Putative Iron Acquisition Systems in Stenotrophomonas maltophilia

 , , and
, , and
Abstract

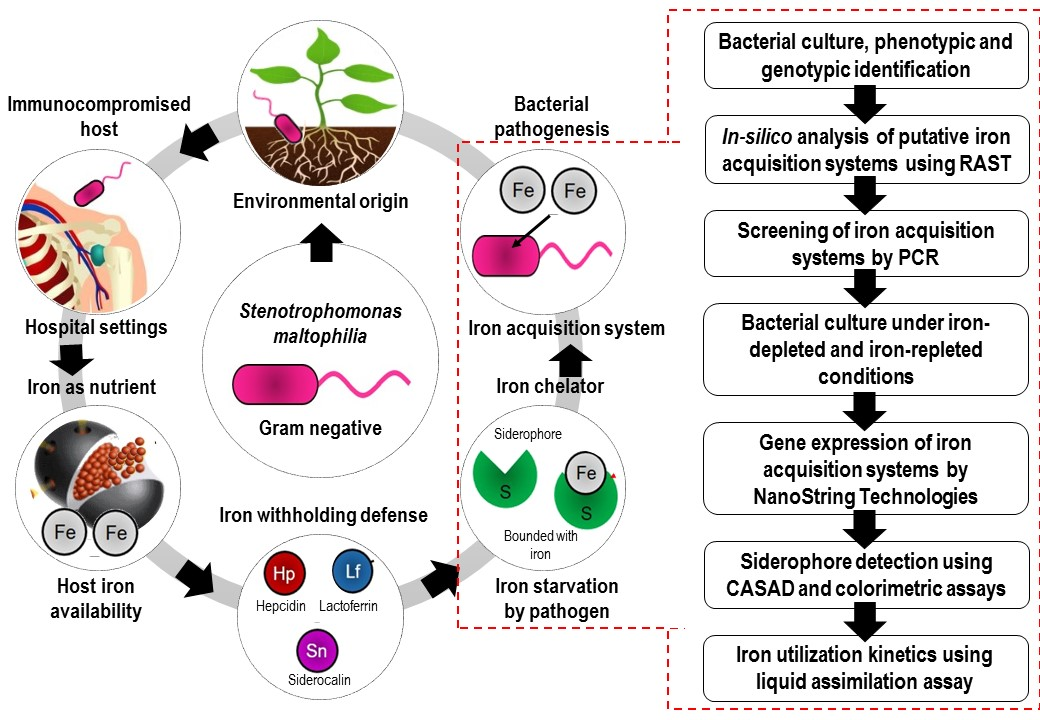
1. Introduction
2. Results
2.1. Putative Iron Acquisition Systems in S. maltophilia
2.2. Distribution of Iron Acquisition Genes and Systems in S. maltophilia
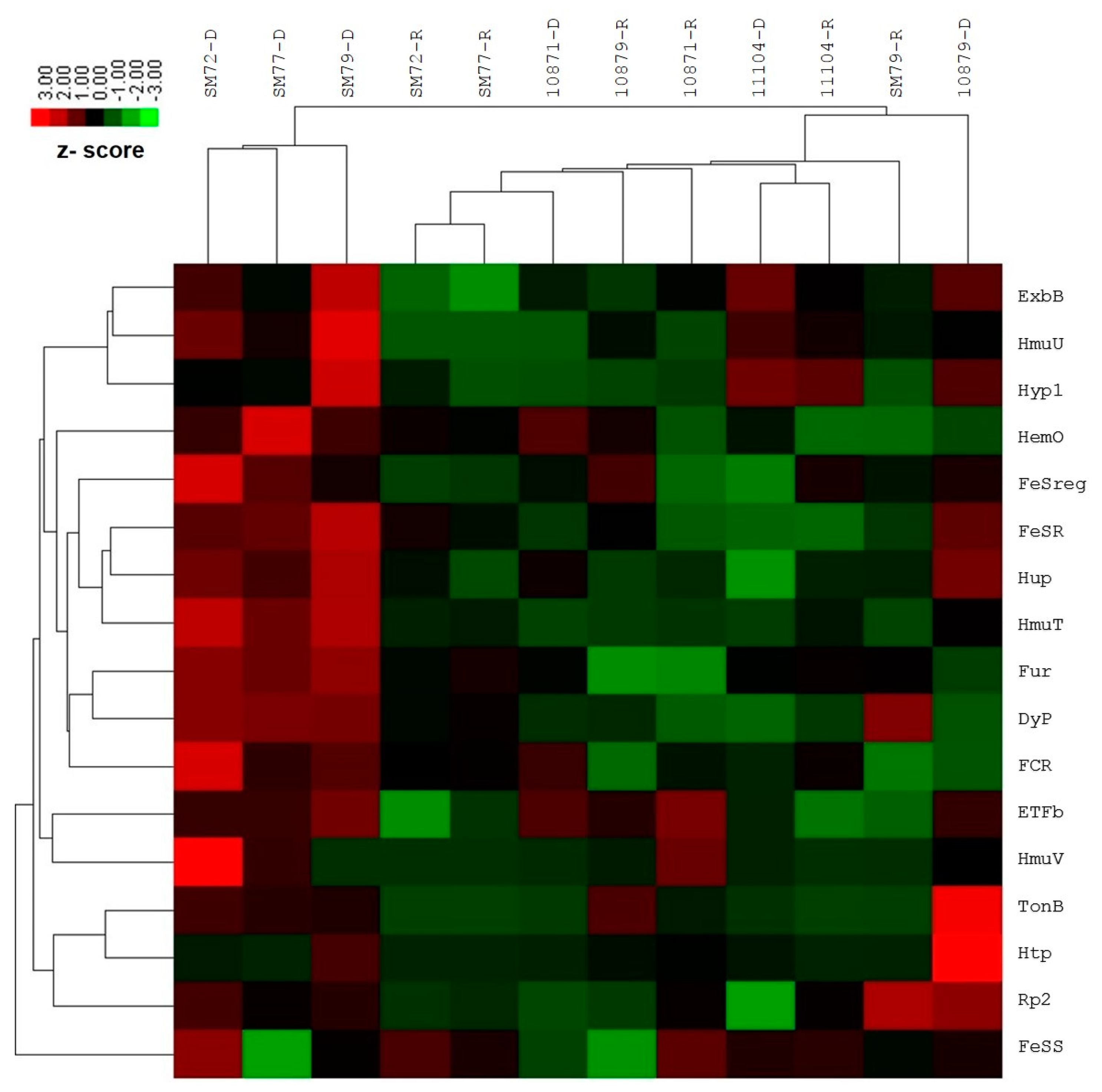
2.3. Expression Profile of the Iron Acquisition System in S. maltophilia
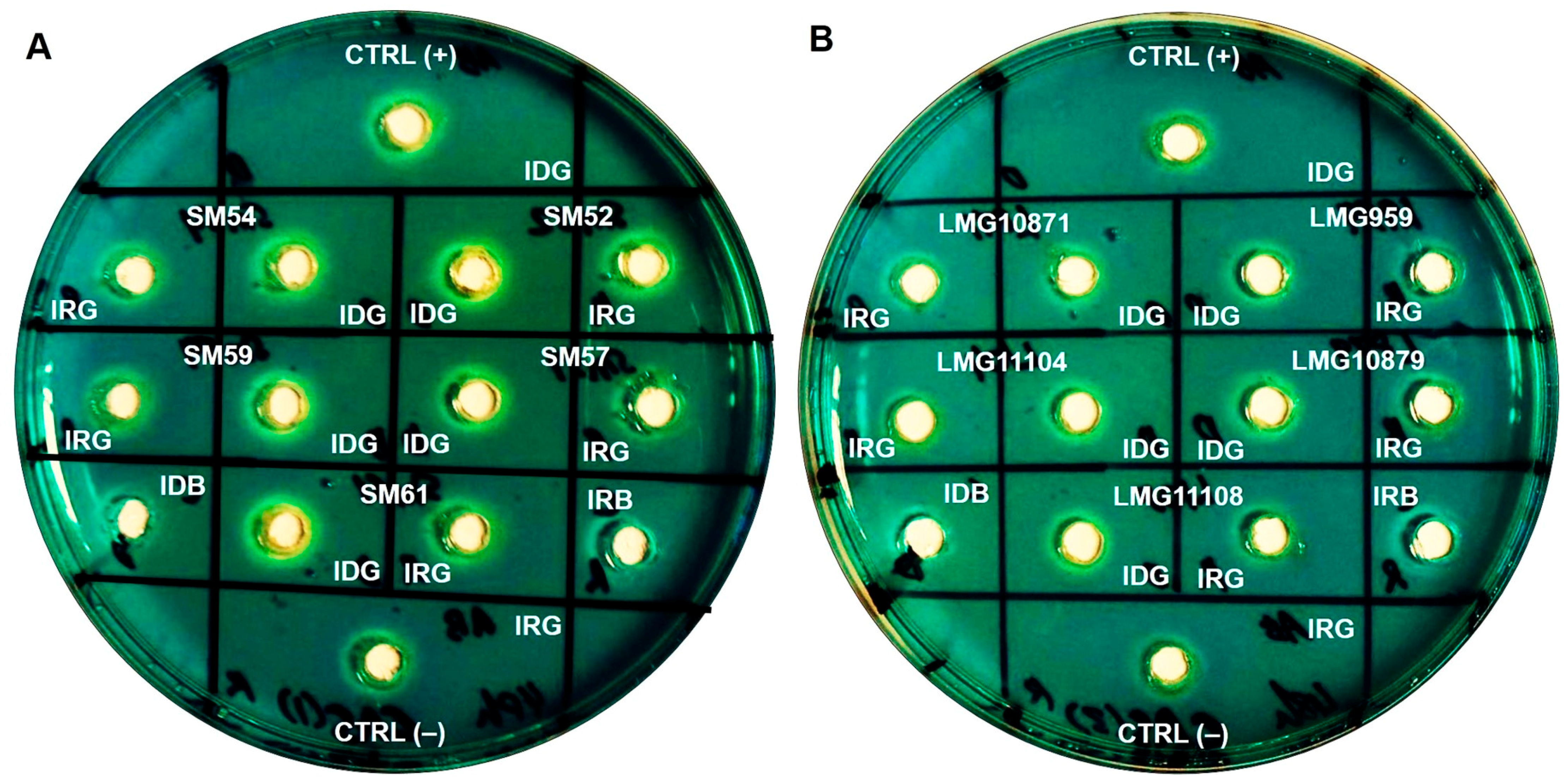
2.4. Siderophore Production and Its Chemical Nature
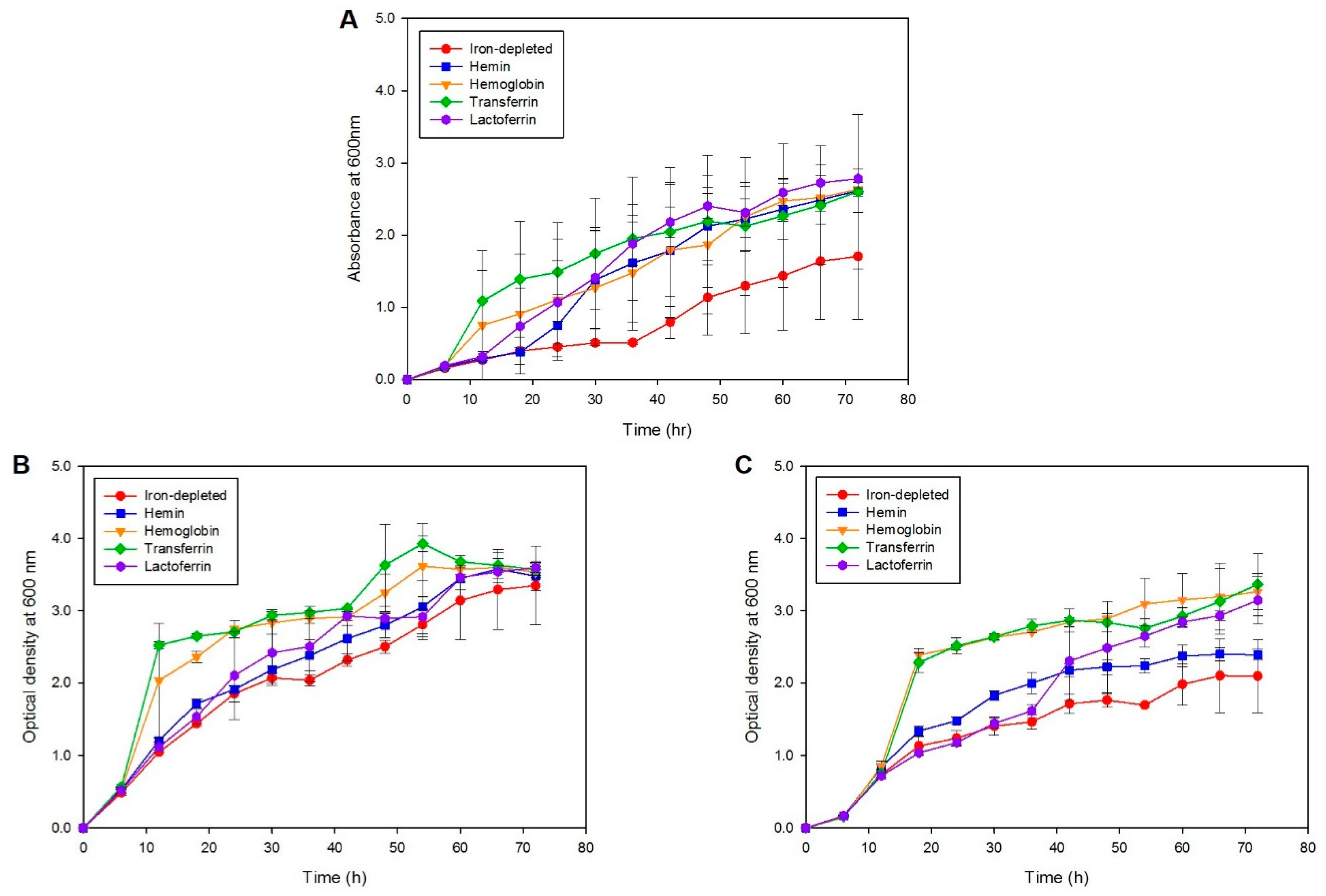
2.5. Iron Source Utilization during Iron Starvation
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Identification and Culture Conditions
4.2. In-Silico Analysis of Putative Iron Acquisition Systems
4.3. Screening of Iron Acquisition Systems by Polymerase Chain Reaction (PCR)
4.4. Bacterial Culture under Iron-Depleted and Iron-Repleted Conditions
4.5. Gene Expression of Iron Acquisition Systems by NanoString Technologies
4.6. Siderophore Detection Using CASAD and Colorimetric Assays
4.7. Iron Utilization Kinetics Using Liquid Assimilation Assay
4.8. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Symeonidis, A.; Marangos, M. Iron and Microbial Growth. In Insight and Control of Infectious Disease in Global Scenario; InTech: London, UK, 2012; pp. 289–330. [Google Scholar]
- Braun, V.; Hantke, K. Recent insights into iron import by bacteria. Curr. Opin. Chem. Biol. 2011, 15, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Skaar, E.P. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 2010, 6, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Marx, J.J.M. Iron and infection: Competition between host and microbes for a precious element. Best Pract. Res. Clin. Haematol. 2002, 15, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.C.; Garcia-Herrero, A.; Johanson, T.H.; Krewulak, K.D.; Lau, C.K.; Peacock, R.S.; Slavinskaya, Z.; Vogel, H.J. Siderophore uptake in bacteria and the battle for iron with the host; a bird’s eye view. BioMetals 2010, 23, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Braun, V.; Braun, M. Iron transport and signaling in Escherichia coli. FEBS Lett. 2002, 529, 78–85. [Google Scholar] [CrossRef]
- Ecker, D.J.; Passavant, C.W.; Emery, T. Role of two siderophores in Ustilago sphaerogena. Regulation of biosynthesis and uptake mechanisms. BBA Mol. Cell Res. 1982, 720, 242–249. [Google Scholar] [CrossRef]
- Schrettl, M.; Ibrahim-Granet, O.; Droin, S.; Huerre, M.; Latgé, J.P.; Haas, H. The crucial role of the Aspergillus fumigatus siderophore system in interaction with alveolar macrophages. Microbes Infect. 2010, 12, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Hensel, M. Pathogenicity islands in bacterial pathogenesis. Clin. Microbiol. Rev. 2004, 17, 14–56. [Google Scholar] [CrossRef] [PubMed]
- Schryvers, A.B.; Stojiljkovic, I. Iron acquisition systems in the pathogenic Neisseria. Mol. Microbiol. 1999, 32, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Looney, W.J.; Narita, M.; Muhlemann, K. Stenotrophomonas maltophilia: An emerging opportunist human pathogen. Lancet Infect. Dis. 2009, 9, 312–323. [Google Scholar] [CrossRef]
- Brooke, J.S. Stenotrophomonas maltophilia: An emerging global opportunistic pathogen. Clin. Microbiol. Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef] [PubMed]
- Pompilio, A.; Pomponio, S.; Crocetta, V.; Gherardi, G.; Verginelli, F.; Fiscarelli, E.; Dicuonzo, G.; Savini, V.; D’Antonio, D.; Di Bonaventura, G. Phenotypic and genotypic characterization of Stenotrophomonas maltophilia isolates from patients with cystic fibrosis: Genome diversity, biofilm formation, and virulence. BMC Microbiol. 2011, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Al-Anazi, K.A.; Al-Jasser, A.M.; Alsaleh, K. Infections Caused by Stenotrophomonas maltophilia in Recipients of Hematopoietic Stem Cell Transplantation. Front. Oncol. 2014, 4, 231. [Google Scholar] [CrossRef] [PubMed]
- Adamek, M.; Linke, B.; Schwartz, T. Virulence genes in clinical and environmental Stenotrophomas maltophilia isolates: A genome sequencing and gene expression approach. Microb. Pathog. 2014, 67–68, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Crossman, L.C.; Gould, V.C.; Dow, J.M.; Vernikos, G.S.; Okazaki, A.; Sebaihia, M.; Saunders, D.; Arrowsmith, C.; Carver, T.; Peters, N.; et al. The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol. 2008, 9, R74. [Google Scholar] [CrossRef] [PubMed]
- García, C.A.; Alcaraz, E.S.; Franco, M.A.; Rossi, B.N.P. De Iron is a signal for Stenotrophomonas maltophilia biofilm formation, oxidative stress response, OMPs expression, and virulence. Front. Microbiol. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.P.; Lee Wong, A.C. A cyclic AMP receptor protein-regulated cell-cell communication system mediates expression of a FecA homologue in Stenotrophomonas maltophilia. Appl. Environ. Microbiol. 2007, 73, 5034–5040. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Chin, K.H.; Hung, H.C.; Shen, G.H.; Wang, A.H.J.; Chou, S.H. Structure of Stenotrophomonas maltophilia FeoA complexed with zinc: A unique prokaryotic SH3—Domain protein that possibly acts as a bacterial ferrous iron-transport activating factor. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Minkwitz, A.; Berg, G. Comparison of Antifungal Activities and 16S Ribosomal DNA Sequences of Clinical and Environmental Isolates of Stenotrophomonas maltophilia. J. Clin. Microbiol. 2001, 39, 139–145. [Google Scholar] [CrossRef] [PubMed]
- García, C.A.; Passerini De Rossi, B.; Alcaraz, E.; Vay, C.; Franco, M. Siderophores of Stenotrophomonas maltophilia: Detection and determination of their chemical nature. Rev. Argent Microbiol. 2012, 44, 150–154. [Google Scholar] [PubMed]
- Taghavi, S.; Garafola, C.; Monchy, S.; Newman, L.; Hoffman, A.; Weyens, N.; Barac, T.; Vangronsveld, J.; Van Der Lelie, D.D. Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl. Environ. Microbiol. 2009, 75, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Lira, F.; Hernandez, A.; Belda, E.; Sanchez, M.B.; Moya, A.; Silva, F.J.; Martinez, J.L. Whole-genome sequence of Stenotrophomonas maltophilia D457, A clinical isolate and a model strain. J. Bacteriol. 2012, 194, 3563–3564. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.; Han, J.; Lapidus, A.; Cheng, J.-F.; Goodwin, L.; Pitluck, S.; Peters, L.; Ovchinnikova, G.; Teshima, H.; Detter, J.C.; et al. Complete Sequence of Stenotrophomonas Maltophilia Jv3. EMBL/GenBank/DDBJ Databases 2011. submitted. [Google Scholar]
- Neela, V.; Rankouhi, S.Z.R.; van Belkum, A.; Goering, R.V.; Awang, R. Stenotrophomonas maltophilia in Malaysia: Molecular epidemiology and trimethoprim-sulfamethoxazole resistance. Int. J. Infect. Dis. 2012, 16, e603–e607. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Hamat, R.A.; Neela, V. Extracellular enzyme profiling of Stenotrophomonas maltophilia clinical isolates. Virulence 2014, 5, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Davenport, K.W.; Daligault, H.E.; Minogue, T.D.; Broomall, S.M.; Bruce, D.C.; Chain, P.S.; Coyne, S.R.; Gibbons, H.S.; Jaissle, J.; Rosenzweig, C.N.; et al. Complete Genome Sequence of Stenotrophomonas maltophilia Type Strain 810-2 (ATCC 13637). Genome Announc. 2014, 2, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Roscetto, E.; Rocco, F.; Carlomagno, M.S.; Casalino, M.; Colonna, B.; Zarrilli, R.; Di Nocera, P.P. PCR-based rapid genotyping of Stenotrophomonas maltophilia isolates. BMC Microbiol. 2008, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, M.; Iacobino, A.; Prosseda, G.; Fiscarelli, E.; Zarrilli, R.; De Carolis, E.; Petrucca, A.; Nencioni, L.; Colonna, B.; Casalino, M. Stenotrophomonas maltophilia strains from cystic fibrosis patients: Genomic variability and molecular characterization of some virulence determinants. Int. J. Med. Microbiol. 2011, 301, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Rocco, F.; De Gregorio, E.; Colonna, B.; Di Nocera, P.P. Stenotrophomonas maltophilia genomes: A start-up comparison. Int. J. Med. Microbiol. 2009, 299, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Lira, F.; Berg, G.; Martínez, J.L. Double-face meets the bacterial world: The opportunistic pathogen Stenotrophomonas maltophilia. Front. Microbiol. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Grim, C.J.; Kothary, M.H.; Gopinath, G.; Jarvis, K.G.; Jean-Gilles Beaubrun, J.; McClelland, M.; Tall, B.D.; Franco, A.A. Identification and characterization of Cronobacter iron acquisition systems. Appl. Environ. Microbiol. 2012, 78, 6035–6050. [Google Scholar] [CrossRef] [PubMed]
- Kaes, C.; Katz, A.; Hosseini, M.W. Bipyridine: The most widely used ligand. A review of molecules comprising at least two 2,2′-bipyridine units. Chem. Rev. 2000, 100, 3553–3590. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.L.; Hemming, B.C. Iron Chelation in Plants and Soil Microorganisms; Academic Press: Cambridge, MA, USA, 1993. [Google Scholar]
- Payne, S.M. Detection, Isolation and Characterization of Siderophores. Methods Enzymol. 1994, 235, 229–344. [Google Scholar] [CrossRef]
- Navarro Llorens, J.M.; Tormo, A.; Martínez-García, E. Stationary phase in gram-negative bacteria. FEMS Microbiol. Rev. 2010, 34, 476–495. [Google Scholar] [CrossRef] [PubMed]
- Beaume, M.; Hernandez, D.; Docquier, M.; Delucinge-Vivier, C.; Descombes, P.; François, P. Orientation and expression of methicillin-resistant Staphylococcus aureus small RNAs by direct multiplexed measurements using the nCounter of NanoString technology. J. Microbiol. Methods 2011, 84, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Roskot, N.; Smalla, K. Genotypic and Phenotypic Relationships between Clinical and Environmental Isolates of Stenotrophomonas maltophilia. J. Clin. Microbiol. 1999, 37, 3594–3600. [Google Scholar] [PubMed]
- Crowley, D.E. Microbial Siderophores in the plant rhizosphere. In Iron Nutrition in Plants and Rhizospheric Microorganisms; Springer Science & Business Media: Berlin, Germany, 2006; pp. 169–198. ISBN 1402047428. [Google Scholar]
- Dunne, C.; Crowley, J.J.; Moenne-Loccoz, Y.; Dowling, D.N.; De Bruijn, F.J.; O’Gara, F. Biological control of Pythium ultimum by Stenotrophomonas maltophilia W81 is mediated by an extracellular proteolytic activity. Microbiology 1997, 143, 3921–3931. [Google Scholar] [CrossRef]
- Mokracka, J.; Cichoszewska, E.; Kaznowski, A. Siderophore production by Gram-negative rods isolated from human polymicrobial infections. Biol. Lett. 2011, 48, 147–157. [Google Scholar] [CrossRef]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Chhibber, S.; Gupta, A.; Sharan, R.; Gautam, V.; Ray, P. Putative virulence characteristics of Stenotrophomonas maltophilia: A study on clinical isolates. World J. Microbiol. Biotechnol. 2008, 24, 2819–2825. [Google Scholar] [CrossRef]
- Perkins-Balding, D.; Ratliff-Griffin, M.; Stojiljkovic, I. Iron transport systems in Neisseria meningitidis. Microbiol. Mol. Biol. Rev. 2004, 68, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.N.M.; Jones, H.A.; Jones, H.A.; Perry, R.D.; Perry, R.D. Molecular Characterization of the Hemin Uptake Locus (hmu) from Yersinia pestis and Analysis ofhmu Mutants for Hemin and Hemoprotein Utilization. Infect. Immun. 1999, 67, 3879–3892. [Google Scholar] [PubMed]
- Hornung, J.M.; Jones, H.A.; Perry, R.D. The HMU locus of Yersinia pestis is essential for utilization of free haemin and haem-protein complexes as iron sources. Mol. Microbiol. 1996, 20, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Qin, L.; Han, Y.; Qiu, J.; Chen, Z.; Li, B.; Song, Y.; Wang, J.; Guo, Z.; Zhai, J.; et al. Global analysis of iron assimilation and fur regulation in Yersinia pestis. FEMS Microbiol. Lett. 2006, 258, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Stojiljkovic, I.; Hantke, K. Hemin uptake system of Yersinia enterocolitica: Similarities with other TonB-dependent systems in gram-negative bacteria. EMBO J. 1992, 11, 4359–4367. [Google Scholar] [PubMed]
- Braun, V. Bacterial Iron Transport Related to Virulence. Signal. Gene Regul. 2005, 12, 210–233. [Google Scholar] [CrossRef]
- Braun, V.; Hantke, K. Acquisition of Iron by Bacteria. Mol. Microbiol. Heavy Met. 2007, 43, 189–219. [Google Scholar] [CrossRef]
- Kikuchi, G.; Yoshida, T.; Noguchi, M. Heme oxygenase and heme degradation. Biochem. Biophys. Res. Commun. 2005, 338, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Pieracci, F.M.; Barie, P.S. Iron and the Risk of Infection. Surg. Infect. 2005, 6, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Manganaro, L.; O’Brien, J.; Trottier, S.J.; Parkar, N.; Veremakis, C. Impact of allogenic packed red blood cell transfusion on nosocomial infection rates in the critically ill patient. Crit. Care Med. 2002, 30, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.W.; Ramos, P.L.; Ferreira, C.A.S.; de Oliveira, S.D. A specific polymerase chain reaction method to identify Stenotrophomonas maltophilia. Mem. Inst. Oswaldo Cruz 2013, 108, 390–391. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA 6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Owczarzy, R.; Tataurov, A.V.; Wu, Y.; Manthey, J.A.; McQuisten, K.A.; Almabrazi, H.G.; Pedersen, K.F.; Lin, Y.; Garretson, J.; McEntaggart, N.O.; et al. IDT SciTools: A suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 2008, 36, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Louden, B.C.; Haarmann, D.; Lynne, A. Use of Blue Agar CAS Assay for Siderophore Detection. J. Microbiol. Biol. Educ. 2011, 12, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.M. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr. Protoc. Mol. Biol. 2011, 1–17. [Google Scholar] [CrossRef]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Solis, N.V.; Filler, S.G.; Mitchell, A.P. Gene Expression Profiling of Infecting Microbes Using a Digital Bar-coding Platform. J. Vis. Exp. 2016, 107, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Lim, Y.; Lee, S.E.; Yang, N.W.; Rhee, J.H. CAS agar diffusion assay for the measurement of siderophores in biological fluids. J. Microbiol. Methods 2001, 44, 89–95. [Google Scholar] [CrossRef]
- Schwyn, B.; Neilands, J.B. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 1987, 160, 47–56. [Google Scholar] [CrossRef]
- Pal, R.B.; Gokarn, K. Siderophores and pathogenecity of microorganisms. J. Biosci. Technol. 2010, 1, 127–134. [Google Scholar]
- Machuca, A.; Milagres, A.M.F. Use of CAS-agar plate modified to study the effect of different variables on the siderophore production by Aspergillus. Lett. Appl. Microbiol. 2003, 36, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Atkin, C.L.; Neilands, J.B.; Phaff, H.J. Rhodotorulic Acid from Species of Leucosporidium, Rhodosporidium, Rhodotorula, Sporidiobolus, and Sporobolomyces, and a New Alanine-Containing Ferrichrome from Cryptococcus melibiosum. J. Bacteriol. 1970, 103, 722–733. [Google Scholar] [PubMed]
- Arnow, L.E. Colorimetric determination of the components of 3,4-dihydroxyphenylalaninetyrosine mixtures. J. Biol. Chem. 1937, 531–537. [Google Scholar]
- Watts, R.E.; Totsika, M.; Challinor, V.L.; Mabbett, A.N.; Ulett, G.C.; De Voss, J.J.; Schembri, M.A. Contribution of siderophore systems to growth and urinary tract colonization of asymptomatic bacteriuria Escherichia coli. Infect. Immun. 2012, 80, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, C.W.; Tomaras, A.P.; Connerly, P.L.; Tolmasky, M.E.; Crosa, J.H.; Actis, L.A. The siderophore-mediated iron acquisition systems of Acinetobacter baumannii ATCC 19606 and Vibrio anguillarum 775 are structurally and functionally related. Microbiology 2004, 150, 3657–3667. [Google Scholar] [CrossRef] [PubMed]
- Kvitko, B.H.; Goodyear, A.; Propst, K.L.; Dow, S.W.; Schweizer, H.P. Burkholderia pseudomallei known siderophores and hemin uptake are dispensable for lethal murine melioidosis. PLoS Negl. Trop. Dis. 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Genco, C.A.; Chen, C.Y.; Arko, R.J.; Kapczynski, D.R.; Morse, S.A. Isolation and characterization of a mutant of Neisseria gonorrhoeae that is defective in the uptake of iron from transferrin and haemoglobin and is avirulent in mouse subcutaneous chambers. J. Gen. Microbiol. 1991, 137, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Hunt, D.J.; Richardson, A.R.; Stojiljkovic, I. Use of Heme Compounds as Iron Sources by Pathogenic Neisseriae Requires the Product of the hemO Gene. J. Bacteriol. 2000, 182, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Stojiljkovic, I.; Srinivasan, N. Neisseria meningitidis tonB, exbB, and exbD genes: Ton-dependent utilization of protein-bound iron in Neisseriae. J. Bacteriol. 1997, 179, 805–812. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain I.D. | Biological Source | Geographical Source | GenBank Accession Number | References |
|---|---|---|---|---|
| In-silico | ||||
| K279a | Clinical (Blood infection) | Bristol, UK | AE016879 | [16] |
| R551-3 | Environmental (Poplar tree endophyte) | Washington, USA | CP001111 | [22] |
| D457 | Clinical | Mostoles, Spain | HE798556 | [23] |
| JV3 | Environmental (Rhizosphere) | Brazil | CP002986 | [24] |
| Clinical | ||||
| SM1 to SM 101 | Various specimens | Malaysia | — | [25,26] |
| CS17 | Blood | Malaysia | — | [26] |
| CS24 | Wound swab | Malaysia | — | [26] |
| ATCC13637 | Pleural fluid of a patient with oral carcinoma | Stafford, England | CP008838 | [27] |
| Environmental | ||||
| LMG959 | Rice paddy | Japan | — | [28] |
| LMG10871 | Soil | Japan | — | [28] |
| LMG10879 | Rice paddy | Japan | — | [28] |
| LMG11104 | Roots | Unknown | — | [29] |
| LMG11108 | Roots | Unknown | — | [29] |
| Targets a | Functional Role a | Locus Tag b | BLAST Identity c |
|---|---|---|---|
| Subsystem: Iron siderophore sensor & receptor systems | |||
| FeSreg | Sigma factor ECF subfamily | SMLT_RS12950 | 98% (13637) |
| FeSR | Iron siderophore receptor protein | SMLT_RS18575 | 99% (D457) |
| FeSS | Iron siderophore sensor protein | SMLT_RS18580 | 99% (K279a) |
| Subsystem: Heme, hemin uptake and utilization systems in Gram-positives | |||
| HemO/HO | Heme oxygenase, associated with heme uptake | SMLT_RS18565 | 100% (13637) |
| HmuV | Heme ABC transporter, ATPase component | SMLT_RS11325 | 99% (K279a) |
| Hyp1 | Hypothetical protein related to heme utilization | SMLT_RS19415 | 98% (1337) |
| HmuU | Heme ABC transporter, permease protein | SMLT_RS11320 | 92% (K279a) |
| HmuT | Heme ABC transporter, cell surface heme and hemoprotein receptor | SMLT_RS11315 | 97% (D457) |
| Subsystem: Heme, hemin uptake and utilization systems in Gram-negatives | |||
| Rp2 | Outer membrane receptor proteins, mostly Fe transport | SMLT_RS18050 | 95% (D457) |
| Hup | Hemin uptake protein | SMLT_RS03780 | 100% (D457) |
| ETFb | Electron transfer flavoprotein, beta subunit | SMLT_RS03080 | 100% (D457) |
| TonB | Ferric siderophore transport system, periplasmic binding protein | SMLT_RS21345 | 99% (D457) |
| ExbB | Ferric siderophore transport system, biopolymer transport protein | SMLT_RS07890 | 99% (K279a) |
| Htp | Hemin transport protein | SMLT_RS03790 | 99% (K279a) |
| FCR | TonB-dependent hemin, ferrichrome receptor | SMLT_RS03785 | 99% (K279a) |
| Subsystem: Encapsulating protein DyP-type peroxidase and ferritin-like protein oligomers | |||
| DyP | Predicted dye-decolorizing peroxidase, encapsulated subgroup | SMLT_RS00875 | 95% (K279a) |
| Subsystem: Oxidative stress | |||
| Fur | Ferric uptake regulation protein (FUR) | SMLT_RS09600 | 96% (K279a) |
| Targets | Abbrev. (RAST) | Primer Pairs | Target Sequence (5′–3′) | Amplicon Size (bp) | Ta (°C) |
|---|---|---|---|---|---|
| Iron siderophore sensor & receptor system | |||||
| Sigma factor ECF subfamily | FeSreg | FeSreg-F FeSreg-R | TTCATCGCGCGCTATCTC AGGATGGTCCGGGTGAT | 275 | 50 |
| Iron siderophore receptor protein | FeSR | FeSR-F FeSR-R | CAATCGCAGCGTACCTACC CGGCCACGTTGAAGAACT | 271 | 51 |
| Iron siderophore sensor protein | FeSS | FeSS-F FeSS-R | ACGTCGTGCAGAACGTAAC GGGTTTCCACCAGGTCATC | 237 | 51 |
| Heme, hemin uptake and utilization systems in Gram-positives | |||||
| Heme oxygenase, associated with heme uptake | HemO/HO | HemO-F HemO-R | CAGCAATTTCGCCCGTTTC GCTTGGCAGCCATCTTGTA | 281 | 51 |
| Heme ABC transporter, ATPase component | HmuV | HmuV-F HmuV-R | GAAGCTGCATGAGGTGGT TCTACGCTGAAGGCGAAAC | 273 | 51 |
| Hypothetical protein related to heme utilization | Hyp1 | Hyp1-F Hyp1-R | GGCATCGTCGGCATCTT ACTTCACCCAGGCAATCG | 247 | 51 |
| Heme ABC transporter, permease protein | HmuU | HmuU-F HmuU-R | ACGCCATTGGACATGCT AACAAGCCCAGCGGAAT | 299 | 50 |
| Heme ABC transporter, cell surface heme and hemoprotein receptor | HmuT | HmuT-F HmuT-R | CATGCGCCACGACTGAT CATCACCCAGACCCGATTG | 300 | 51 |
| Heme, hemin uptake and utilization systems in Gram-negatives | |||||
| Outer membrane receptor proteins, mostly Fe transport | Rp2 | Rp2-F Rp2-R | AACGCATGCCCGACTAC CTGGCTCATGCCCATCAT | 221 | 51 |
| Hemin uptake protein | Hup | Hup-F Hup-R | ATGCTCATGAATGCTCAACC TACTTGGTCAGGATCAGCTTG | 200 | 51 |
| Electron transfer flavoprotein, beta subunit | ETFb | ETFb-F ETFb-R | CCTGGAAACGCTGGAAGT CCTTGACCATCACACCCTT | 215 | 51 |
| Ferric siderophore transport system, periplasmic binding protein | TonB | TonB-F TonB-R | CGCGAGAACCGCATGTAT TCCTCGGCGTCCTTCTT | 314 | 51 |
| Ferric siderophore transport system, biopolymer transport protein | ExbB | ExbB-F ExbB-R | GAGCGTTTCTGGTCCCTTC CCCAGTGCGTTCAGGAAT | 251 | 51 |
| Hemin transport protein | Htp | Htp-F Htp-R | CACCGTGTTGTGCCTGTA GCCTCGCTATCGTGTTCC | 234 | 51 |
| TonB-dependent hemin, ferrichrome receptor | FCR | FCR-F FCR-R | CGGAAATGAAGGCCGGTATC CCATTCGATGTAGCGCTTGT | 380 | 51 |
| Encapsulating protein DyP-type peroxidase and ferritin-like protein oligomers | |||||
| Predicted dye-decolorizing peroxidase, encapsulated subgroup | DyP | DyP-F DyP-R | GTGCTGAAGGTGAAGGATGA TGCACGGATGTGGTACAG | 258 | 50 |
| Oxidative stress related to iron uptake | |||||
| Ferric uptake regulation protein FUR | Fur | Fur-F Fur-R | TGACCGCCGAAGACATCTA GCGAGTGCTCTTCCAGTTC | 279 | 51 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalidasan, V.; Azman, A.; Joseph, N.; Kumar, S.; Awang Hamat, R.; Neela, V.K. Putative Iron Acquisition Systems in Stenotrophomonas maltophilia. Molecules 2018, 23, 2048. https://doi.org/10.3390/molecules23082048
Kalidasan V, Azman A, Joseph N, Kumar S, Awang Hamat R, Neela VK. Putative Iron Acquisition Systems in Stenotrophomonas maltophilia. Molecules. 2018; 23(8):2048. https://doi.org/10.3390/molecules23082048
Chicago/Turabian StyleKalidasan, V., Adleen Azman, Narcisse Joseph, Suresh Kumar, Rukman Awang Hamat, and Vasantha Kumari Neela. 2018. "Putative Iron Acquisition Systems in Stenotrophomonas maltophilia" Molecules 23, no. 8: 2048. https://doi.org/10.3390/molecules23082048
APA StyleKalidasan, V., Azman, A., Joseph, N., Kumar, S., Awang Hamat, R., & Neela, V. K. (2018). Putative Iron Acquisition Systems in Stenotrophomonas maltophilia. Molecules, 23(8), 2048. https://doi.org/10.3390/molecules23082048