
A Dynamic Overview of Antimicrobial Peptides and Their Complexes


Abstract
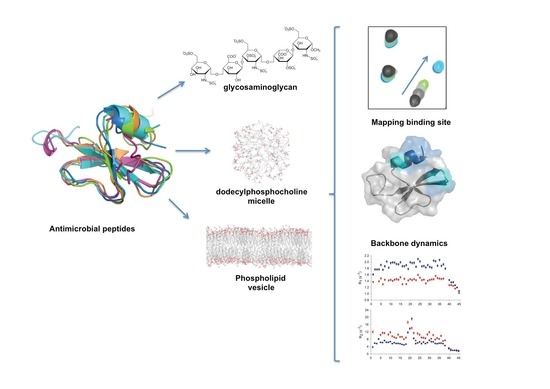
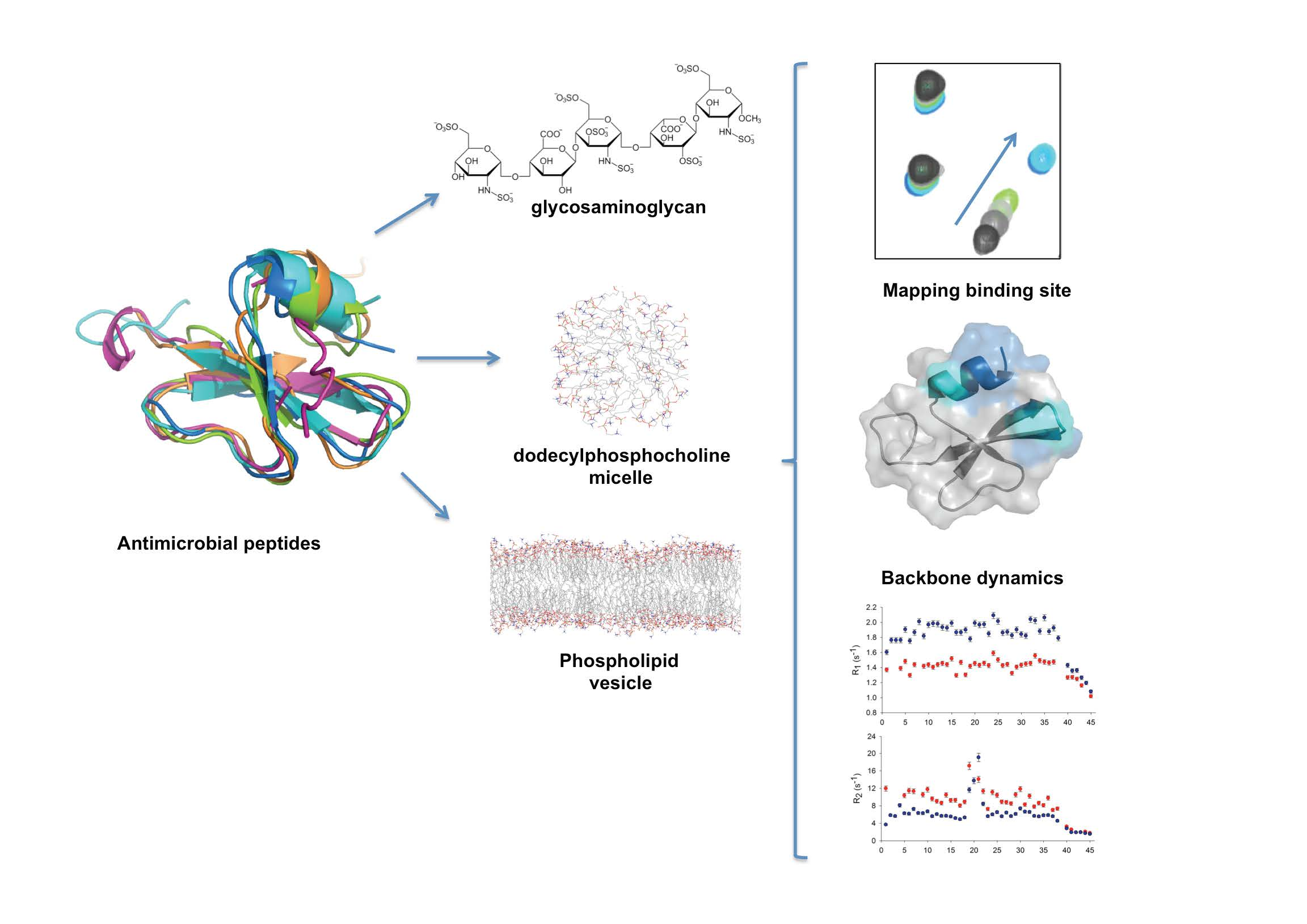{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Antimicrobial Peptides
2.1. Conformational Dynamics of Defensins
2.2. Sugar Cane Defensin 5
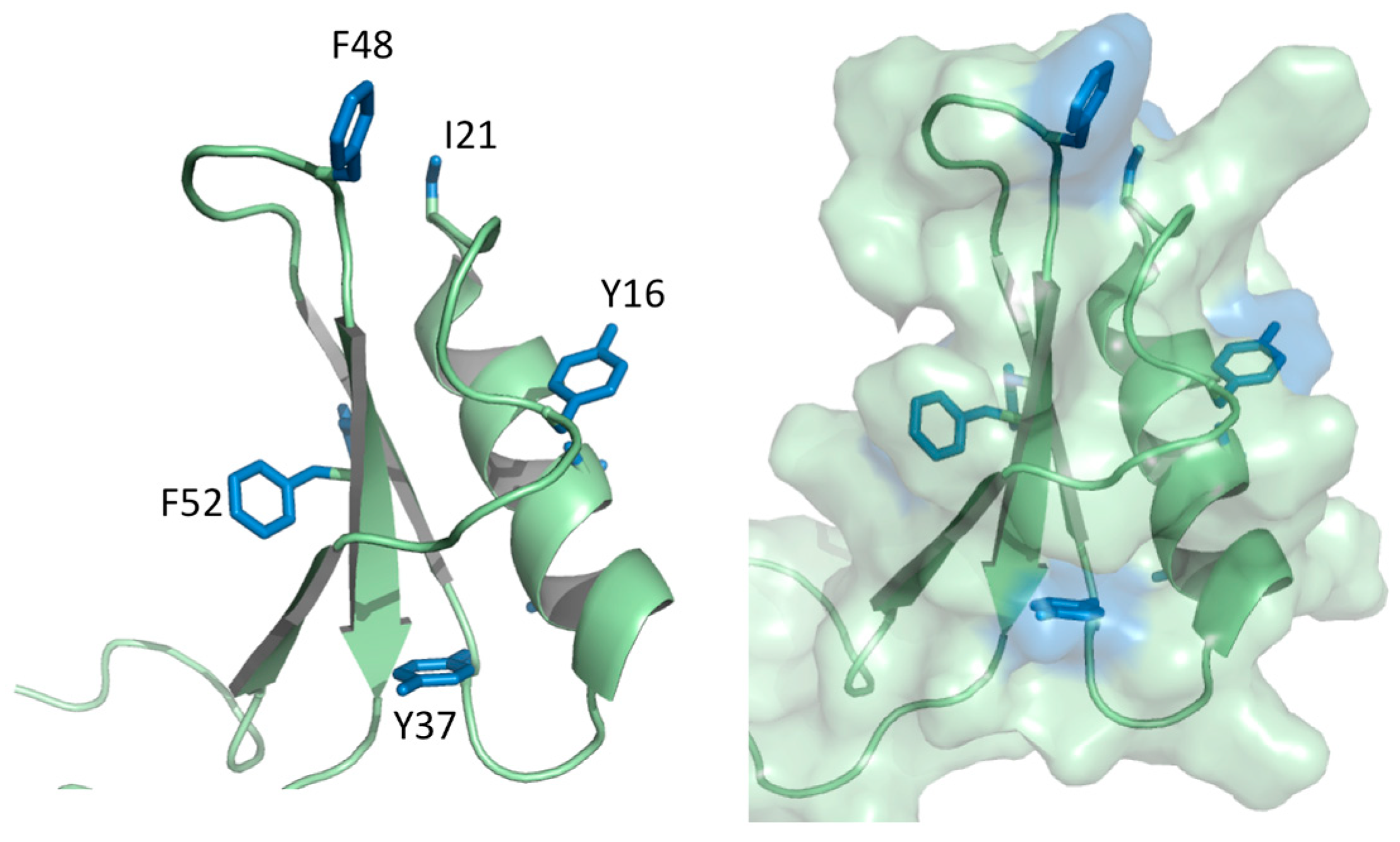
2.3. Dynamics Obtained Using Molecular Dynamics Simulations
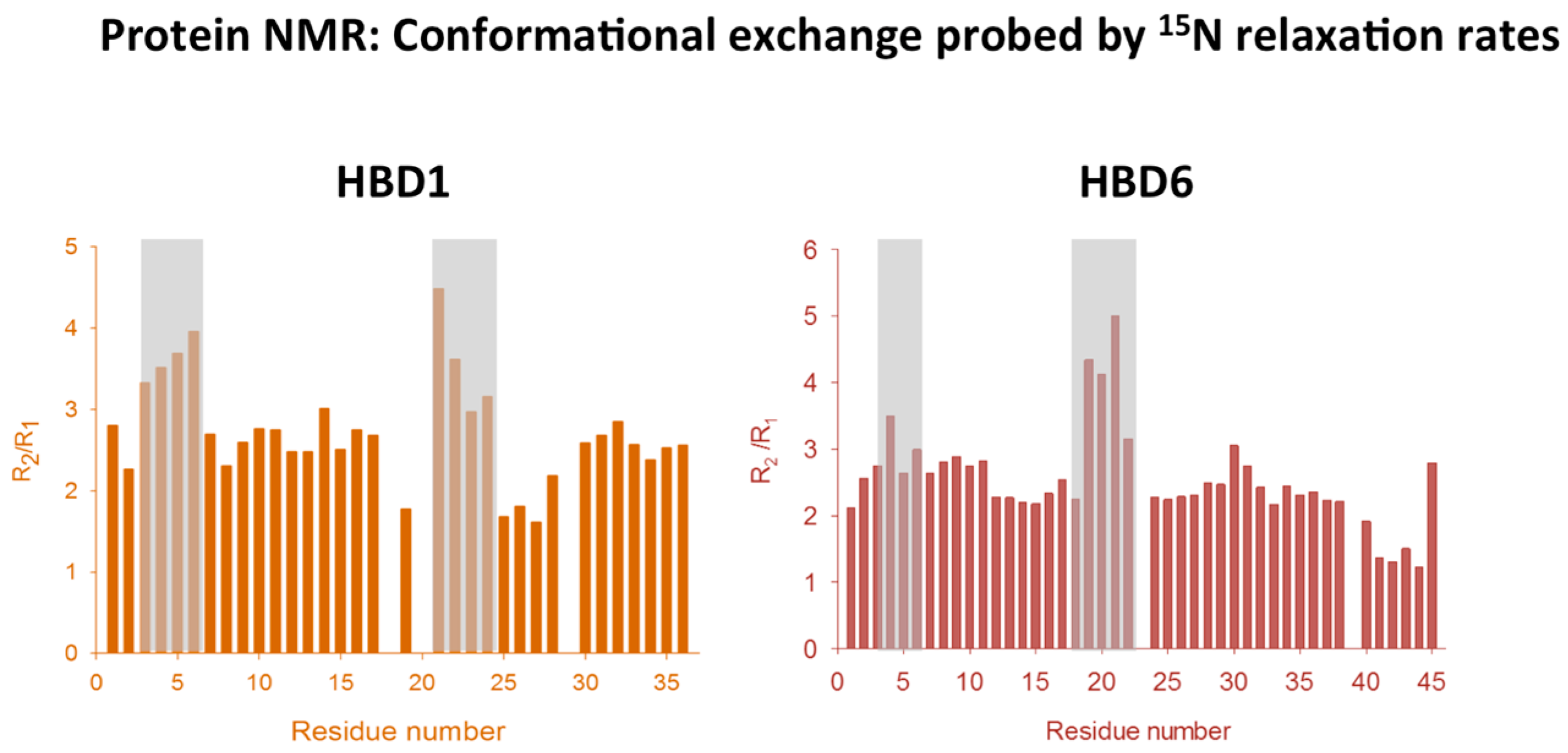
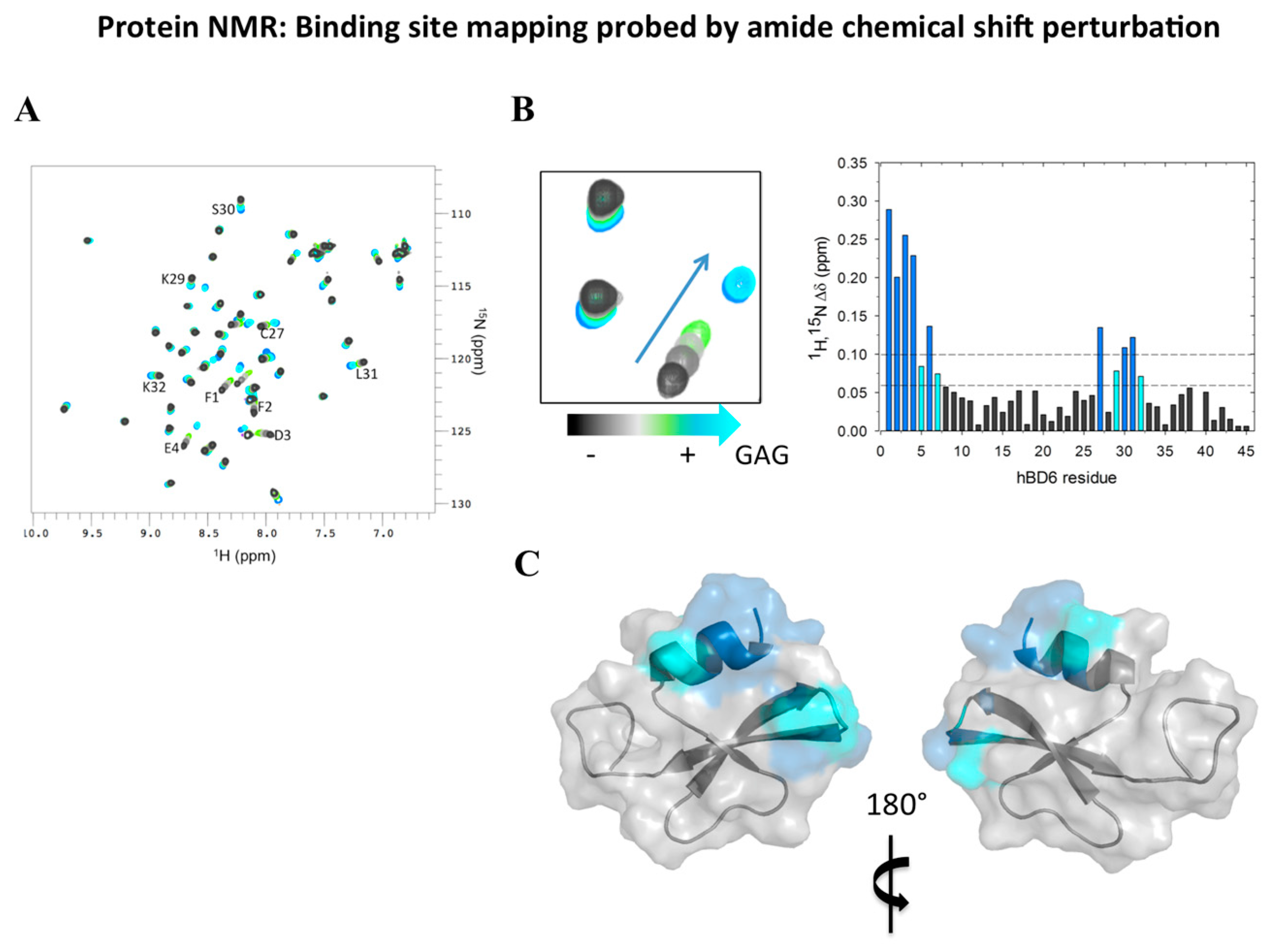
2.4. Analyzing Defensin Complexes Using NMR
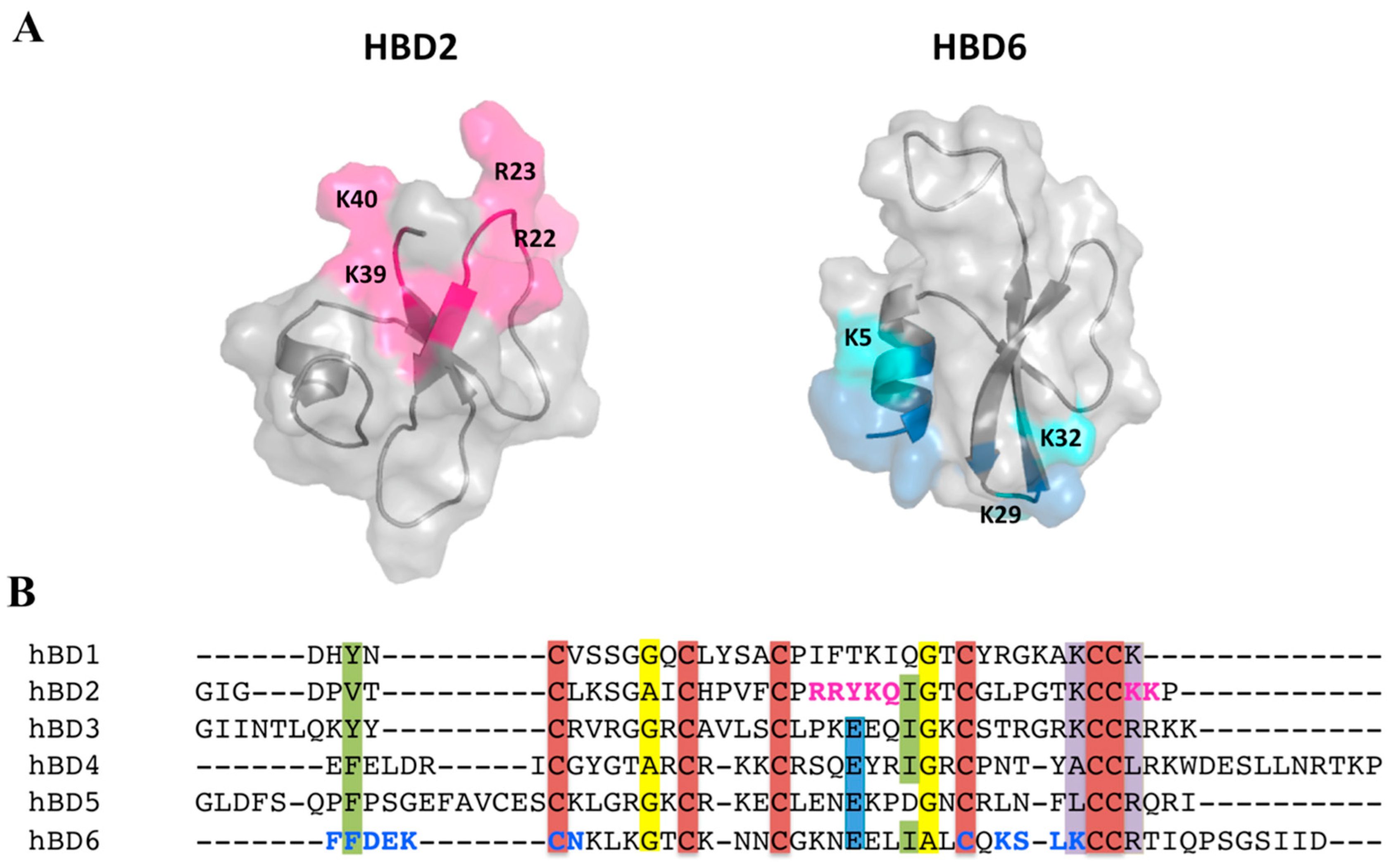
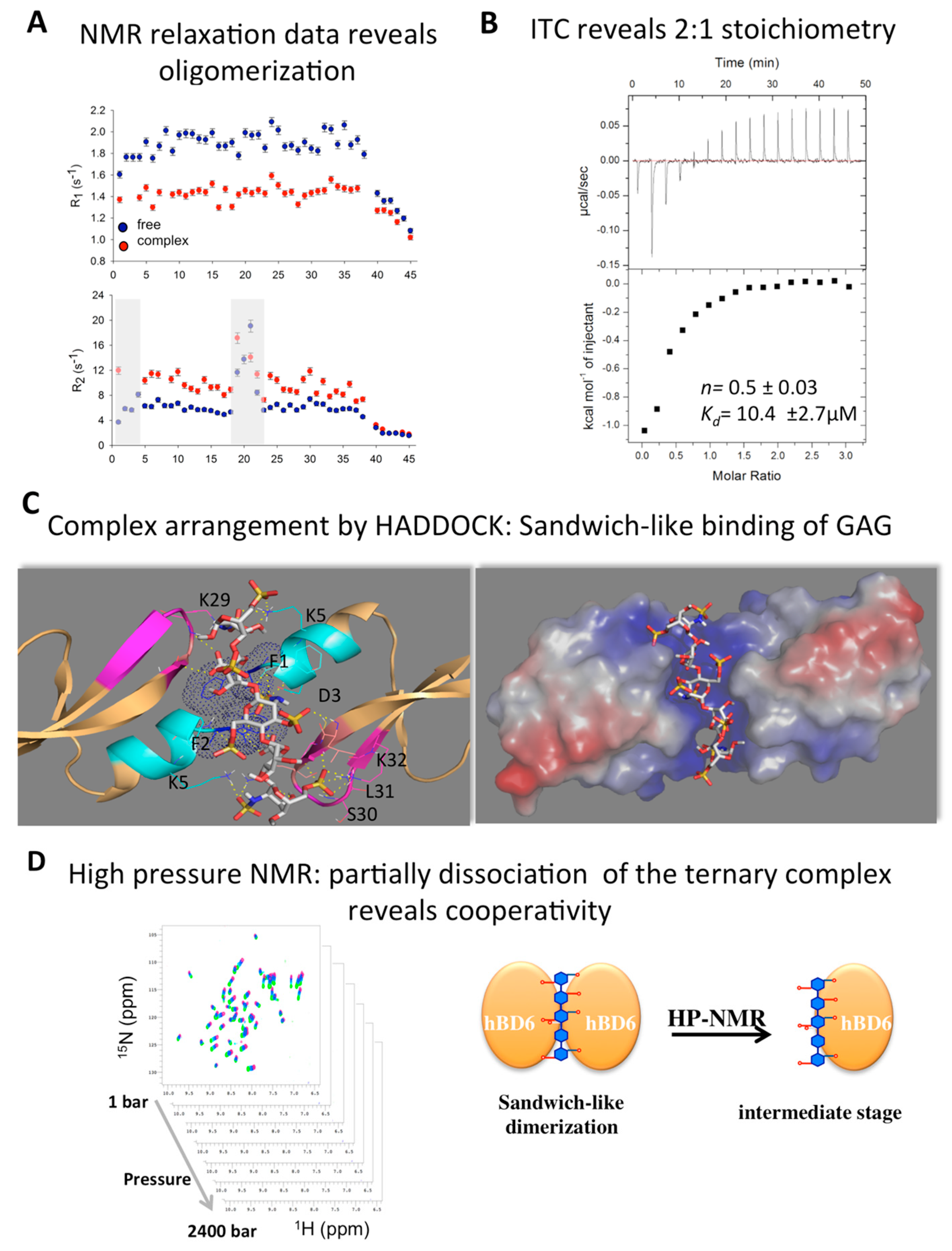
2.5. Defensin Interactions with Glycosaminoglycans and the Role of Dimerization
2.6. Modeling Defensin Complexes Using High Ambiguity Driven Docking
2.7. Challenges to Understanding AMPs and Membrane Interaction
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- James, L.C.; Tawfik, D.S. Conformational diversity and protein evolution—A 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003, 28, 361–368. [Google Scholar] [CrossRef]
- Copperman, J.; Guenza, M.G. Mode localization in the cooperative dynamics of protein recognition. J. Chem. Phys. 2016, 145, 015101. [Google Scholar]
- Okazaki, K.I.; Takada, S. Dynamic energy landscape view of coupled binding and protein conformational change: Induced-fit versus population-shift mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 11182–11187. [Google Scholar] [CrossRef] [PubMed]
- Clore, G. Interplay between conformational selection and induced fit in multidomain protein–ligand binding probed by paramagnetic relaxation enhancement. Biophys. Chem. 2014, 186, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mobley, D.L.; Dill, K.A. Binding of Small-Molecule Ligands to Proteins: “What You See” Is Not Always “What You Get”. Structure 2009, 17, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.; Nussinov, R.; Wright, P. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Monod, J.; Wyman, J.; Changeux, J.P. On the nature of allosteric transitions—A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Nussinov, R.; Ma, B.; Tsai, C.J. Multiple conformational selection and induced fit events take place in allosteric propagation. Biophys. Chem. 2014, 186, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D. Application of a theory of enzime specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Popovych, N.; Sun, S.; Ebright, R.H.; Kalodimos, C.G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006, 13, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, A.; Velyvis, A.; Zoltsman, G.; Rosenzweig, R.; Bouvignies, G.; Kay, L.E. Conserved conformational selection mechanism of Hsp70 chaperone-substrate interactions. eLife 2018, 7, e32764. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.; Vogel, H. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1999, 1462, 11–28. [Google Scholar] [CrossRef]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14-9. [Google Scholar] [CrossRef] [PubMed]
- Travkova, O.G.; Moehwald, H.; Brezesinski, G. The interaction of antimicrobial peptides with membranes. Adv. Colloid Interface Sci. 2017, 247, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Neto, F.; Valente, A.P.; Almeida, F.C.L. Modeling the interaction of dodecylphosphocholine micelles with anticoccidial peptide PW2 guided by NMR data. Molecules 2013, 18, 10056–10080. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.P.; Miyamoto, C.; Almeida, F.C.L. Implications of protein conformational diversity for binding and development of new biological active compounds. Curr. Med. Chem. 2006, 13, 3697–3703. [Google Scholar] [CrossRef] [PubMed]
- Mattar, E.H.; Almehdar, H.A.; Yacoub, H.A.; Uversky, V.N.; Redwan, E.M. Antimicrobial potentials and structural disorder of human and animal defensins. Cytokine Growth Factor Rev. 2016, 28, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I. Primate defensins. Nat. Rev. Microbiol. 2004, 2, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Ouellette, A.J. Mammalian defensins in the antimicrobial immune response. Nat. Immunol. 2005, 6, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Hazlett, L.; Wu, M. Defensins in innate immunity. Cell Tissue Res. 2011, 343, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Verma, C.; Seebah, S.; Low, S.M.; Zhou, L.; Liu, S.P.; Li, J.; Beuerman, R.W. Defensins: Antimicrobial peptides for therapeutic development. Biotechnol. J. 2007, 11, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Biragyn, A.; Kwak, L.W.; Oppenheim, J.J. Mammalian defensins in immunity: More than just microbicidal. Trends Immunol. 2002, 23, 291–296. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Ganz, T. Defensins of vertebrate animals. Curr. Opin. Immunol. 2002, 1, 96–102. [Google Scholar] [CrossRef]
- Schibli, D.J.; Hunter, H.N.; Aseyev, V.; Starner, T.D.; Wiencek, J.M.; McCray, P.B., Jr.; Tack, B.F.; Vogel, H.J. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J. Biol. Chem. 2002, 277, 8279–8289. [Google Scholar] [CrossRef] [PubMed]
- Schutte, B.C.; Mitros, J.P.; Bartlett, J.A.; Walters, J.D.; Jia, H.P.; Welsh, M.J.; Casavant, T.L.; McCray, P.B., Jr. Discovery of five conserved beta-defensin gene clusters using a computational search strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Biragyn, A.; Hoover, D.M.; Lubkowski, J.; Oppenheim, J.J. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu. Rev. Immunol. 2004, 22, 181–215. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Nagase, T.; Makita, R.; Fukuhara, S.; Tomita, T.; Tominaga, T.; Kurihara, H.; Ouchi, Y. Identification of multiple novel epididymis-specific beta-defensin isoforms in humans and mice. J. Immunol. 2002, 169, 2516–2523. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Godzik, A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 2003, 19, ii246–255. [Google Scholar] [CrossRef] [PubMed]
- Hiroaki, H. Recent applications of isotopic labeling for protein NMR in drug discovery. Expert Opin. Drug Discov. 2013, 8, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Sugiki, T.; Fujiwara, T.; Kojima, C. Latest approaches for efficient protein production in drug discovery. Expert Opin. Drug Discov. 2014, 9, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- De Paula, V.S.; Gomes, N.S.; Lima, L.G.; Miyamoto, C.A.; Monteiro, R.Q.; Almeida, F.C.; Valente, A.P. Structural basis for the interaction of human β-defensin 6 and its putative chemokine receptor CCR2 and breast cancer microvesicles. J. Mol. Biol. 2013, 425, 4479–4495. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Nguyen, L.T.; Gopal, R.; Aizawa, T.; Vogel, H.J. Overexpression of Antimicrobial, Anticancer, and Transmembrane Peptides in Escherichia coli through a Calmodulin-Peptide Fusion System. J. Am. Chem. Soc. 2016, 138, 11318–11326. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E. Antimicrobial Peptides: An Introduction. Methods Mol. Biol. 2017, 1548, 3–22. [Google Scholar] [PubMed]
- LaVallie, E.R.; DiBlasio, E.A.; Kovacic, S.; Grant, K.L.; Schendel, P.F.; McCoy, J.M. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology 1993, 11, 187–193. [Google Scholar] [CrossRef] [PubMed]
- De Medeiros, L.N.; Angeli, R.; Sarzedas, C.G.; Barreto-Bergter, E.; Valente, A.P.; Kurtenbach, E.; Almeida, F.C.L. Backbone dynamics of the antifungal Psd1 pea defensin and its correlation with membrane interaction by NMR spectroscopy. Biochim. Biophys. Acta 2010, 1798, 105–113. [Google Scholar] [CrossRef] [PubMed]
- De Paula, V.S.; Razzera, G.; Barreto-Bergter, E.; Almeida, F.C.L.; Valente, A.P. Portrayal of complex dynamic properties of sugarcane defensin 5 by NMR: Multiple motions associated with membrane interaction. Structure 2011, 19, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Mittermaier, A.K.; Kay, L.E. Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 2009, 34, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Korzhnev, D.M.; Religa, T.L.; Banachewicz, W.; Fersht, A.R.; Kay, L.E. A Transient and Low-Populated Protein-Folding Intermediate at Atomic Resolution. Science 2010, 329, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- De Paula, V.S.; Pomin, V.H.; Valente, A.P. Unique properties of human β-defensin 6 (hBD6) and glycosaminoglycan complex: Sandwich-like dimerization and competition with the chemokine receptor 2 (CCR2) binding site. J. Biol. Chem. 2014, 289, 22969–22979. [Google Scholar] [CrossRef] [PubMed]
- Machado, L.E.S.F.; De Paula, V.S.; Pustovalova, Y.; Bezsonova, I.; Valente, A.P.; Korzhnev, D.M.; Almeida, F.C.L. Conformational Dynamics of a Cysteine-Stabilized Plant Defensin Reveals Evolutionary Mechanism to Expose Hydrophobic Residues. Biochemistry 2018, in press. [Google Scholar]
- Sharadadevi, A.; Nagaraj, R. A Molecular dynamics study of human defensins HBD-1 and HNP-3 in water. J. Biomol. Struct. Dyn. 2010, 27, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.M.; Chertov, O.; Lubkowski, J. The structure of human b-defensin-1. J. Biol. Chem. 2001, 276, 39021–39026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Different dynamics and pathway of disulfide bonds reduction of two human defensins, a molecular dynamics simulation study. Proteins 2017, 85, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Toubar, R.A.; Zhmurov, A.; Barsegov, V.; Marx, K.A. Comparative simulation studies of native and single-site mutant human beta-defensin-1 peptides. J. Biomol. Struct. Dyn. 2013, 31, 174–194. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Williams, J.G.; Campbell, S.L. Protein–protein interaction analysis by nuclear magnetic resonance spectroscopy. Methods Mol. Biol. 2004, 261, 79–92. [Google Scholar] [PubMed]
- Palmer, A.G. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004, 104, 3623–3640. [Google Scholar] [CrossRef] [PubMed]
- Hoogewerf, A.J.; Kuschert, G.S.; Proudfoot, A.E.; Borlat, F.; Clark-Lewis, I.; Power, C.A.; Wells, T.N. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 1997, 36, 13570–13578. [Google Scholar] [CrossRef] [PubMed]
- Crown, S.E.; Yu, Y.; Sweeney, M.D.; Leary, J.A.; Handel, T.M. Heterodimerization of CCR2 chemokines and regulation by glycosaminoglycan binding. J. Biol. Chem. 2006, 281, 25438–25446. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Salanga, C.L.; Handel, T.M. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: The role of structural dynamics in function. Exp. Cell Res. 2011, 317, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.S.; Blaum, B.S.; Vargues, T.; DeCecco, M.; Deakin, J.A.; Lyon, M.; Barran, P.E.; Campopiano, D.J.; Uhrín, D. Interaction of human β-defensin 2 (HBD2) with glycosaminoglycans. Biochemistry 2010, 49, 10486–10495. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Fritchley, S.; Borlat, F.; Shaw, J.P.; Vilbois, F.; Zwahlen, C.; Trkola, A.; Marchant, D.; Clapham, P.R.; Wells, T.N.C. The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J. Biol. Chem. 2001, 276, 10620–10626. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, J.I.; Morton, C.J.; Plaxco, K.W.; Campbell, I.D.; Dobson, C.M. Folding kinetics of the SH3 domain of PI3 kinase by real-time NMR combined with optical spectroscopy. J. Mol. Biol. 1998, 276, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.D.J.; Boelens, R.; Bonvin, A.M.J.J. Data-driven docking for the study of biomolecular complexes. FEBS J. 2005, 272, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.; Bonvin, A.M.J.J. Modeling Protein–Protein Complexes Using the HADDOCK Webserver, Modeling Protein Complexes with HADDOCK. Methods Mol. Biol. 2014, 1137, 163–179. [Google Scholar] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Bonvin, A.M.J.J.; Winkler, G.S.; Van Schaik, F.M.A.; Timmers, H.T.M.; Boelens, R. Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure 2004, 12, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Van Drogen-Petit, A.; Zwahlen, C.; Peter, M.; Bonvin, A.M. Insight into molecular interactions between two PB1 domains. J. Mol. Biol. 2004, 336, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Järvå, M.; Phan, T.K.; Lay, F.T.; Caria, S.; Kvansakul, M.; Hulett, M.D. Human β-defensin 2 kills Candida albicans through phosphatidylinositol 4,5-bisphosphate-mediated membrane permeabilization. Sci. Adv. 2018, 4, eaat0979. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.; Yount, N. Mechanisms of Antimicrobial Peptide Action and Resistance. Phamacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Yedery, R.; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.; Ferre, R.; Castanho, M. Antimicrobial peptides: Linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 2009, 7, 245–280. [Google Scholar] [CrossRef] [PubMed]
- Marquette, A.; Bechinger, B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Salnikov, E.S. The membrane interactions of antimicrobial peptides revealed by solid-state NMR spectroscopy. Chem. Phys. Lipids 2012, 165, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Cady, S.D.; Tang, M.; Waring, A.J.; Lehrer, R.I.; Hong, M. Membrane-dependent oligomeric structure and pore formation of a β-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA 2006, 103, 16242–16247. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Hong, M. Structure and mechanism of beta-hairpin antimicrobial peptides in lipid bilayers from solid-state NMR spectroscopy. Mol. Biosyst. 2009, 5, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Waring, A.; Hong, M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptíde from solid-state NMR. J. Am. Chem. Soc. 2007, 129, 11438–11446. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, F.; Ramamoorthy, A.; Barany, G.; Veglia, G. On the role of NMR spectroscopy for characterization of antimicrobial peptides. Methods Mol. Biol. 2013, 1063, 159–180. [Google Scholar] [PubMed]
- Respondek, M.; Madl, T.; Göbl, C.; Golser, R.; Zangger, K. Mapping the Orientation of Helices in Micelle-Bound Peptides by Paramagnetic Relaxation Waves. J. Am. Chem. Soc. 2007, 129, 5228–5234. [Google Scholar] [CrossRef] [PubMed]
- Schibli, D.; Hwang, P.; Vogel, H. Structure of the antimicrobial peptide tritrpticin bound to micelles: A distinct membrane-bound peptide fold. Biochemistry 1999, 38, 16749–16755. [Google Scholar] [CrossRef] [PubMed]
- Schibli, D.J.; Nguyen, L.T.; Vogel, H.J. Structure-function analysis of tritrpticin analogs: Potential relationships between antimicrobial activities, model membrane interactions, and their micelle-bound NMR structures. Biophys. J. 2006, 91, 4413–4426. [Google Scholar] [CrossRef] [PubMed]
- Arias, M.; Jensen, K.V.; Vogel, H.J. Hydroxy-tryptophan containing derivatives of tritrpticin: Modification of antimicrobial activity and membrane interactions. Biochim. Biophys. Acta 2015, 1848, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Bozelli, J.; Sasahara, E.T.; Pinto, M.R.; Nakaie, C.R.; Schreier, S. Effect of head group and curvature on binding of the antimicrobial peptide tritrpticin to lipid membranes. Chem. Phys. Lipids 2012, 165, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.L.; Moraes, A.; Nakaie, C.R.; Almeida, F.C.L.; Valente, A.P. Structural and dynamics insights of the interaction between Tritrpticin and micelles: An NMR study. Biophys. J. 2016, 111, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, T.; Wei, D.; Strandberg, E.; Ulrich, A.S.; Ulmschneider, J.P. How reliable are molecular dynamics simulations of membrane active antimicrobial peptides? Biochim. Biophys. Acta 2014, 1838, 2280–2288. [Google Scholar] [CrossRef] [PubMed]
- Deleu, M.; Crowet, J.M.; Nasir, M.N.; Lins, L. Complementary biophysical tools to investigate lipid specificity in the interaction between bioactive molecules and the plasma membrane: A review. Biochim. Biophys. Acta 2014, 1838, 3171–3190. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, C.H.; Hu, D.; Ulmschneider, M.B.; Ulmschneider, J.P. Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 2016, 7, 13535. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, J.P.; Ulmschneider, M.B. Molecular Dynamics Simulations Are Redefining Our View of Peptides Interacting with Biological Membranes. Acc. Chem. Res. 2018, 51, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, S.W.; Cho, A.E. Molecular Insights into the Adsorption Mechanism of Human β-Defensin-3 on Bacterial Membranes. Langmuir 2016, 32, 1782–1790. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Paula, V.S.; Valente, A.P. A Dynamic Overview of Antimicrobial Peptides and Their Complexes. Molecules 2018, 23, 2040. https://doi.org/10.3390/molecules23082040
De Paula VS, Valente AP. A Dynamic Overview of Antimicrobial Peptides and Their Complexes. Molecules. 2018; 23(8):2040. https://doi.org/10.3390/molecules23082040
Chicago/Turabian StyleDe Paula, Viviane Silva, and Ana Paula Valente. 2018. "A Dynamic Overview of Antimicrobial Peptides and Their Complexes" Molecules 23, no. 8: 2040. https://doi.org/10.3390/molecules23082040
APA StyleDe Paula, V. S., & Valente, A. P. (2018). A Dynamic Overview of Antimicrobial Peptides and Their Complexes. Molecules, 23(8), 2040. https://doi.org/10.3390/molecules23082040