
Safe Synthesis of 4,7-Dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine and Its SNAr Reactions


 ,
, 
Abstract
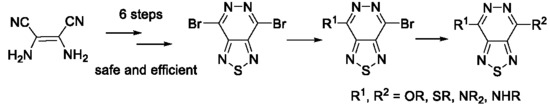
1. Introduction
2. Results and Discussion
2.1. Aromatic Nucleophilic Substitution in Bromo Derivatives of [1,2,5]thiadiazolo[3,4-d]pyridazine
2.1.1. Oxygen Nucleophiles
2.1.2. Sulfur Nucleophiles
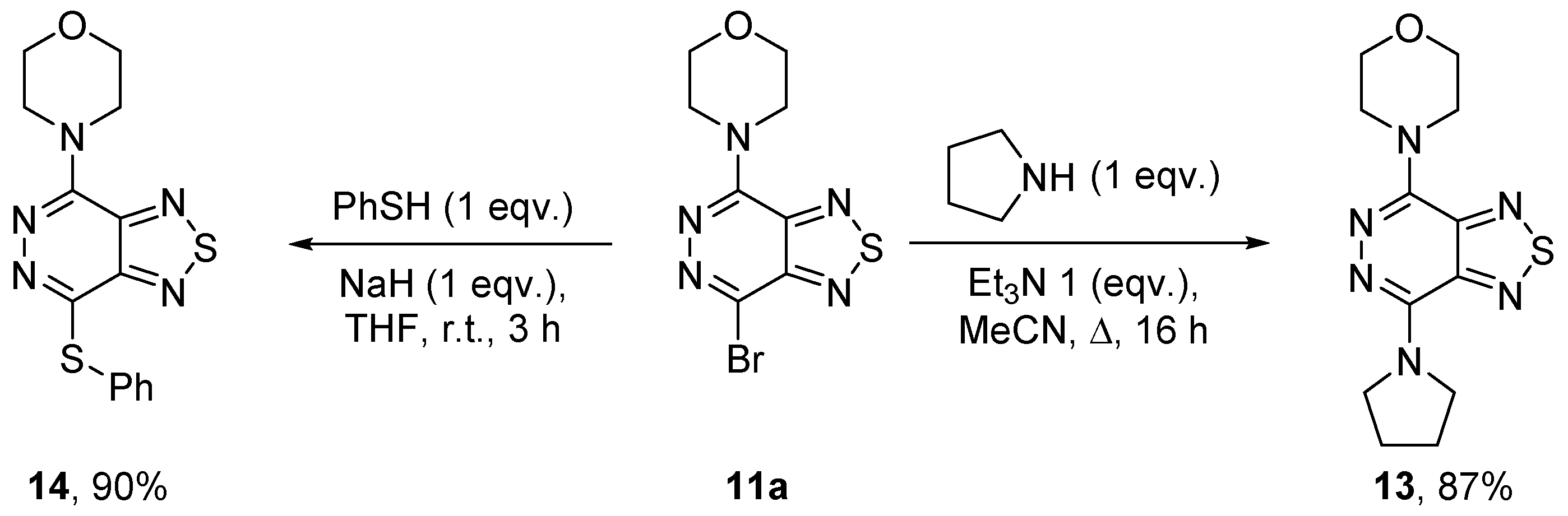
2.1.3. Nitrogen Nucleophiles
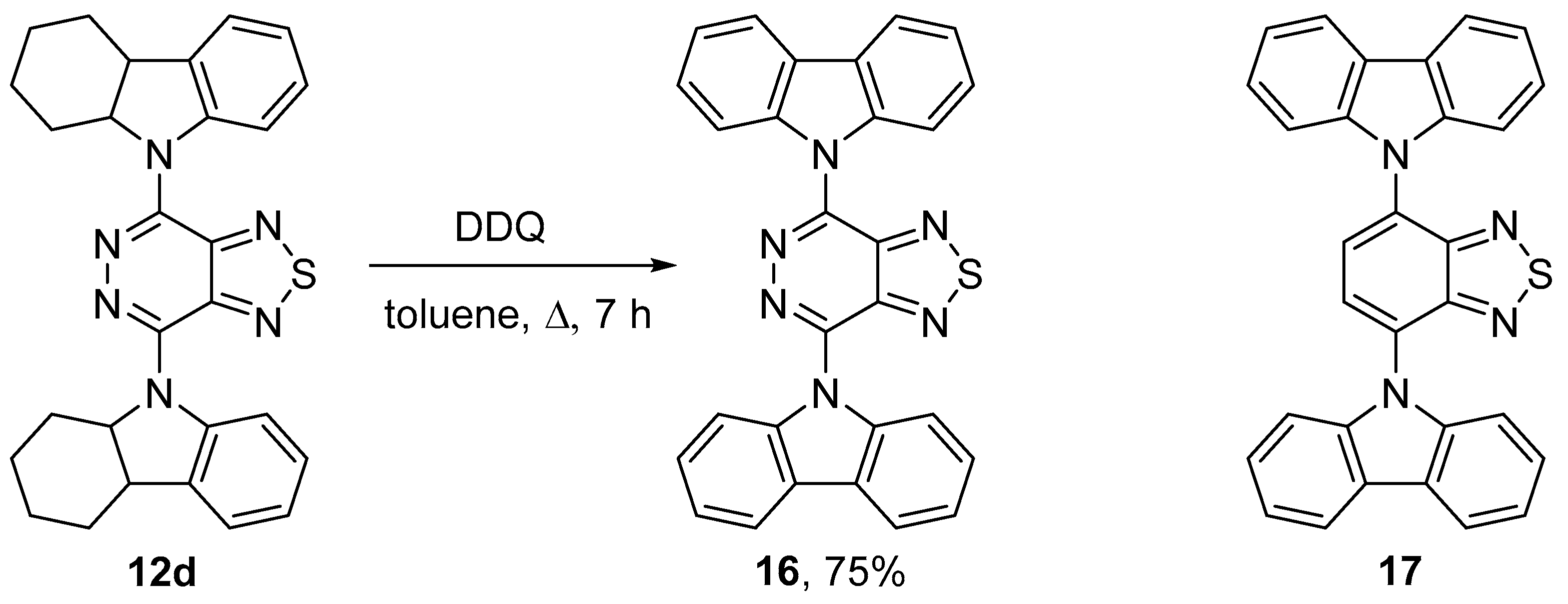
2.2. Cross-Couplings Based on the Buchwald-Hartwig and Ullmann Techniques—Synthesis of Mono- and Bis(9H-carbazol-9-yl)[1,2,5]thiadiazolo[3,4-d]pyridazines
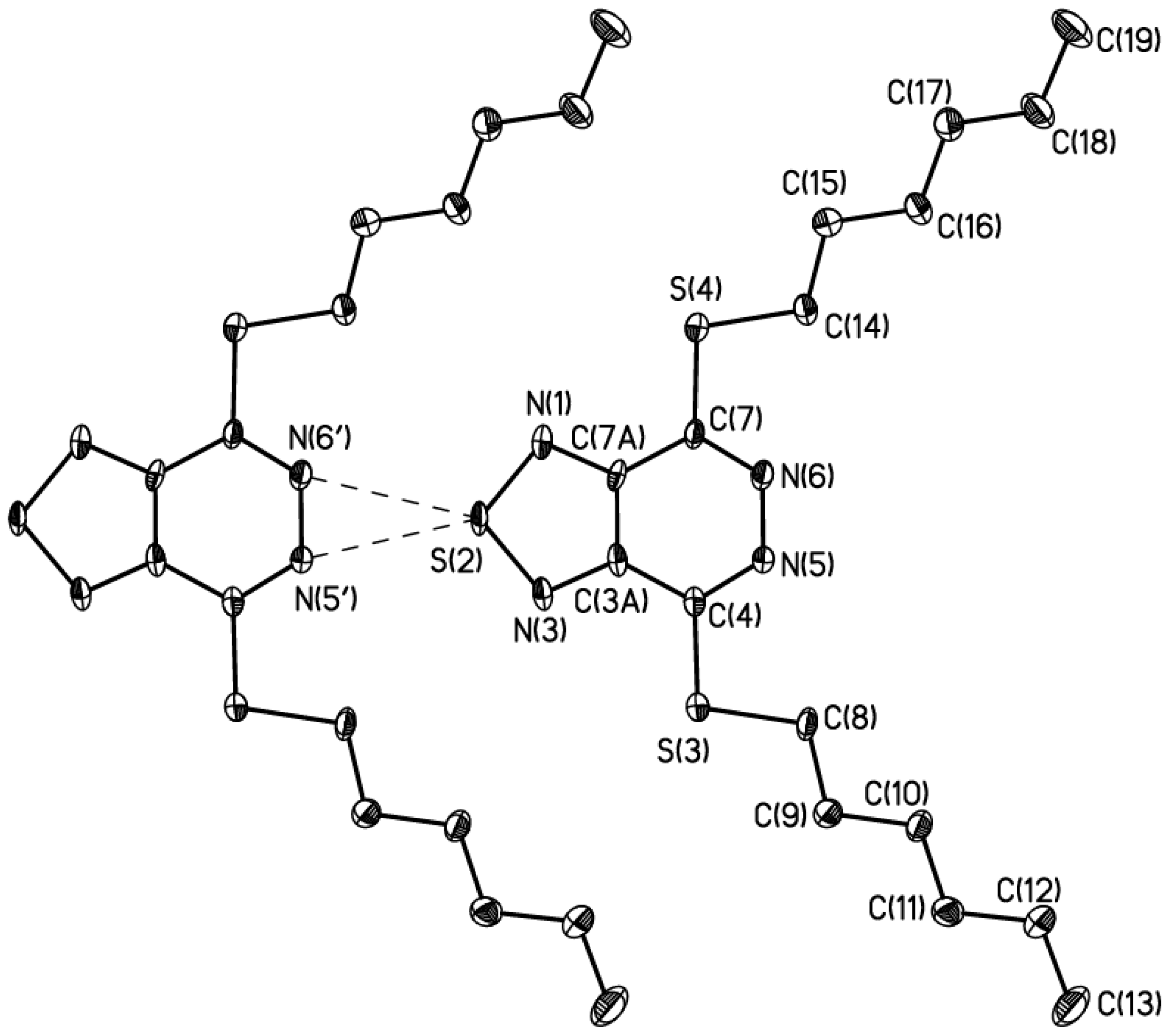
3. X-Ray Analysis
4. Experimental Section
4.1. General Information
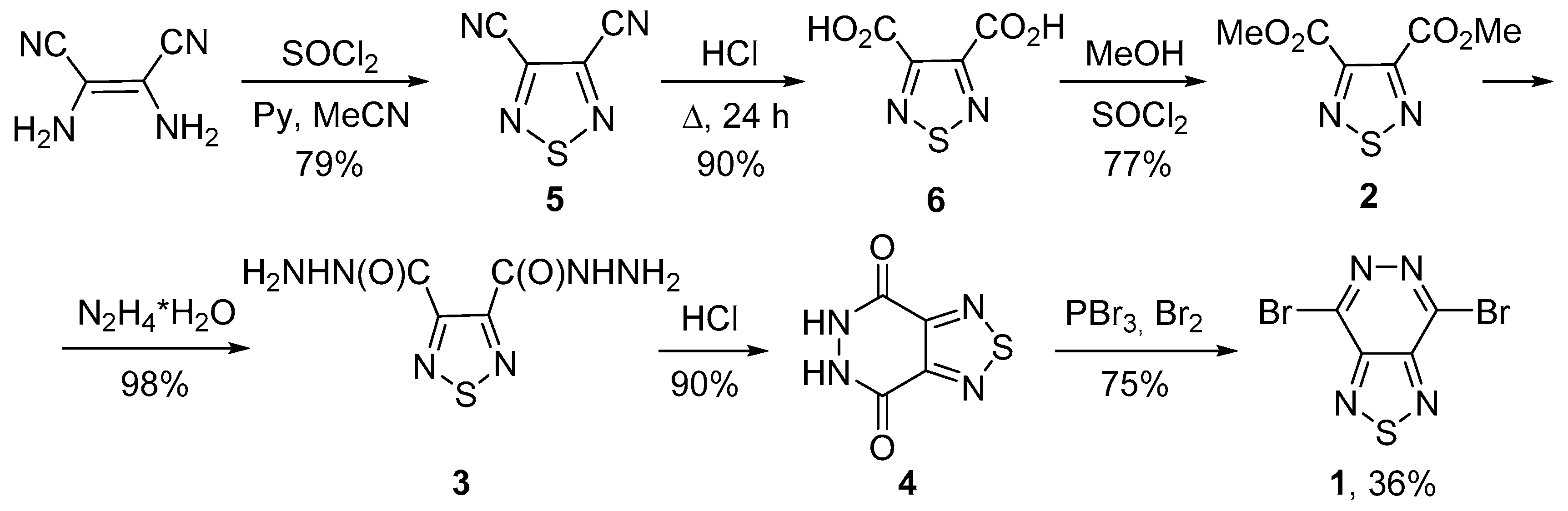
4.2. 4,7-Dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine (1)
4.2.1. 1,2,5-Thiadiazole-3,4-dicarbonitrile (5)
4.2.2. 1,2,5-Thiadiazole-3,4-dicarboxylic Acid (6)
4.2.3. Dimethyl 1,2,5-thiadiazole-3,4-dicarboxylate (2)
4.2.4. [1,2,5]Thiadiazole-3,4-dicarboxylic Acid Dihydrazide (3)
4.2.5. 5,6-Dihydro[1,2,5]thiadiazolo[3,4-d]pyridazine-4,7-dione (4)
4.2.6. 4,7-Dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine (1)
4.3. Reaction of 4,7-dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine (1) with H2O
4.4. Reaction of 4,7-dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine (1) with MeOH
4.5. 4-Bromo-7-methoxy[1,2,5]thiadiazolo[3,4-d]pyridazine (8a)
4.6. 4-Bromo-7-phenoxy[1,2,5]thiadiazolo[3,4-d]pyridazine (8b)
4.7. 4,7-Dimethoxy[1,2,5]thiadiazolo[3,4-d]pyridazine (9a)
4.8. 4,7-Diphenoxy[1,2,5]thiadiazolo[3,4-d]pyridazine (9b)
4.9. General Procedure for the Reaction of 4,7-dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine (1) with Thiols
4.9.1. 4,7-Bis(phenylthio)[1,2,5]thiadiazolo[3,4-d]pyridazine (10a)
4.9.2. 4,7-Bis(hexylthio)[1,2,5]thiadiazolo[3,4-d]pyridazine (10b)
4.9.3. 4,7-Bis(dodecylthio)[1,2,5]thiadiazolo[3,4-d]pyridazine (10c)
4.10. General Procedure for the Preparation of Mono-Aminated Products 11
4.10.1. 4-(7-Bromo[1,2,5]thiadiazolo[3,4-d]pyridazin-4-yl)morpholine (11a)
4.10.2. 4-Bromo-7-(piperidin-1-yl)[1,2,5]thiadiazolo[3,4-d]pyridazine (11b)
4.10.3. 4-Bromo-7-(pyrrolidin-1-yl)[1,2,5]thiadiazolo[3,4-d]pyridazine (11c)
4.10.4. 4-Bromo-7-(2,3,4,4a-hexahydro-1H-carbazol-9(9aH)-yl)[1,2,5]thiadiazolo[3,4-d]pyridazine (11d)
4.10.5. 4-Bromo-7-(1,3,3a,8b-tetrahydrocyclopenta[b]indol-4(2H)-yl)[1,2,5]thiadiazolo[3,4-d]pyridazine (11e)
4.10.6. 4-Bromo-7-(2,3,4,4a-tetrahydro-1H-1,4-methanocarbazol-9(9aH)-yl)-[1,2,5]thiadiazolo[3,4- d]pyridazine (11f)
4.10.7. 7-Bromo-N-methyl-N-phenyl[1,2,5]thiadiazolo[3,4-d]pyridazin-4-amine (11g)
4.10.8. 7-Bromo-N-cyclohexyl[1,2,5]thiadiazolo[3,4-d]pyridazin-4-amine (11h)
4.10.9. 7-Bromo-N-phenyl[1,2,5]thiadiazolo[3,4-d]pyridazin-4-amine (11i)
4.10.10. 7-Bromo-N-(tert-butyl)[1,2,5]thiadiazolo[3,4-d]pyridazin-4-amine (11j)
4.11. General Procedure for the Preparation of Di-Aminated Products 12
4.11.1. 4,7-Dimorpholino[1,2,5]thiadiazolo[3,4-d]pyridazine (12a)
4.11.2. 4,7-Di(piperidin-1-yl)[1,2,5]thiadiazolo[3,4-d]pyridazine (12b)
4.11.3. 4,7-Bis(2,3,4,4a-tetrahydro-1H-carbazol-9(9aH)-yl)[1,2,5]thiadiazolo[3,4-d]pyridazine (12d)
4.11.4. N4,N7-Diphenyl[1,2,5]thiadiazolo[3,4-d]pyridazine-4,7-diamine (12i)
4.12. 4-(7-(Pyrrolidin-1-yl)[1,2,5]thiadiazolo[3,4-d]pyridazin-4-yl)morpholine (13)
4.13. 4-(7-(Phenylthio)[1,2,5]thiadiazolo[3,4-d]pyridazin-4-yl)morpholine (14)
4.14. 4-(7-(9H-Carbazol-9-yl)[1,2,5]thiadiazolo[3,4-d]pyridazin-4-yl)morpholine (15)
4.14.1. By Buchwald-Hartwig Reaction
4.14.2. By Ullmann Reaction
4.15. 4,7-Di(9H-carbazol-9-yl)[1,2,5]thiadiazolo[3,4-d]pyridazine (16)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parker, T.C.; Dan Patel, D.G.; Moudgil, K.; Barlow, S.; Risko, C.; Brédas, J.-L.; Reynolds, J.R.; Marder, S.R. Heteroannulated acceptors based on benzothiadiazole. Mater. Horiz. 2015, 2, 22–36. [Google Scholar] [CrossRef]
- Knyazeva, E.A.; Rakitin, O.A. Influence of structural factors on the photovoltaic properties of dye-sensitized solar cells. Russ. Chem. Rev. 2016, 85, 1146–1183. [Google Scholar] [CrossRef]
- Zhang, X.; Grätzel, M.; Hua, J. Donor design and modification strategies of metal-free sensitizers for highly-efficient n-type dye-sensitized solar cells. Front. Optoelectron. 2016, 9, 3–35. [Google Scholar] [CrossRef]
- Takimiya, K.; Osaka, I.; Nakano, M. π-Building Blocks for Organic Electronics: Revaluation of “Inductive” and “Resonance” Effects of π-Electron Deficient Units. Chem. Mater. 2014, 26, 587–593. [Google Scholar] [CrossRef]
- Wu, Y.; Zhu, W. Organic sensitizers from D–p–A to D–A–p–A: Effect of the internal electron-withdrawing units on molecular absorption, energy levels and photovoltaic performances. Chem. Soc. Rev. 2013, 42, 2039–2058. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Huang, F.; Cao, Y. Recent development of push–pull conjugated polymers for bulk-heterojunction photovoltaics: Rational design and fine tailoring of molecular structures. J. Mater. Chem. 2012, 22, 10416–10434. [Google Scholar] [CrossRef]
- Pron, A.; Leclerc, M. Imide/amide based π-conjugated polymers for organic electronics. Prog. Polym. Sci. 2013, 38, 1815–1831. [Google Scholar] [CrossRef]
- Lee, C.P.; Li, C.T.; Ho, K.C. Use of organic materials in dye-sensitized solar cells. Mater. Today 2017, 20, 267–282. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Y.; Liu, Y. 25th Anniversary Article: Recent Advances in n-Type and Ambipolar Organic Field-Effect Transistors. Adv. Mater. 2013, 25, 5372–5391. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Facchetti, A.; Marks, T.J. Imide- and Amide-Functionalized Polymer Semiconductors. Chem. Rev. 2014, 114, 8943–9021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zou, J.; Cheuh, C.C.; Yip, H.L.; Jen, A.K.Y. Significant Improved Performance of Photovoltaic Cells Made from a Partially Fluorinated Cyclopentadithiophene/Benzothiadiazole Conjugated Polymer. Macromolecules 2012, 45, 5427–5435. [Google Scholar] [CrossRef]
- Stuart, A.C.; Tumbleston, J.R.; Zhou, H.; Li, W.; Liu, S.; Ade, H.; You, W. Fluorine Substituents Reduce Charge Recombination and Drive Structure and Morphology Development in Polymer Solar Cells. J. Am. Chem. Soc. 2013, 135, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Van Mullekom, H.A.M.; Vekemans, J.; Havinga, E.E.; Meijer, E.W. Developments in the chemistry and band gap engineering of donor–acceptor substituted conjugated polymers. Mater. Sci. Eng. R 2001, 32, 1–40. [Google Scholar] [CrossRef]
- Roncali, J. Molecular Engineering of the Band Gap of π-Conjugated Systems: Facing Technological Applications. Macromol. Rapid Commun. 2007, 28, 1761–1775. [Google Scholar] [CrossRef]
- Velusamy, M.; Thomas, K.R.J.; Lin, J.T.; Hsu, Y.C.; Ho, K.C. Organic Dyes Incorporating Low-Band-Gap Chromophores for Dye-Sensitized Solar Cells. Org. Lett. 2005, 7, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wu, Y.Z.; Lu, X.F.; Zhang, X.; Zhou, G.; Miapeh, F.B.; Zhu, W.H.; Wang, Z.S. Incorporating Benzotriazole Moiety to Construct D-A-π-A Organic Sensitizers for Solar Cells: Significant Enhancement of Open-Circuit Photovoltage with Long Alkyl Group. Chem. Mater. 2011, 23, 4394–4401. [Google Scholar] [CrossRef]
- Pei, K.; Wu, Y.Z.; Wu, W.J.; Zhang, Q.; Chen, B.Q.; Tian, H.; Zhu, W.H. Constructing Organic D–A–p-A-Featured Sensitizers with a Quinoxaline Unit for High-Efficiency Solar Cells: The Effect of an Auxiliary Acceptor on the Absorption and the Energy Level Alignment. Chem. Eur. J. 2012, 18, 8190–8200. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Q.; Wu, Y.Z.; Zhang, Q.; Tian, H.; Zhu, W.H. D-A-π-A Featured Sensitizers Bearing Phthalimide and Benzotriazole as Auxiliary Acceptor: Effect on Absorption and Charge Recombination Dynamics in Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2012, 4, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.Y.; Wu, W.J.; Hua, J.L.; Kong, C.Y.; Long, T.; Tian, H. New Diketopyrrolopyrrole (DPP) Dyes for Efficient Dye-Sensitized Solar Cells. J. Phys. Chem. C 2010, 114, 1343–1349. [Google Scholar] [CrossRef]
- Lu, X.F.; Zhou, G.; Wang, H.; Feng, Q.Y.; Wang, Z.S. Near infrared thieno[3,4-b]pyrazine sensitizers for efficient quasi-solid-state dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2012, 14, 4802–4809. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, K.; Xu, F.; Tang, Z.; Zheng, W.; Zhang, J.; Li, C.; Yu, T.; You, X. Synthesis and photovoltaic performances of donor–π–acceptor dyes utilizing 1,3,5-triazine as π spacers. Tetrahedron Lett. 2011, 52, 6492–6496. [Google Scholar] [CrossRef]
- Chmovzh, T.N.; Knyazeva, E.A.; Mikhalchenko, L.V.; Golovanov, I.S.; Amelichev, S.A.; Rakitin, O.A. Synthesis of 4,7-dibromo derivative of ultrahigh electron-deficient [1,2,5]thiadiazolo[3,4-d]pyridazine heterocycle and its cross-coupling reactions, Eur. J. Org. Chem. Eur. J. Org. Chem. 2018. [Google Scholar] [CrossRef]
- Warren, J.D.; Lee, V.J.; Angier, R.B. Synthesis of 5,8-dihydroxynaphtho[2,3-c][1,2,5]thiadiazole-6,9-dione and 6,9-dihydroxybenzo[g]quinoxaline-5,10-dione. J. Heterocycl. Chem. 1979, 16, 1617–1624. [Google Scholar] [CrossRef]
- Mataka, S.; Takahashi, K.; Yamada, Y.; Tashiro, M. Sulfur nitride in organic chemistry. 6 Preparation of 3,4-disubstituted 1,2,5-thiadiazoles by the reaction of sulfur nitride with acetylenes. J. Heterocycl. Chem. 1979, 16, 1009–1015. [Google Scholar] [CrossRef]
- Kolb, H.C.; Bennani, Y.L.; Sharpless, K.B. Short and practical syntheses of (R)-(−)-carnitine and (R)-(−)-γ-amino-β-hydroxybutyric acid (GABOB). Tetrahedron Asymmetry 1993, 4, 133–141. [Google Scholar] [CrossRef]
- Biagi, G.; Giorgi, I.; Livi, O.; Manera, C.; Scartoni, V.; Betti, L.; Giannaccini, G.; Lucacchini, A. 1,2,3-Triazolo[4,5-d]pyridazines: Part VI. New 1-substituted-4-amino derivatives and their affinity towards A1 and A2A adenosine receptors. Il Farm. 1999, 54, 615–623. [Google Scholar] [CrossRef]
- Biagi, G.; Giorgi, I.; Livi, O.; Scartoni, V.; Velo, S.; Martini, C.; Senatore, G.; Barili, P.L. 1,2,3-Triazole[4,5-d]pyridazines. 4. Preparation and adenosine receptor-binding of new 4 and/or 7 aminoderivatives. Il Farm. 1995, 50, 99–105. [Google Scholar]
- Miller-Moslin, K.; Peukert, S.; Jain, R.K.; McEwan, M.A.; Karki, R.; Llamas, L.; Yusuff, N.; He, F.; Li, Y.; Sun, Y.; et al. 1-Amino-4-benzylphthalazines as Orally Bioavailable Smoothened Antagonists with Antitumor Activity. J. Med. Chem. 2009, 52, 3954–3968. [Google Scholar] [CrossRef] [PubMed]
- Carbon, J.A. The Preparation of Several 4-Substituted Imidazo[4,5-d]pyridazines as Possible Purine Antimetabolites. J. Am. Chem. Soc. 1958, 80, 6083–6088. [Google Scholar] [CrossRef]
- Ni, F.; Wu, Z.; Zhu, Z.; Chen, T.; Wu, K.; Zhong, C.; An, K.; Wei, D.; Ma, D.; Yang, C. Teaching an old acceptor new tricks: Rationally employing 2,1,3-benzothiadiazole as input to design a highly efficient red thermally activated delayed fluorescence emitter. J. Mater. Chem. C 2017, 5, 1363–1368. [Google Scholar] [CrossRef]
- Jung, D.; Thirupathaiah, B.; Lee, E.; Kwon, G.; Kim, C.; Seo, S. Synthesis and Characterization of Benzothiadiazole Derivatives as Organic Semiconductors for Organic Thin-Film Transistors. J. Nanosci. Nanotechnol. 2016, 16, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.; Gautam, P. Tuning of the HOMO–LUMO gap of donor-substituted symmetrical and unsymmetrical benzothiadiazoles. Org. Biomol. Chem. 2014, 12, 5448–5457. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, N.; Wright, I.A.; Bozic-Weber, B.; Constable, E.C.; Housecroft, C.E. Dye-sensitized solar cells with hole-stabilizing surfaces: “inorganic” versus “organic” strategies. RSC Adv. 2015, 5, 37906–37915. [Google Scholar] [CrossRef]
- Ye, Q.; Chen, S.; Zhu, D.; Lu, X.; Lu, Q. Preparation of aggregation-induced emission dots for long-term two-photon cell imaging. J. Mater. Chem. B 2015, 3, 3091–3097. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Y.; Wang, Y.; Ai, L.; Ouyang, X.; Han, L.; Ge, Z. Anthradithiophene-benzothiadiazole-based small molecule donors for organic solar cells. New J. Chem. 2013, 37, 3627–3633. [Google Scholar] [CrossRef]
- Misra, R.; Gautam, P.; Mobin, S.M. Aryl-Substituted Unsymmetrical Benzothiadiazoles: Synthesis, Structure, and Properties. J. Org. Chem. 2013, 78, 12440–12452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.H.; Wang, Z.M.; Chen, M.D.; Guo, S.L.; Zheng, Y.X. Syntheses and photoluminescence properties of rhenium(I) complexes based on dipyrido[3,2-a:2′,3′-c]phenazine derivatives with carbazole moiety. J. Coord. Chem. 2013, 66, 958–965. [Google Scholar] [CrossRef]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2007. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Ananyev, I.V.; Karnoukhova, V.A.; Dmitrienko, A.O.; Lyssenko, K.A. Toward a Rigorous Definition of a Strength of Any Interaction Between Bader’s Atomic Basins. J. Phys. Chem. A 2017, 121, 4517–4522. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADABS v2008/1, Bruker/Siemens Area Detector Absorption Correction Program; Bruker AXS: Madison, WI, USA, 2008. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. A. 2015, 71–78. [Google Scholar] [CrossRef]
- Duan, X.G.; Duan, X.-L.; Rees, C.W.; Yue, T.Y. Reaction of trithiazyl trichloride with alkenes and alkynes. J. Chem. Soc. Perkin Trans. 1 1997, 2597–2601. [Google Scholar] [CrossRef]
- Sekikawa, I. Oxidation of 5-Methyl-2,1,3-benzothiadiazole with Potassium Permanganate. Bull. Chem. Soc. Japan 1960, 33, 1229–1231. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Reagent (eqv.) | Solvent | Temp. (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | H2O (excess) | H2O | 25 | 30 | 7 (75) |
| 2 | H2O (1) | CHCl3 | 25 | 30 | 7 (80) |
| 3 | H2O (2) | CHCl3 | 25 | 30 | 7 (82) |
| 4 | H2O (1) | CHCl3 | 60 | 20 | 7 (75) |
| 5 | H2O (2) | CHCl3 | 60 | 20 | 7 (77) |
| 6 | MeOH (excess) | CHCl3 | 25 | 16 | 7 (78) |
| 7 | MeONa (1) | MeOH | 25 | 6 | 8a (80) |
| 8 | MeONa (2) | MeOH | 25 | 24 | 9a (82) |
| 9 | MeONa (2) | MeOH | 64 | 6 | 9a (70) |
| 10 | PhOH (1) | THF | 25 | 8 | - * |
| 11 | PhOH (1) | DMF | 25 | 8 | - * |
| 12 | PhONa (1) | THF | 25 | 8 | 8b (80) |
| 13 | PhONa (2) | THF | 25 | 8 | 8b (78) |
| 14 | PhONa (2) | THF | 60 | 6 | 8b (74) |
| 15 | PhONa (1) | DMF | 25 | 8 | 8b (70) |
| 16 | PhONa (2) | DMF | 25 | 8 | 8b (73) |
| 17 | PhONa (2) | DMF | 90 | 6 | 9b (69) |

| Entry | RSH (equiv.) | Solvent | Base (equiv.) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | a (1) | CHCl3 | - | 6 | 10a (45) * |
| 2 | a (2) | CHCl3 | - | 7 | 10a (78) |
| 3 | a (2) | THF | - | 4 | 10a(80) |
| 4 | a (2) | MeCN | - | 4 | 10a (76) |
| 5 | a (2) | DMF | - | 1 | 10a (77) |
| 6 | a (2) | THF | NaH (2) | 3 | 10a (85) |
| 7 | b (2) | THF | NaH (2) | 3 | 10b (90) |
| 8 | c (2) | THF | NaH (2) | 4 | 10c (88) |

| Entry | Morpholine (eqv.) | Solvent | Base (eqv.) | Temp. (°C) | Time (h) | Yields (%) | |
|---|---|---|---|---|---|---|---|
| 11a | 12a | ||||||
| 1 | 2 | MeOH | - | 25 | 6 | 79 | 0 |
| 2 | 4 | MeOH | - | 25 | 12 | 78 | 0 |
| 3 | 2 | CH2Cl2 | - | 25 | 4 | 82 | 0 |
| 4 | 4 | CH2Cl2 | - | 25 | 10 | 83 | 0 |
| 5 | 1 | CH2Cl2 | Et3N (1) | 25 | 4 | 86 | 0 |
| 6 | 2 | MeCN | - | 25 | 3 | 80 | 0 |
| 7 | 4 | MeCN | - | 25 | 6 | 82 | 0 |
| 8 | 1 | MeCN | Et3N (1) | 25 | 3 | 81 | 0 |
| 9 | 2 | DMF | - | 25 | 0.5 | 84 | 0 |
| 10 | 4 | DMF | - | 25 | 2 | 84 | 0 |
| 11 | 1 | DMF | Et3N (1) | 25 | 0.5 | 82 | 0 |
| 12 | 2 | CHCl3 | Et3N (2) | 61 | 50 | 0 | 70 |
| 13 | 2 | MeCN | Et3N (2) | 80 | 30 | 0 | 87 |
| 14 | 2 | DMF | Et3N (2) | 80 | 20 | 0 | 82 |

| Entry | Amine (eqv.) | Solvent | Base (eqv.) | Temp. (°C) | Time (h) | Yields (%) |
|---|---|---|---|---|---|---|
| 1 | a (1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11a (86) |
| 2 | b (1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11b (82) |
| 3 | c (1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11c (81) |
| 4 | d (1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11d (85) |
| 5 | e (1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11e (82) |
| 6 | f (1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11f (79) |
| 7 | g (1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11g (80) |
| 8 | h (1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11h (85) |
| 9 | i (1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11i (80) |
| 10 | j(1) | CH2Cl2 | Et3N (1) | 25 | 4 | 11j (75) |
| 11 | a (2) | MeCN | Et3N (2) | 80 | 30 | 12a (87) |
| 12 | b (2) | MeCN | Et3N (2) | 80 | 30 | 12b (85) |
| 13 | d (2) | MeCN | Et3N (2) | 80 | 10 | 12d (90) |
| 14 | i (2) | MeCN | Et3N (2) | 80 | 30 | 12i (78) |

| Entry | Catalyst (mol%) | Ligand (mol%) | Solvent | Temp. (°C) | Time (h) | 15, Yield (%) |
|---|---|---|---|---|---|---|
| 1 | Pd2(dba)3 (10) | - | toluene | 111 | 12 | decomp. |
| 2 | Pd(OAc)2 (10) | - | toluene | 111 * | 0.25 | decomp. |
| 3 | Pd2(dba)3 (10) | XPhos (5) | toluene | 111 | 12 | 5 |
| 4 | Pd(OAc)2 (10) | XPhos (5) | toluene | 111 | 12 | 8 |
| 5 | Pd2(dba)3 (10) | XPhos (5) | Toluene | 111 * | 0.5 | 20 |
| 6 | Pd(OAc)2 (10) | XPhos (5) | toluene | 111 * | 0.5 | 40 |
| 7 | CuI (10) | DMEDA (5) | dioxane/H2O 3:1 | 100 | 5 | 30 |
| 8 | CuI (10) | DMEDA (5) | dioxane/H2O 3:1 | 100 * | 0.17 | 37 |
| 10b | 16 | |
|---|---|---|
| Chemical formula | C16H26N4S3 | C28H16N6S, 0.5 (CH2Cl2) |
| Formula weight (g mol−1) | 370.59 | 510.99 |
| Temperature (K) | 120 | 120 |
| Crystal system | Monoclinic | Triclinic |
| Space group | C2/c | P-1 |
| a (Å) | 22.221(5) | 9.646(2) |
| b (Å) | 7.5589(16) | 12.785(3) |
| c (Å) | 35.127(7) | 18.575(4) |
| α (°) | 90 | 83.824(5) |
| β (°) | 104.940(11) | 89.837(5) |
| γ (°) | 90 | 89.640(5) |
| V (Å3) | 5701(2) | 2277.3(8) |
| Z/Z’ | 12/1.5 | 4/2 |
| dcalc (g cm–3) | 1.295 | 1.490 |
| μ(MoKα) | 3.95 | 2.93 |
| 2Θmax | 56 | 58 |
| Reflns. Collected/unique | 31,479/6870 | 28,461/12,099 |
| Observed reflns [I > 2σ(I)] | 3287 | 6756 |
| Rint(I) | 0.0843 | 0.0957 |
| R1(F2) | 0.0737 | 0.0689 |
| wR2 | 0.2052 | 0.1932 |
| GOF | 1.022 | 1.043 |
| Δρmin/Δρmax | −0.416/0.939 | −0.812/1.049 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chmovzh, T.N.; Knyazeva, E.A.; Lyssenko, K.A.; Popov, V.V.; Rakitin, O.A. Safe Synthesis of 4,7-Dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine and Its SNAr Reactions. Molecules 2018, 23, 2576. https://doi.org/10.3390/molecules23102576
Chmovzh TN, Knyazeva EA, Lyssenko KA, Popov VV, Rakitin OA. Safe Synthesis of 4,7-Dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine and Its SNAr Reactions. Molecules. 2018; 23(10):2576. https://doi.org/10.3390/molecules23102576
Chicago/Turabian StyleChmovzh, Timofey N., Ekaterina A. Knyazeva, Konstantin A. Lyssenko, Vadim V. Popov, and Oleg A. Rakitin. 2018. "Safe Synthesis of 4,7-Dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine and Its SNAr Reactions" Molecules 23, no. 10: 2576. https://doi.org/10.3390/molecules23102576
APA StyleChmovzh, T. N., Knyazeva, E. A., Lyssenko, K. A., Popov, V. V., & Rakitin, O. A. (2018). Safe Synthesis of 4,7-Dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine and Its SNAr Reactions. Molecules, 23(10), 2576. https://doi.org/10.3390/molecules23102576