Rutacecarpine Inhibits Angiogenesis by Targeting the VEGFR2 and VEGFR2-Mediated Akt/mTOR/p70s6k Signaling Pathway

Abstract
1. Introduction
2. Results
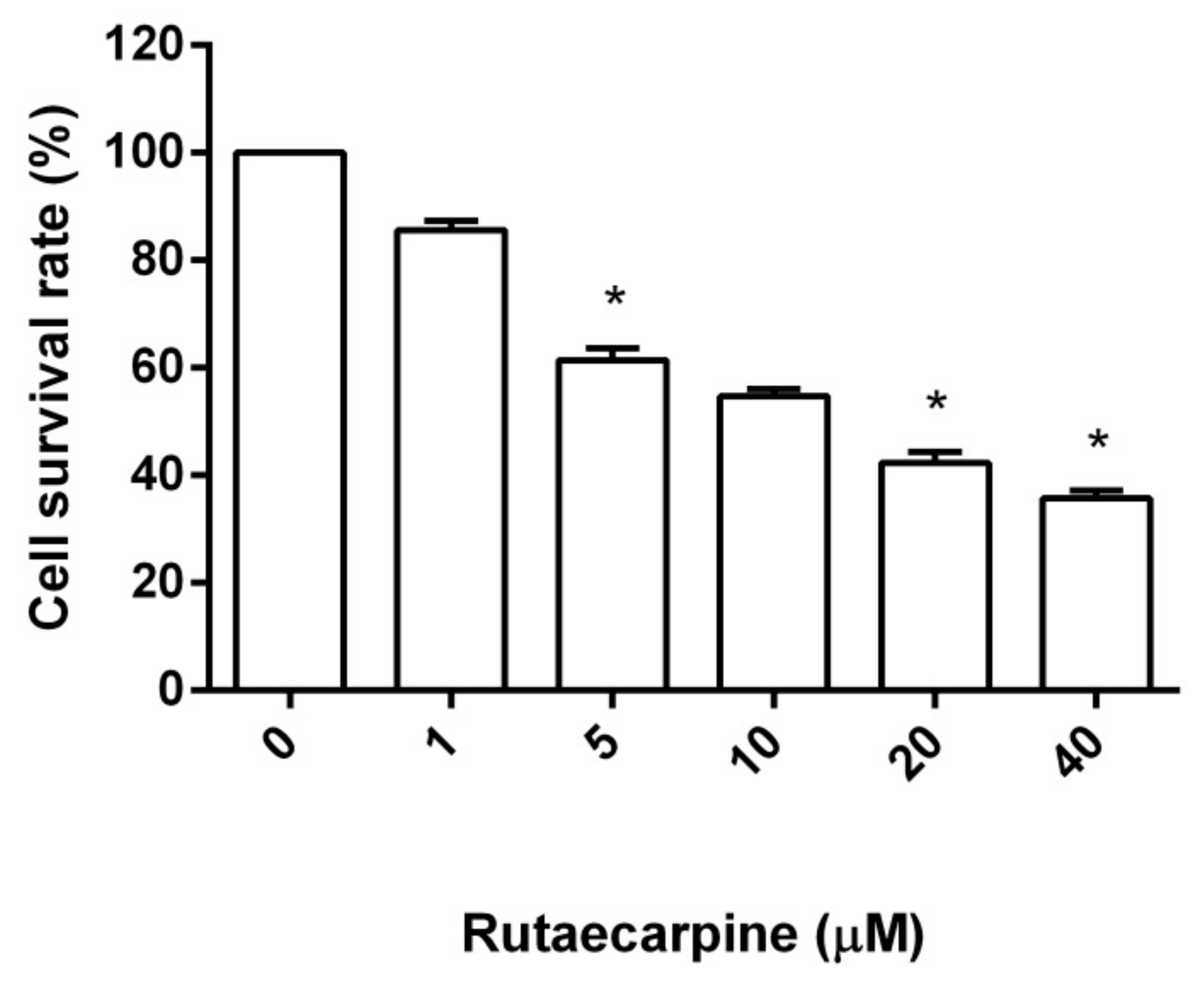
2.1. Cytotoxicity Against HUVECs
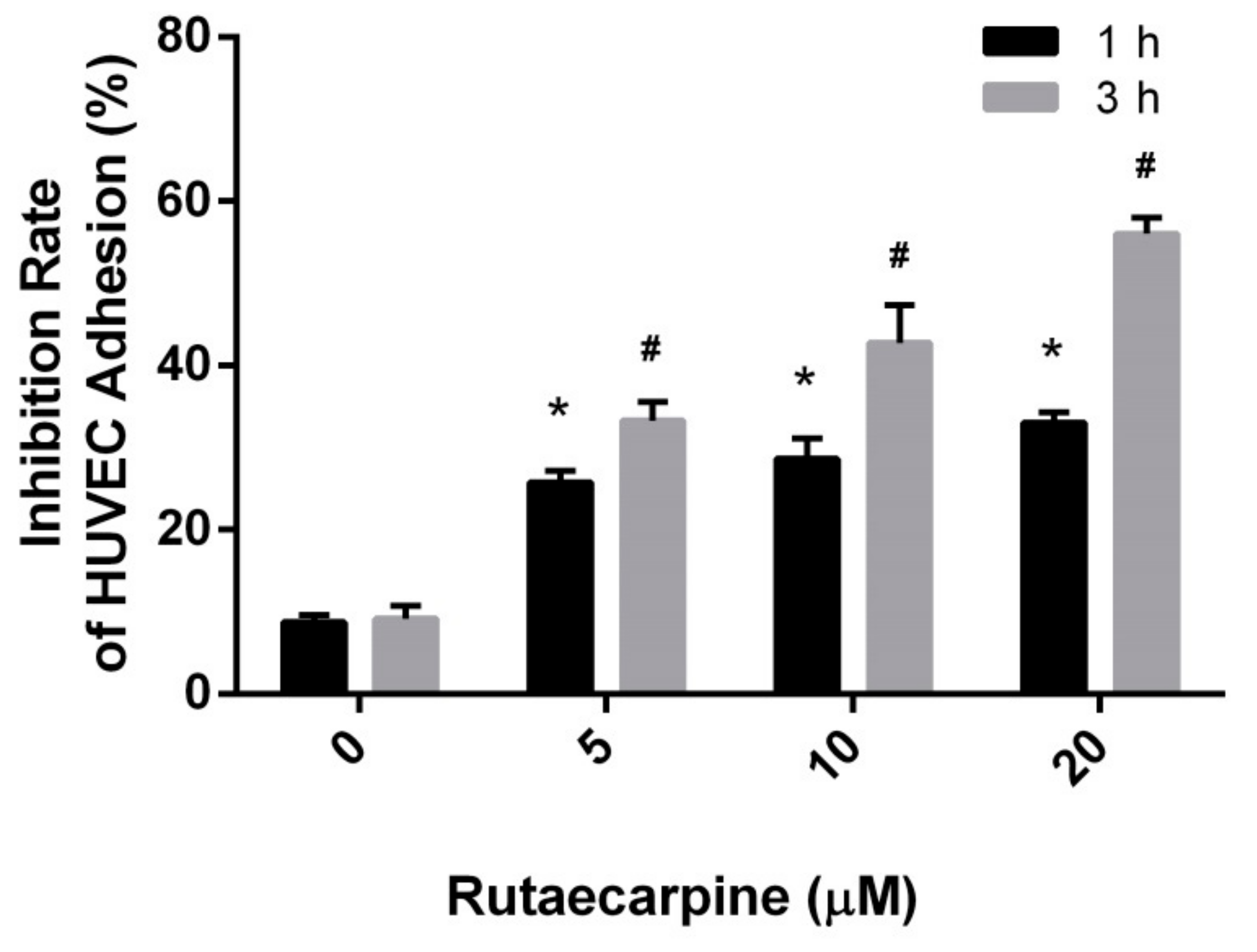
2.2. Inhibition of the Adhesion of HUVECs
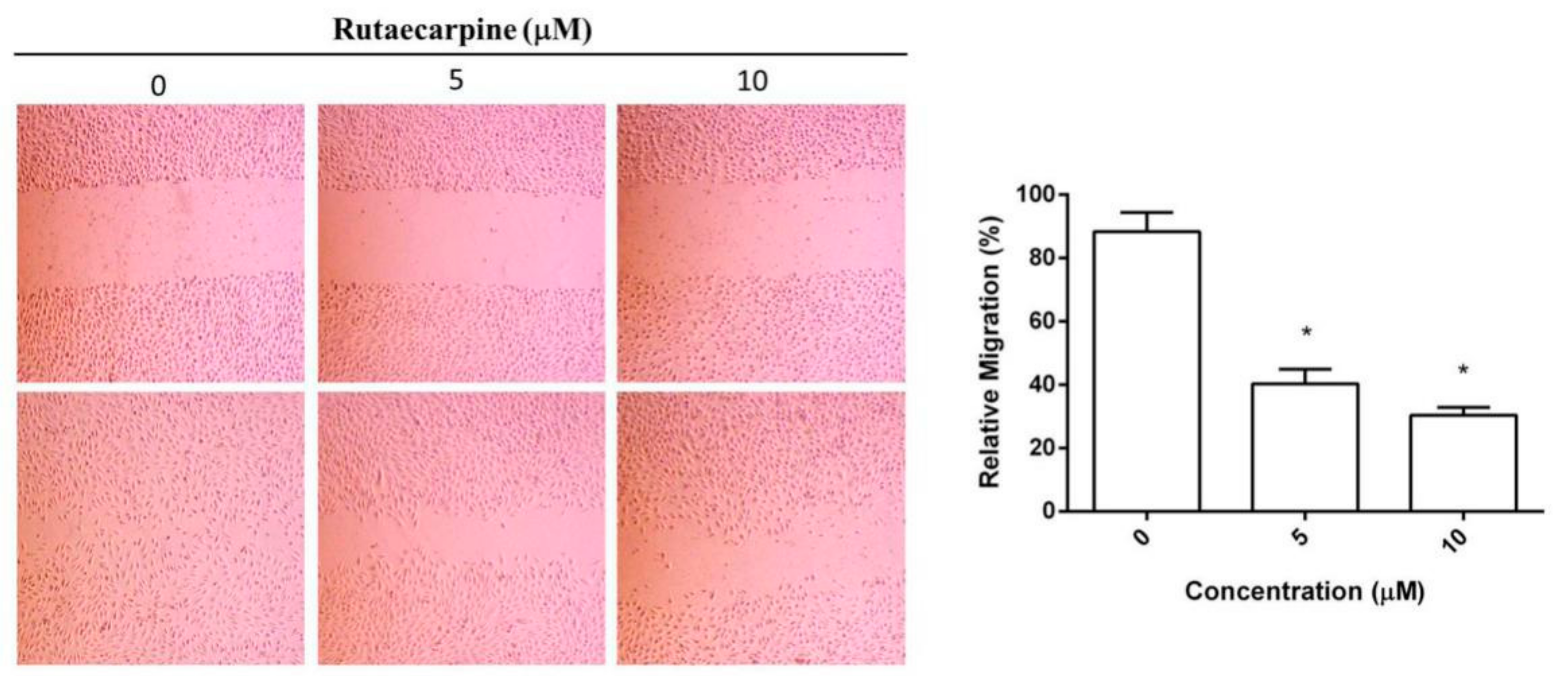
2.3. Inhibition on the Migration of HUVECs
2.4. Ru Inhibited Angiogenesis In Vivo
2.5. Target Prediction
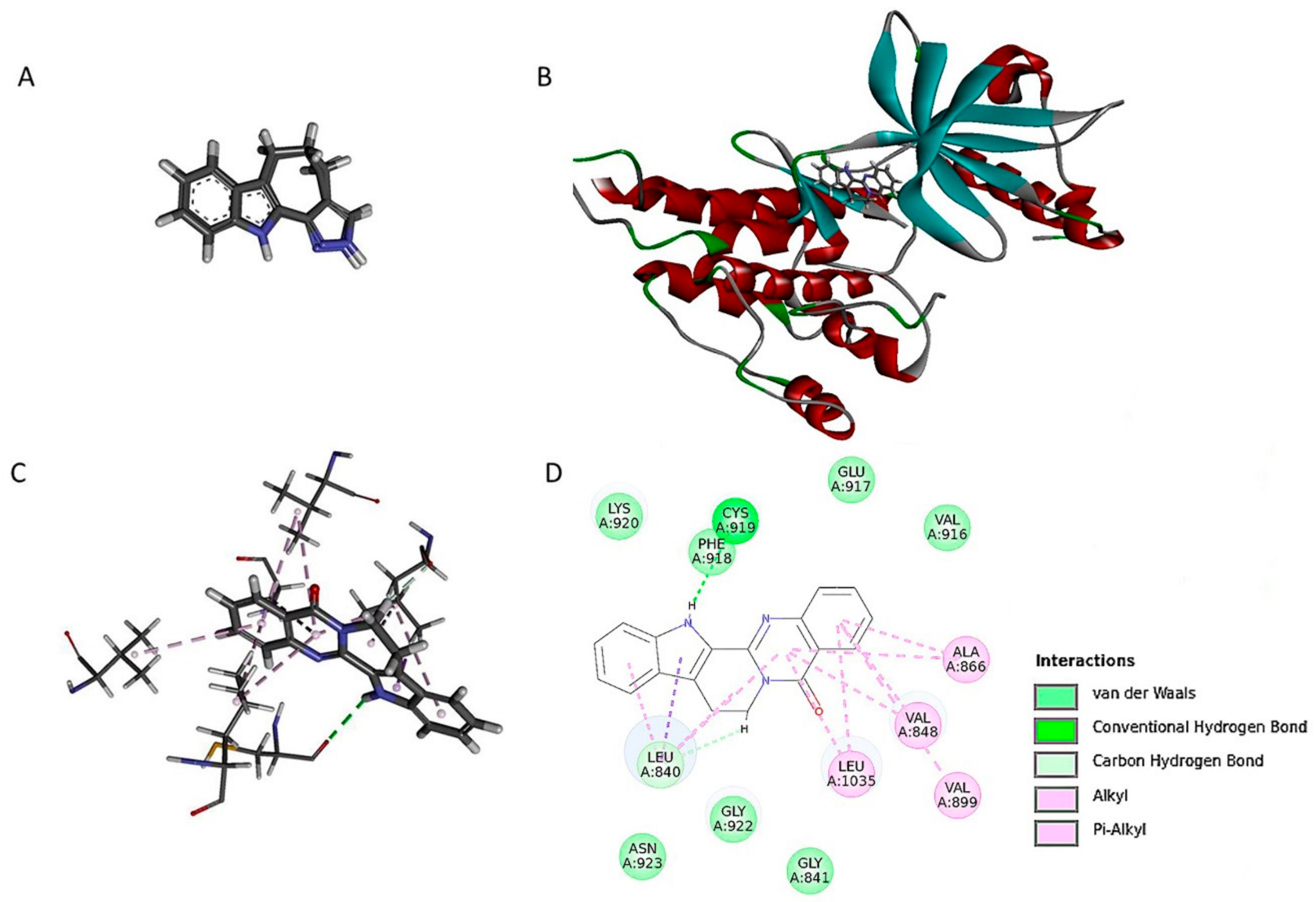
2.6. Ru Directly Bound to VEGFR2 by Molecular Docking and SPR Assay
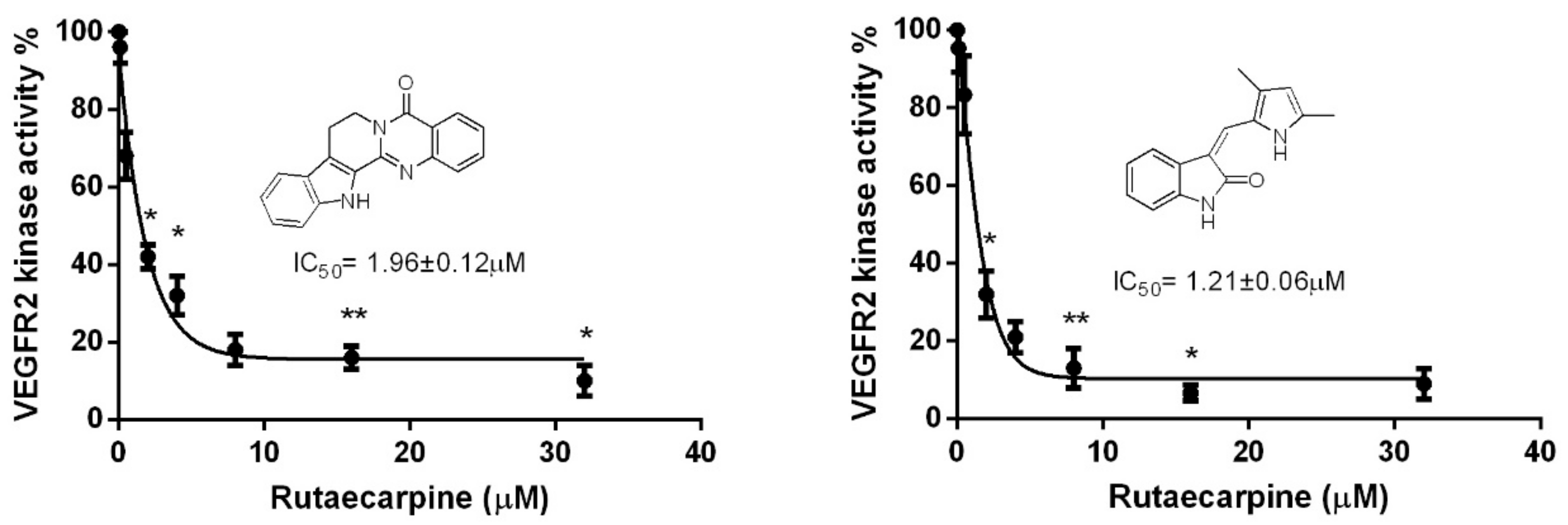
2.7. Ru Attenuates VEGFR2 Tyrosine Kinase Activity
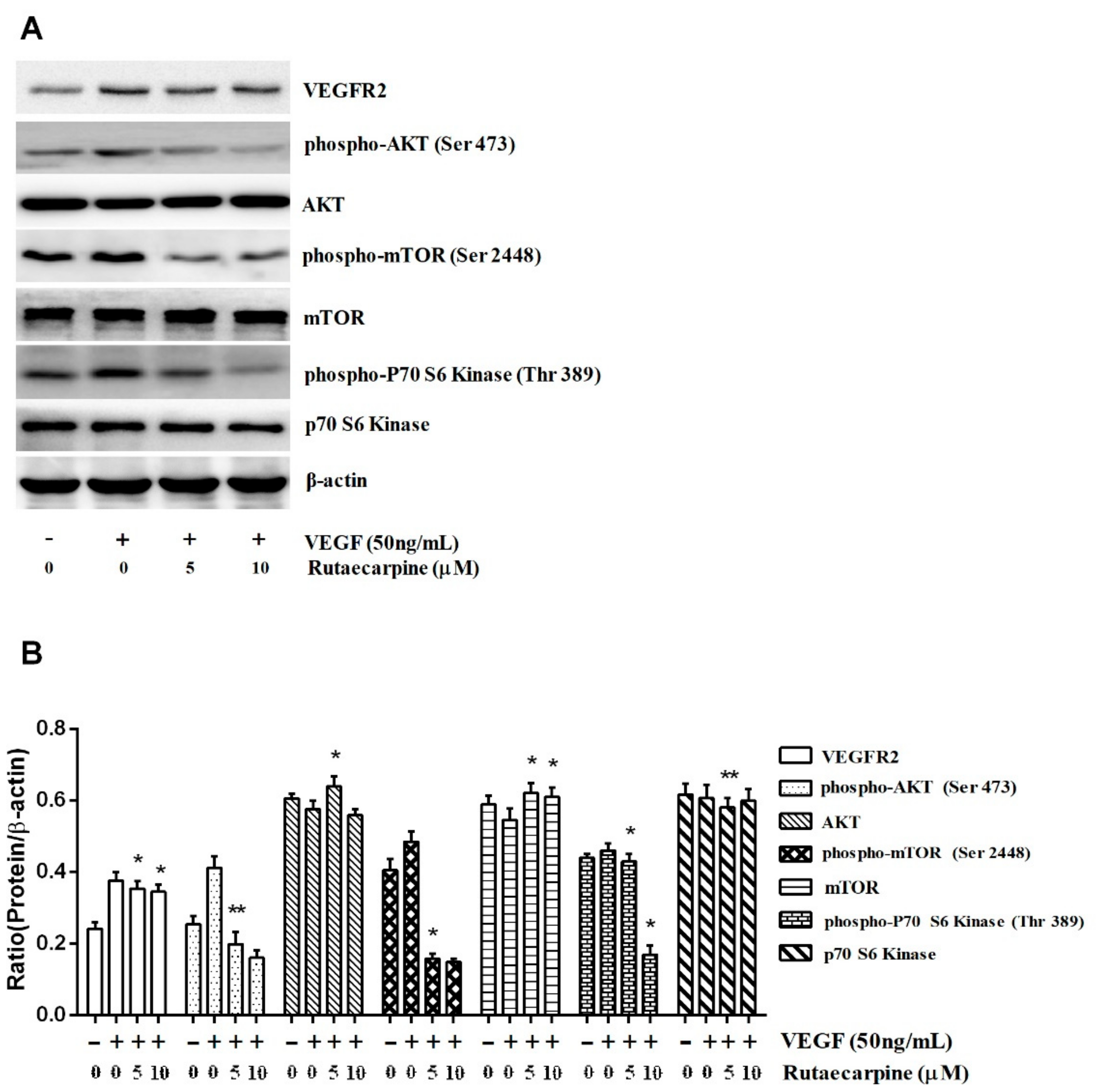
2.8. Ru Inhibits the Activation of VEGFR2-Mediated Akt/mTOR/p70S6K Signaling in HUVECs
3. Materials and Methods
3.1. Materials
3.2. Cell Line and Cell Culture
3.3. Cell Viability Assay
3.4. Extracellular Matrix Adhesion Assay
3.5. Wound Healing Migration Assay
3.6. Chick CAM Assay
3.7. Molecular Modeling
3.8. SPR Assay
3.9. Western Blotting
3.10. Statistical Analysis
4. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Onrubia, M.; Cusido, R.M.; Ramircz, K. Bioprocessing of plant in vitro systems for the mass production of pharmaceutically important metabolites: Paclitaxel and its derivatives. Curr. Med. Chem. 2013, 20, 880–891. [Google Scholar] [PubMed]
- Torre, N.G.; Turner, H.E.; Wass, J.A. Angiogenesis in prolactinom as regulation and relationship with tumour behaviour. Pituitary 2005, 8, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Roguin, A.; Levy, A.P. Angiogenesis—An update. Peditar. Endocrinol. 2005, 2, 391–398. [Google Scholar]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Pandya, N.M.; Dhalla, N.S.; Santani, D.D. Angiogenesis—a new target for future therapy. Vasc. Pharmacol. 2006, 44, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Tumor angiogenesis and therapy. Biomed. Pharmacother. 2005, 59, 340–343. [Google Scholar] [CrossRef]
- Doll, J.A.; Soff, G.A. Angiostatin. Cancer Treat. Res. 2005, 126, 175–204. [Google Scholar] [PubMed]
- Wang, F.; Yamauchi, M.; Muramatsu, M.; Osawa, T.; Tsuchida, R.; Shibuya, M. RACK1 regulates VEGF/Flt1-mediated cell migration via activation of a PI3K/Akt pathway. J. Biol. Chem. 2011, 286, 9097–9106. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. VEGF-VEGFR Signals in Health and Disease. Biomol. Ther. 2014, 22, 1. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Lin, R.C.; Paulus, S. Anti-proliferative effects of evodiamine and Ru on human ovarian cancer cell line SKOV3. Biol. Reprod. 2010, 83, 134–140. [Google Scholar] [CrossRef]
- Chen, M.C.; Yu, C.H.; Wang, S.W.; Pu, H.F.; Kan, S.F.; Lin, L.C.; Chi, C.W.; Ho, L.L.; Lee, C.H.; Wang, P.S. Anti-proliferative effects of evodiamine on human thyroid cancer cell line ARO. J. Cell. Biochem. 2010, 110, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, X.; Wu, D.; Zhang, M.; Ran, G.; Bi, Y.; Huang, H. Growth inhibition and induction of apoptosis in SGC-7901 human gastric cancer cells by evodiamine. Mol. Med. Rep. 2014, 9, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.G.; Lin, S.; Lee, C.C.; Chen, E.; Lin, L.C.; Wang, B.W.; Tsai, S.C. Evodiamine inhibits in vitro angiogenesis: Implication for antitumorgenicity. Life Sci. 2006, 78, 2234–2243. [Google Scholar] [CrossRef] [PubMed]
- Rollinger, J.M.; Schuster, D.; Danzl, B.; Schwaiger, S.; Markt, P.; Schmidtke, M.; Gertsch, J.; Raduner, S.; Wolber, G.; Lange, T.; et al. In silico target fishing forrationalized ligand discovery exemplified on constituents of Ruta graveolens. Planta Med. 2009, 75, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Tan, X.L.; Yan, X.; Liu, H.B. In silico profiling for secondary metabolites from Lepidium meyenii (maca) by the pharmacophore and ligand-shape-based joint approach. Chin. Med. 2016, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, C.; Adasme, F.; Alzate-Morales, J.H; Vergara-Jaque, A.; Kniess, T.; Caballero, J. Study of differences in the VEGFR2 inhibitory activities between semaxanib and SU5205 using 3D-QSAR, docking, and molecular dynamics simulations. J. Mol. Graph. Model. 2012, 32, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Risau, W. Mechanism of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. CSH Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Mhaske, S.B.; Argade, N.P. The chemistry of recently isolated naturally occurring quinazolinone alkaloids. Tetrahedron 2006, 62, 9787–9826. [Google Scholar] [CrossRef]
- Jia, S.J.; Hu, C.P. Pharmacological effects of Ru as a cardiovascular protective agent. Molecules 2010, 15, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.L.; Chao, H.L.; Wang, S.W.; Wang, P.S. Effects of evodiamine and Ru on the secretion of corticosterone by zona fasciculata-reticularis cells in male rats. J. Cell. Biochem. 2009, 108, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Lee, S.J.; Lee, S.; Chae, S.; Han, M.D.; Mar, W.; Nam, K.W. Rutecarpine ameliorates bodyweight gain through the inhibition of orexigenic neuropeptides NPY and AgRP in mice. Biochem. Biophys. Res. Commun. 2009, 89, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Pan, S.L.; Liao, C.H.; Huang, D.Y.; Guh, J.H.; Peng, C.Y.; Chang, Y.L.; Teng, C.M. Evodiamine represses hypoxia-induced inflammatory proteins expression and hypoxia-inducible factor 1alpha accumulation in RAW264.7. Shock 2009, 32, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, Y.; Kontani, Y.; Kobayashi, Y.; Sato, Y.; Mori, N.; Yamashita, H. Evodiamine improves diet-induced obesity in a uncoupling protein-1-independent manner: Involvement of antiadipogenic mechanism and extracellularly regulated kinase/mitogen-activated protein kinase signaling. Endocrinology 2008, 149, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Son, J.K.; Chang, H.W.; Jahng, Y. Progress in Studies on Rutaecarpine. II.—Synthesis and Structure-Biological Activity Relationships. Molecules 2015, 20, 10800–10821. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Yamada, S.; Koizumi, K.; Sakurai, H.; Saiki, I. Tumour-derived fibroblast growth factor-2 exerts lymphangiogenic effects through Akt/mTOR/p70S6kinase pathway in rat lymphatic endothelial cells. Eur. J. Cancer 2007, 43, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tan, D.; Zhang, Z.; Liang, J.J.; Brown, R.E. Activation of Akt-mTOR-p70S6K pathway in angiogenesis in hepatocellular carcinoma. Oncol. Rep. 2008, 20, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley Ras, L.C. PI (3) K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, H.; Guba, M.; Kleespies, A.; Jauch, K.W.; Bruns, C.J. Role of mTOR in solid tumor systems: A therapeutical target against primary tumor growth, metastases, and angiogenesis. Cancer Metast. Rev. 2007, 26, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Eliceiri, B.P.; Puente, X.S.; Hood, J.D.; Stupack, D.G.; Schlaepfer, D.D.; Huang, X.Z.; Sheppard, D.; Cheresh, D.A. Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J. Cell Biol. 2002, 157, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound is available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | PDB ID | Putative Target | Fit Value |
|---|---|---|---|
| 1 | 3jpv-06-s | Proto-oncogene serine/threonine-protein kinase Pim-1 | 0.91901 |
| 2 | 2ivv-06-s | Proto-oncogene tyrosine-protein kinase receptor RET precursor | 0.883419 |
| 3 | 3geq-05-s | Proto-oncogene tyrosine-protein kinase Src | 0.876451 |
| 4 | 3vid-01-s | VEGFR2 | 0.868609 |
| 5 | 3ad4-01-s | Proto-oncogene tyrosine-protein kinase LCK | 0.847169 |
| 6 | 3ad4-02-s | Proto-oncogene tyrosine-protein kinase LCK | 0.844494 |
| 7 | 3p0n-06-s | Tankyrase-2 | 0.825883 |
| 8 | 3qru-04-s | Cyclin-dependent kinase 2 | 0.824893 |
| 9 | 3hrr-03-s | Aflatoxin biosynthesis polyketide synthase | 0.82355 |
| 10 | 3tiz-04-s | Cyclin-dependent kinase 2 | 0.802779 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, L.; Wu, M.; Li, Z. Rutacecarpine Inhibits Angiogenesis by Targeting the VEGFR2 and VEGFR2-Mediated Akt/mTOR/p70s6k Signaling Pathway. Molecules 2018, 23, 2047. https://doi.org/10.3390/molecules23082047
Ji L, Wu M, Li Z. Rutacecarpine Inhibits Angiogenesis by Targeting the VEGFR2 and VEGFR2-Mediated Akt/mTOR/p70s6k Signaling Pathway. Molecules. 2018; 23(8):2047. https://doi.org/10.3390/molecules23082047
Chicago/Turabian StyleJi, Lijun, Mingfei Wu, and Zeng Li. 2018. "Rutacecarpine Inhibits Angiogenesis by Targeting the VEGFR2 and VEGFR2-Mediated Akt/mTOR/p70s6k Signaling Pathway" Molecules 23, no. 8: 2047. https://doi.org/10.3390/molecules23082047
APA StyleJi, L., Wu, M., & Li, Z. (2018). Rutacecarpine Inhibits Angiogenesis by Targeting the VEGFR2 and VEGFR2-Mediated Akt/mTOR/p70s6k Signaling Pathway. Molecules, 23(8), 2047. https://doi.org/10.3390/molecules23082047