma | P21/n |
| a/Å | 9.6580 (12) | 9.4547 (7) | 15.5888 (3) | 20.2224 (14) | 18.003 (3) |
| b/Å | 10.9397 (12) | 11.4917 (8) | 16.3917 (4) | 15.2835 (11) | 9.5828 (16) |
| c/Å | 15.5479 (17) | 27.2316 (19) | 24.7330 (10) | 5.8839 (4) | 21.764 (4) |
| α/° | 91.094 (4) | 93.099 (6) | 71.062 (6) | 90 | 90 |
| β/° | 91.4028 (15) | 95.470 (6) | 74.004 (7) | 90 | 106.755 (4) |
| γ/° | 113.824 (3) | 90.116 (6) | 62.683 (6) | 90 | 90 |
| U/A3 | 1501.5 (3) | 2940.9 (4) | 5250.6 (4) | 1818.5 (2) | 3595.3 (11) |
| Z | 2 | 4 | 6 | 4 | 4 |
| µ/cm−1 | 15.493 | 25.389 | 11.207 | 20.608 | 10.868 |
| Reflections collected | 27118 | 23358 | 64396 | 13393 | 42195 |
| Independent reflections | 6636 | 10652 | 19067 | 1718 | 6570 |
| Rint | 0.0389 | 0.0979 | 0.0618 | 0.1034 | 0.0613 |
| R1 [I > 2σ (I)] | 0.0294 | 0.0658 | 0.0614 | 0.0403 | 0.0429 |
| wR2 | 0.0819 | 0.1741 | 0.1713 | 0.1218 | 0.1066 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, G.; Cordes, D.B.; Du, J.; Slawin, A.M.Z.; Woollins, J.D. Diverse Derivatives of Selenoureas: A Synthetic and Single Crystal Structural Study. Molecules 2018, 23, 2143. https://doi.org/10.3390/molecules23092143
Hua G, Cordes DB, Du J, Slawin AMZ, Woollins JD. Diverse Derivatives of Selenoureas: A Synthetic and Single Crystal Structural Study. Molecules. 2018; 23(9):2143. https://doi.org/10.3390/molecules23092143
Chicago/Turabian StyleHua, Guoxiong, David B. Cordes, Junyi Du, Alexandra M. Z. Slawin, and J. Derek Woollins. 2018. "Diverse Derivatives of Selenoureas: A Synthetic and Single Crystal Structural Study" Molecules 23, no. 9: 2143. https://doi.org/10.3390/molecules23092143
APA StyleHua, G., Cordes, D. B., Du, J., Slawin, A. M. Z., & Woollins, J. D. (2018). Diverse Derivatives of Selenoureas: A Synthetic and Single Crystal Structural Study. Molecules, 23(9), 2143. https://doi.org/10.3390/molecules23092143