Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A


 ,
,
Abstract
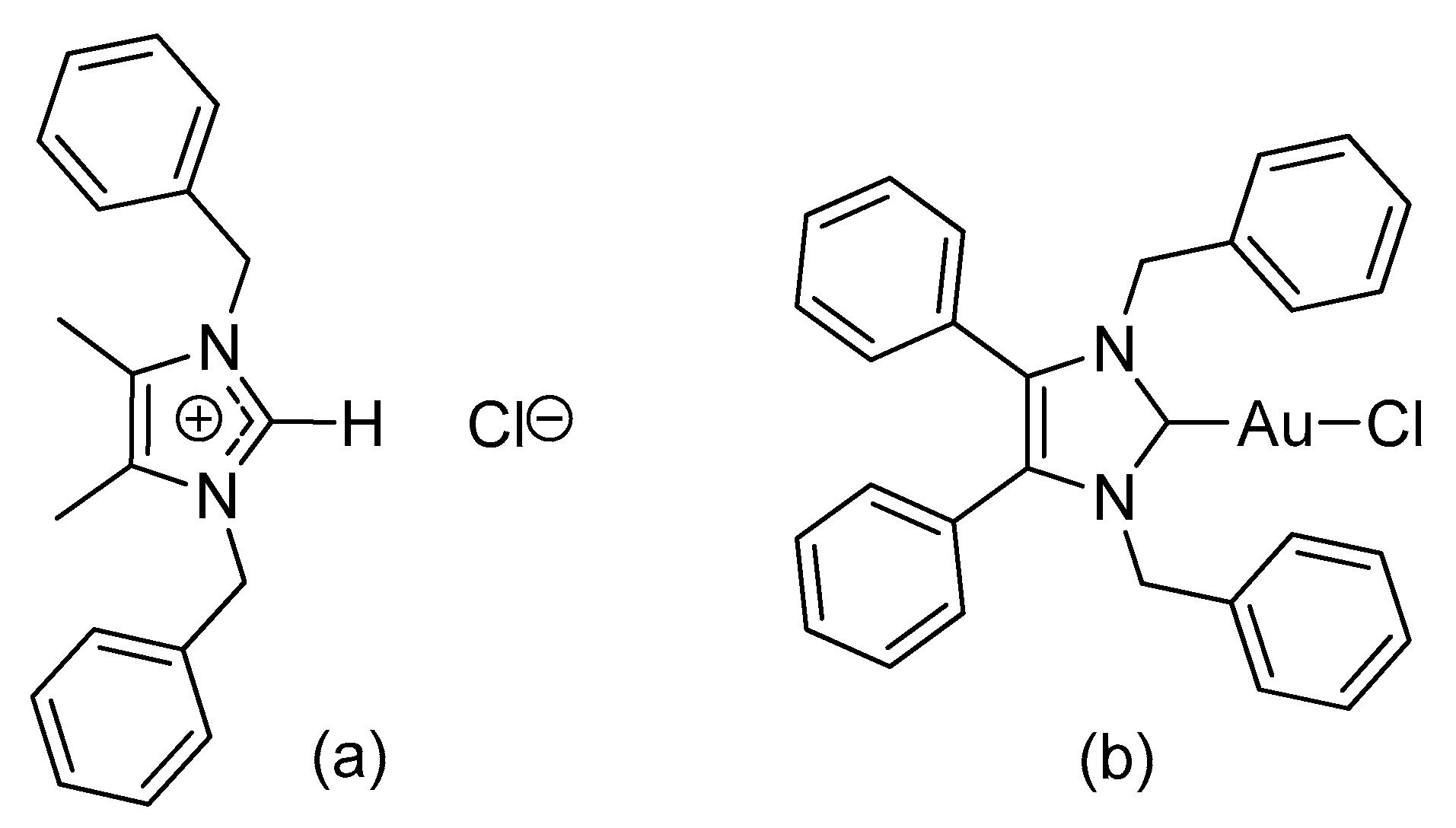
1. Introduction
2. Results and Discussion
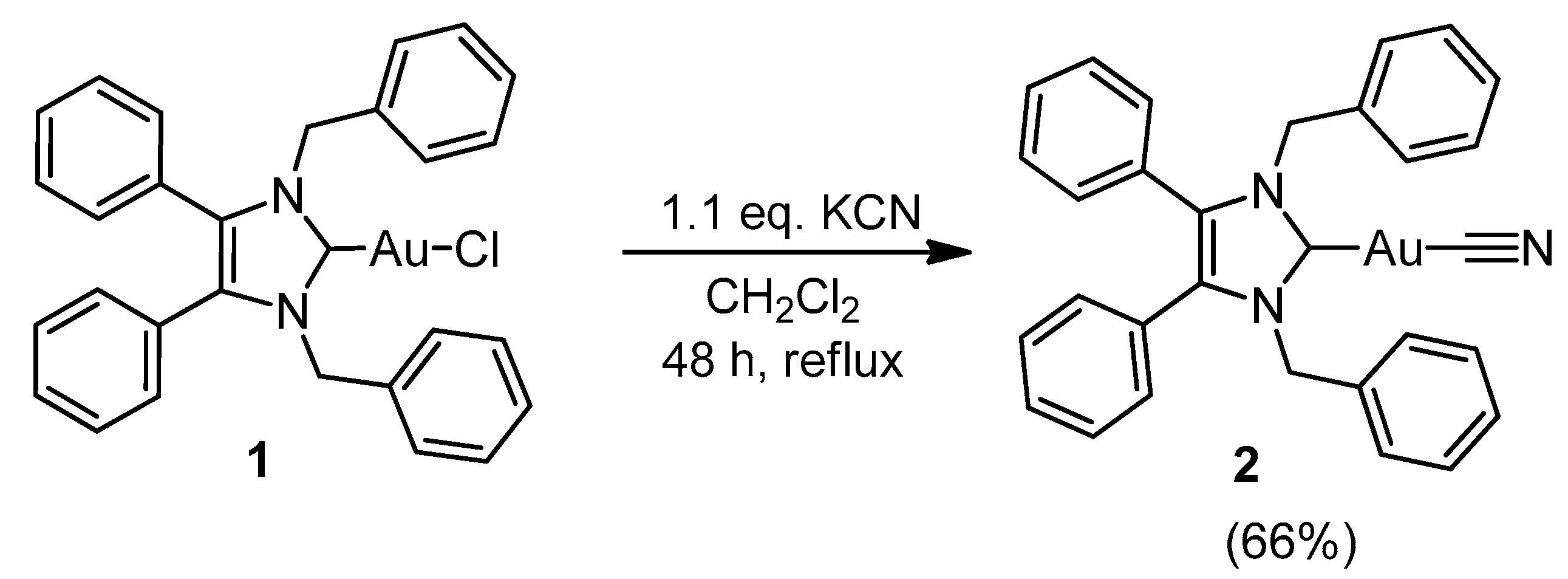
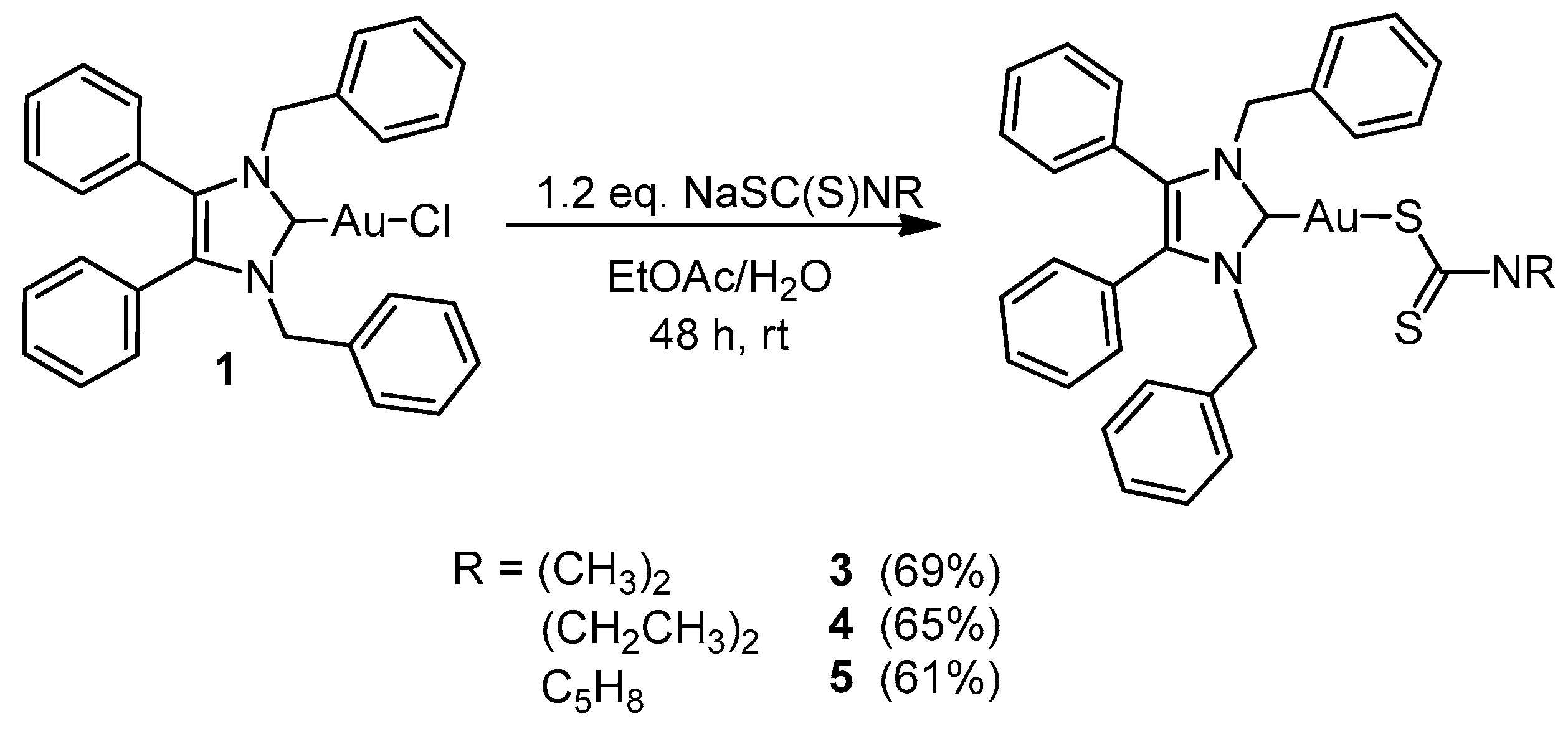
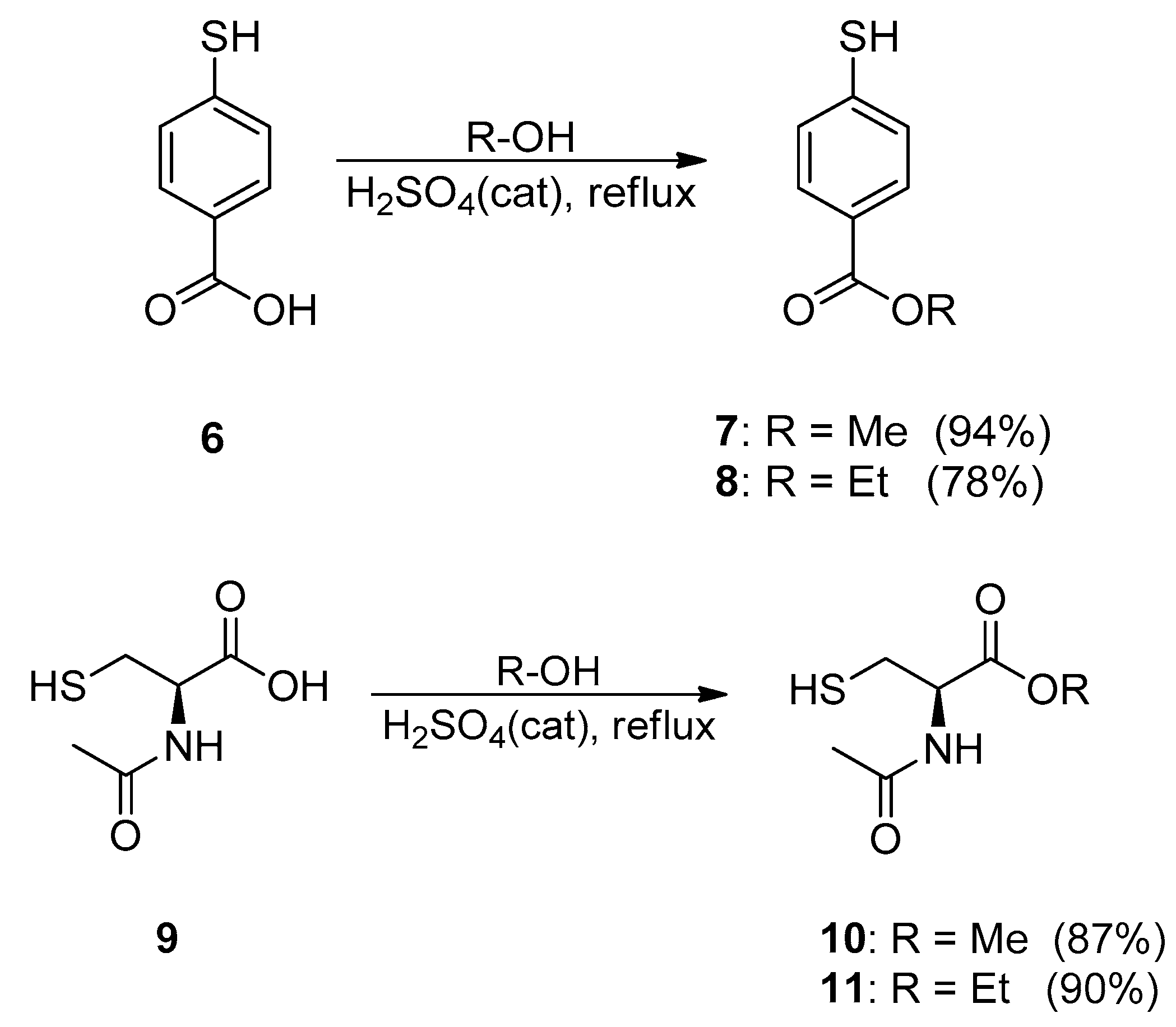
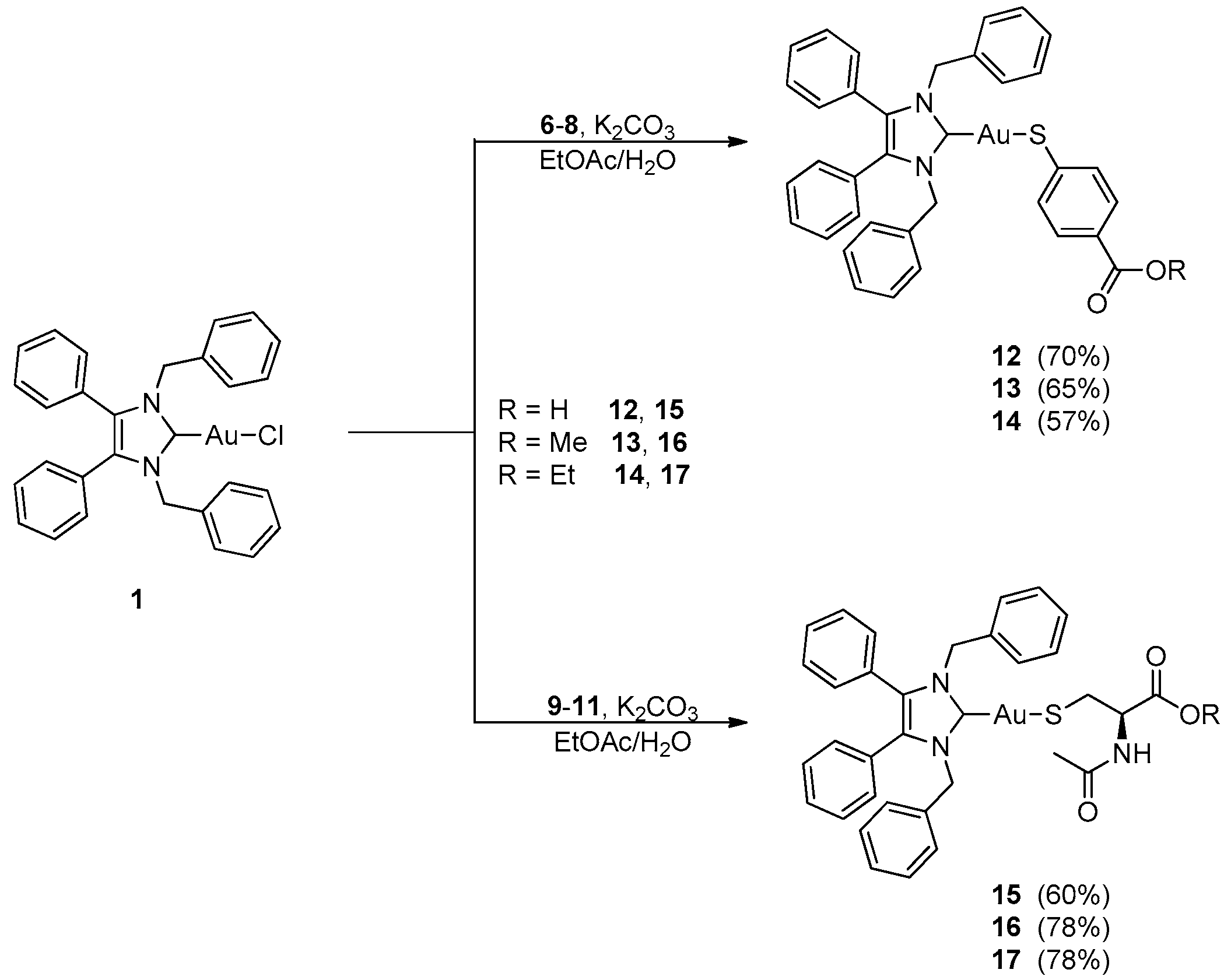
2.1. Synthesis and Characterisation
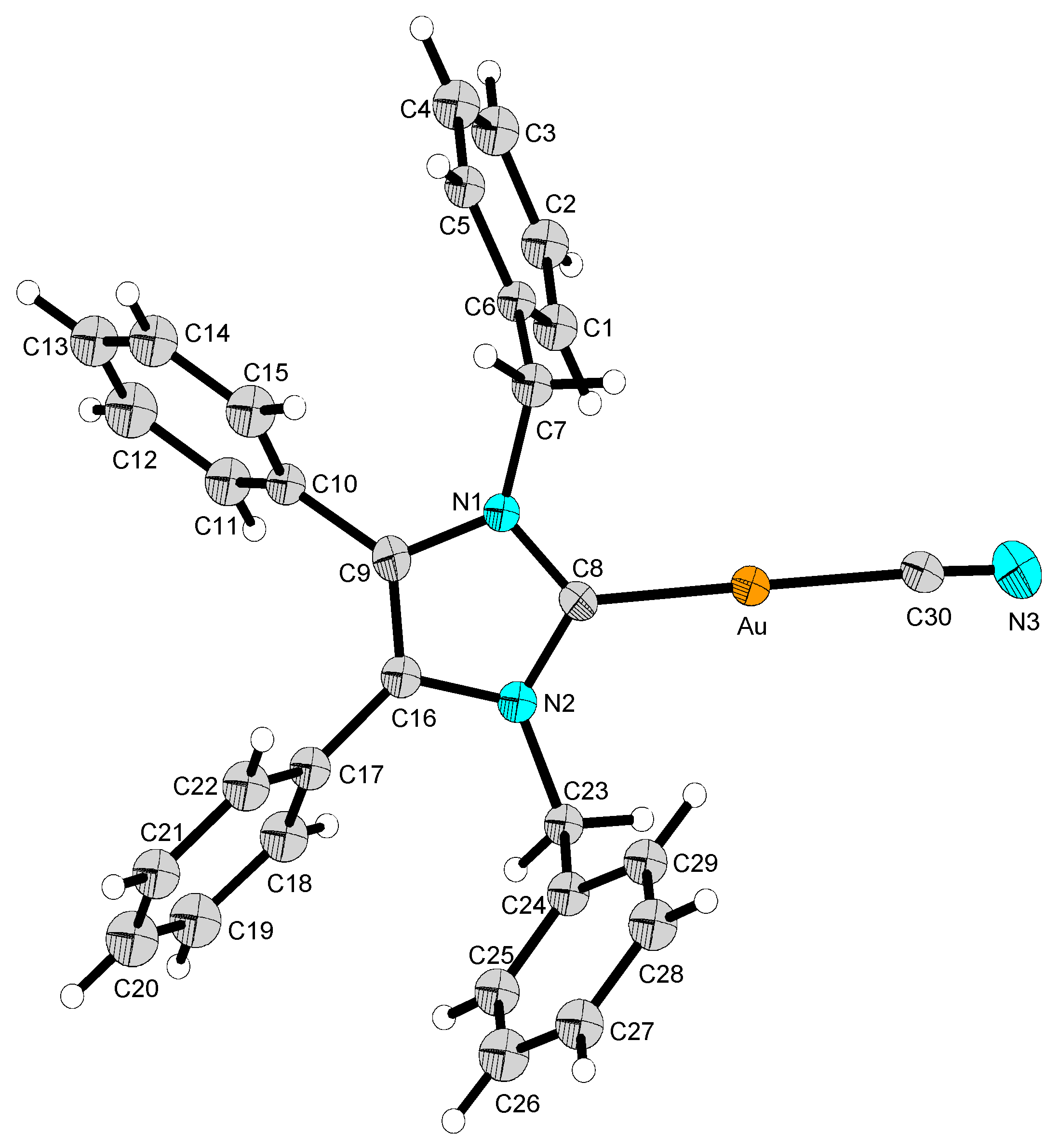
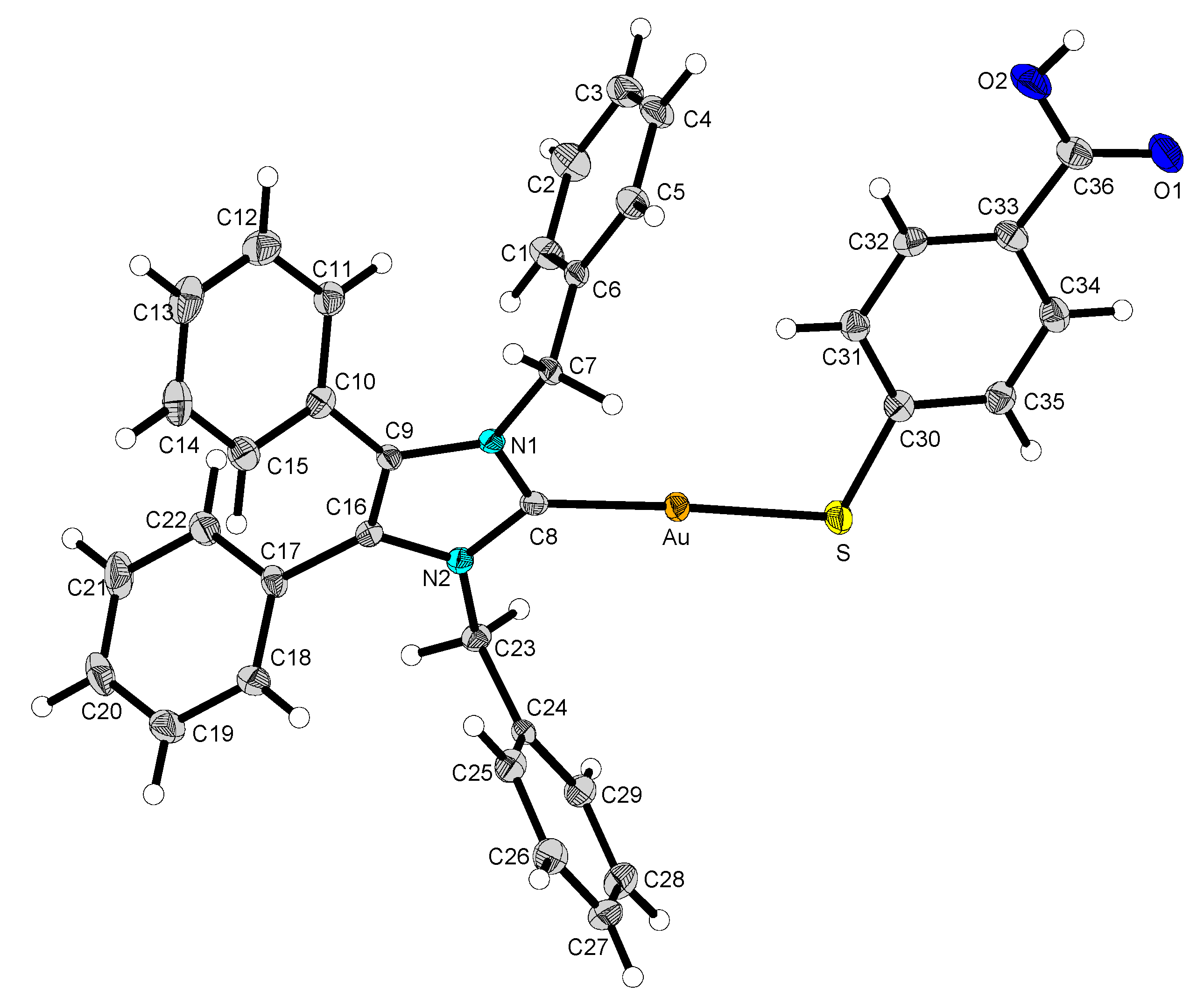
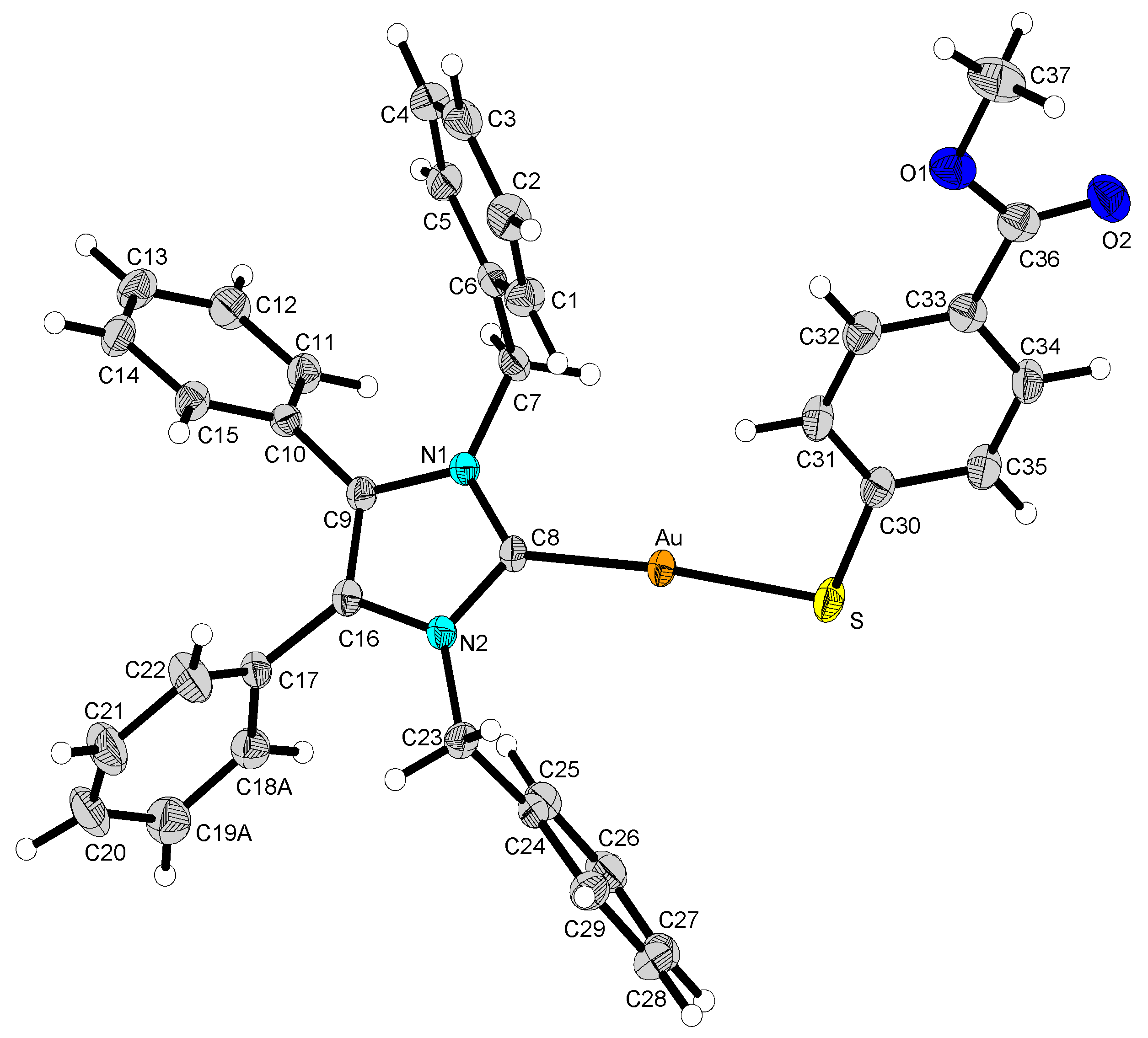
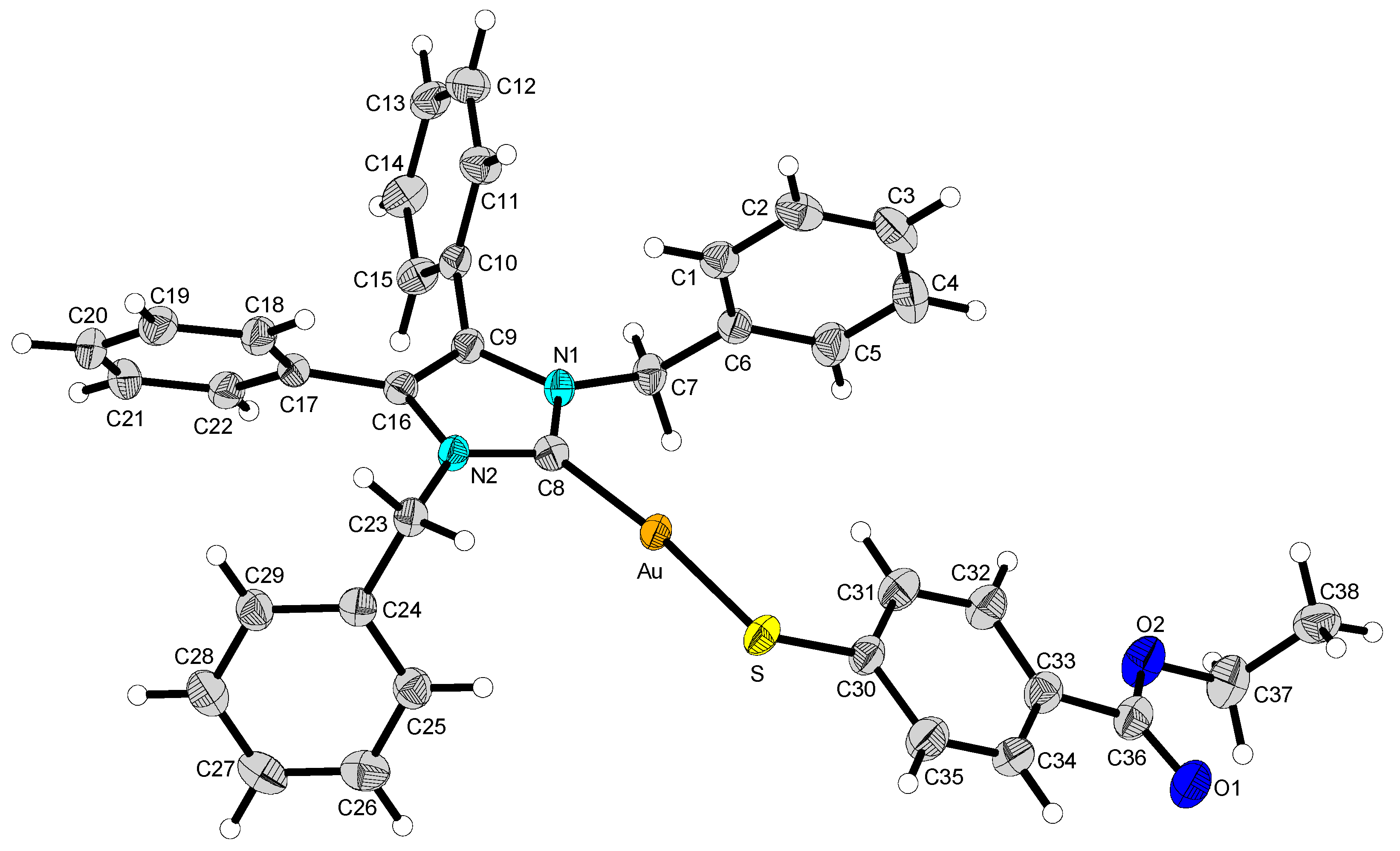
2.2. Structural Discussion
2.3. Biological Evaluation
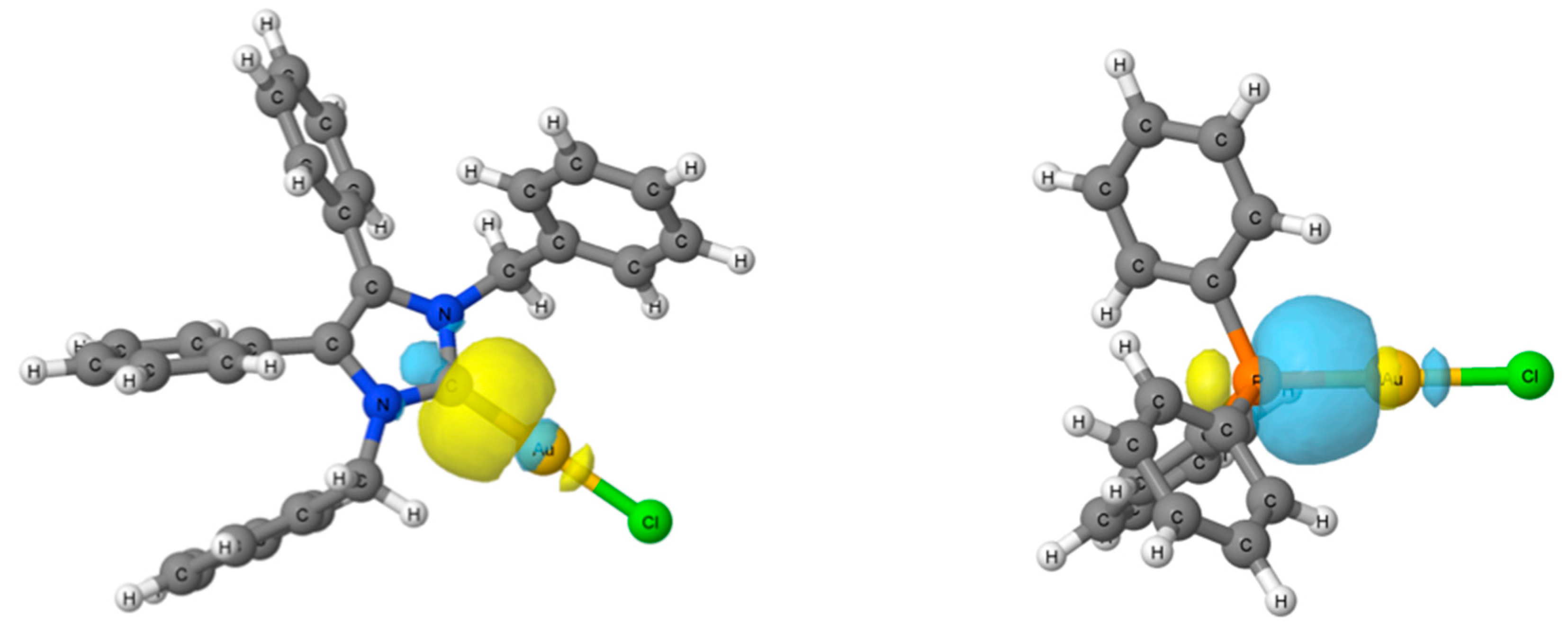
2.4. Computational Results
3. Materials and Methods
3.1. General Conditions
3.2. Synthesis
3.2.1. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) Chloride (1)
3.2.2. (1,3-Dibenzyl-4,5-diphenyl-2-ylidene)gold(I) Cyanide (2)
3.2.3. General Procedure for NHC-Au(I) Complexes 3–5
(1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) Dimethyldithiocarbamate (3)
(1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) Diethyldithiocarbamate (4)
(1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) Pyrrolidinedithiocarbamate (5)
3.2.4. General Procedure for 7–8, 10–11
Methyl-p-mercaptobenzoate (7)
Ethyl-p-mercaptobenzoate (8)
N-Acetyl-l-cysteine Methyl Ester (10)
N-acetyl-l-cysteine Ethyl Ester (11)
3.2.5. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) p-Mercaptobenzoic Acid (12)
3.2.6. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-methyl-p-mercaptobenzoate (13)
3.2.7. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-ethyl-p-mercaptobenzoate (14)
3.2.8. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-N-acetyl-l-cysteine (15)
3.2.9. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-N-acetyl-l-cysteine Methyl Ester (16)
3.2.10. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-N-acetyl-l-cysteine Ethyl Ester (17)
3.3. Structure Determination
3.4. MTT-Based Proliferation Assay
3.5. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, D.H.; Smith, W.E. The chemistry of the gold drugs used in the treatment of rheumatoid arthritis. Chem. Soc. Rev. 1980, 9, 217–240. [Google Scholar] [CrossRef]
- Eisler, R. Chrysotherapy: A synoptic review. Inflamm. Res. 2003, 52, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, F.; Muller-Bunz, H.; Smith, R.; Streciwilk, W.; Zhu, X.; Tacke, M. Novel ruthenium(II) and gold(I) NHC complexes: Synthesis, characterization, and evaluation of their anticancer properties. Organometallics 2013, 32, 5551–5560. [Google Scholar] [CrossRef]
- Baker, M.V.; Barnard, P.J.; Berners-Price, S.J.; Brayshaw, S.K.; Hickey, J.L.; Skelton, B.W.; White, A.H. Synthesis and structural characterisation of linear Au(I) N-Heterocyclic carbene complexes: New analogues of the Au(I) phosphine drug auranofin. J. Organomet. Chem. 2005, 690, 24–25. [Google Scholar] [CrossRef]
- Zhao, W.; Ferro, V.; Baker, M.V. Carbohydrate-N-heterocyclic carbene metal complexes: Synthesis, catalysis and biological studies. Coord. Chem. Rev. 2007, 339, 1–16. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.A.; Patil, S.A.; Patil, R.; Keri, R.S.; Balakrishna, G.R.; Tacke, M. N-heterocylic carbene metal complexes as bio-organometallic antimicrobial and anticancer drugs. Future Med. Chem. 2015, 7, 1305–1333. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, M.; Santini, C.; Pellei, M. Recent advances in medicinal applications of coinage-metal (Cu and Ag) N-heterocyclic carbene complexes. Curr. Top. Med. Chem. 2016, 16, 2995–3017. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Kitanovic, I.; Alborzinia, H.; Can, S.; Kitanovic, A.; Onambele, L.A.; Stefanopoulou, M.; Geldmacher, Y.; Sheldrick, W.S.; Wolber, G.; et al. Benzimidazol-2-ylidene gold(I) complexes are thioredoxin reductase inhibitors with multiple antitumor properties. J. Med. Chem. 2010, 53, 8608–8618. [Google Scholar] [CrossRef] [PubMed]
- Muenzner, J.K.; Biersack, B.; Albrecht, A.; Rehm, T.; Lacher, U.; Milius, W.; Casini, A.; Zhang, J.; Ott, I. Ferrocenyl-coupled N-heterocyclic carbene complexes of gold (I): A successful approach to multinuclear anticancer drugs. Chem. Eur. J. 2016, 22, 18953–18962. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Zheng, B.L.; He, K.; Zheng, Q.Y. Imidazole alkaloids from lepidium meyenii. J. Nat. Prod. 2003, 66, 1101–1103. [Google Scholar] [CrossRef] [PubMed]
- Riduan, S.N.; Zhang, Y. Imidazolium salts and their polymeric materials for biological applications. Chem. Soc. Rev. 2013, 42, 9055–9070. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Can, S.; Kitanovic, I.; Alborzinia, H.; Stefanopoulou, M.; Kokoschka, M.; Mönchgesang, S.; Sheldrick, W.S.; Wölfl, S.; Ott, I. Comparative in vitro evaluation of N-heterocyclic carbene gold(I) complexes of the benzimidazolylidene type. J. Med. Chem. 2011, 54, 8646–8657. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gust, R. Update on metal N-heterocyclic carbene complexes as potential anti-tumor metallodrugs. Coord. Chem. Rev. 2016, 329, 191–213. [Google Scholar] [CrossRef]
- Ott, I. On the Medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Zou, T.; Lum, C.T.; Lok, C.N.; To, W.P.; Low, K.H.; Che, C.M. A binuclear gold(I) complex with mixed bridging diphosphine and bis(N-heterocyclic carbene) ligands shows favorable thiol reactivity and inhibits tumor growth and angiogenesis in vivo. Angew. Chem. Int. Ed. 2014, 53, 5810–5814. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Lum, C.T.; Lok, C.; Zhang, J.; Che, C. Chemical biology of anticancer gold(III) and gold(I) complexes. Chem. Soc. Rev. 2015, 44, 8786–8801. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.; Marzo, I.; Cativiela, C.; Laguna, A.; Gimeno, M.C. Highly cytotoxic bioconjugated gold(I) complexes with cysteine-containing dipeptides. Chem. Eur. J. 2015, 21, 11088–11095. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayana, D.N. Vibrational Spectroscopy: Theory and Applications, 1st ed.; New Age International (P) Ltd.: New Delhi, India, 2004. [Google Scholar]
- Jamaluddin, N.A.; Baba, I. Synthesis and structural characterization of new dithiocarbamate complexes from Sb(III) and Bi(III). AIP Conf. Proc. 2013, 1571, 789–794. [Google Scholar]
- Durgaprasad, G.; Sathyanarayana, D.N.; Patel, C.C. Normal coordinate analysis of dialkylditlhioearbamate and its selenium analogue. Can. J. Chem. 1969, 47, 631–635. [Google Scholar] [CrossRef]
- Pakiari, A.H.; Jamshidi, Z. Nature and strength of M−S Bonds (M = Au, Ag, and Cu) in binary alloy gold clusters. J. Phys. Chem. A 2010, 114, 9212–9221. [Google Scholar] [CrossRef] [PubMed]
- Kokkin, D.L.; Zhang, R.; Steimle, T.C.; Wyse, I.A.; Pearlman, B.W.; Varberg, T.D. Au–S bonding revealed from the characterization of diatomic gold sulfide, AuS. J. Phys. Chem. A 2015, 119, 11659–11667. [Google Scholar] [CrossRef] [PubMed]
- Hormann, A.L.; Bennett, D.W.; Shaw, C.F.; Reiff, W.M. Solid-state structure and solution equilibria of cyano(Triethylphosphine)gold(I). Inorg. Chem. 1986, 25, 3953–3957. [Google Scholar] [CrossRef]
- Tacke, M.; Dada, O.; O’Beirne, C.; Zhu, X.; Müller-Bunz, H. The non-isomorphous crystal structures of NHC–Au–Cl and NHC–Au–Br (NHC is 1,3-dibenzyl-4,5-diphenylimidazol-2-ylidene). Acta Crystallogr. Sect. C Struct. Chem. 2016, 72, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Dada, O.; Curran, D.; O’Beirne, C.; Müller-Bunz, H.; Zhu, X.; Tacke, M. Synthesis and cytotoxicity studies of novel NHC–gold(I) pseudohalides and thiolates. J. Organomet. Chem. 2017, 840, 30–37. [Google Scholar] [CrossRef]
- Novoa, A.; Eierhoff, T.; Topin, J.; Varrot, A.; Barluenga, S.; Imberty, A.; Roemer, W.; Winssinger, N. A LecA ligand identified from a galactoside-conjugate array inhibits host cell invasion by Pseudomonas aeruginosa. Angew. Chem. 2014, 53, 8885–8889. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Cohen, S.M. Nucleophile recognition as an alternative inhibition mode for benzoic acid based carbonic anhydrase inhibitors. Chem. Commun. 2012, 48, 5259–5261. [Google Scholar] [CrossRef] [PubMed]
- Sala, O.; Santschi, N.; Jungen, S.; Lüthi, H.P.; Iannuzzi, M.; Hauser, N.; Togni, A. S-trifluoromethylation of thiols by hypervalent iodine reagents: A joint experimental and computational study. Chem. Eur. J. 2016, 22, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, G.J.L.; Grayson, E.J.; Thompson, S.; Chalker, J.M.; Errey, J.C.; El Oualid, F.; Claridge, T.D.W.; Davis, B.G. From disulfide-to thioether-linked glycoproteins. Angew. Chem. 2008, 47, 2244–2247. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, R.K.; Picard, M.A.; Beyreuther, K.; Wiessler, M. Synthesis of antioxidative and anti-inflammatory drugs glucoconjugates. Carbohydr. Res. 2000, 325, 72–80. [Google Scholar] [CrossRef]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr. Sect. A 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn-Sham global-hybrid exchange-correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. supplementary functions for gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio effective core potentials for molecular calculations: Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gassian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 15 April 2018).
Sample Availability: Samples of the compounds 1–5 and 12–17 are available from the authors. |












{kind=link}
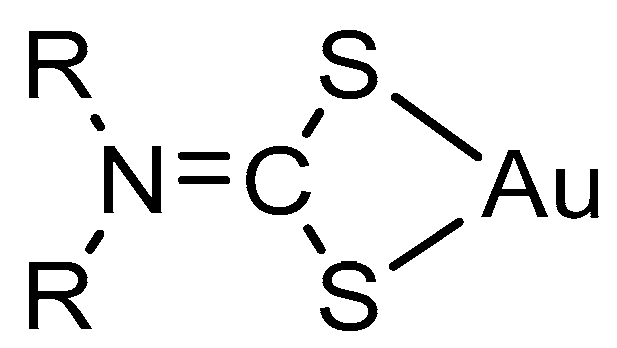{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 | 12 | 13 | 14 | |
|---|---|---|---|---|
| Empirical Formula | C30H24AuN3 | C36H29N2O2SAu | C37H31N2O2SAu | C38H33N2O2SAu |
| Formula Weight (g·mol−1) | 623.49 | 750.54 | 764.66 | 778.69 |
| Temperature (K) | 100(2) | 100(2) | 100(2) | 100(2) |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group | P21/m (#11) | P (#2) | P (#2) | C2/c (#15) |
| Unit cell dimensions | ||||
| a (Å) | 12.8150(7) | 9.3234(3) | 8.89830(6) | 26.1234(3) |
| b (Å) | 6.4797(3) | 10.4210(3) | 12.12378(8) | 10.2154(1) |
| c (Å) | 15.6802(8) | 16.0388(5) | 15.5000(1) | 23.9038(3) |
| α (°) | 90 | 75.663(3) | 103.1637(6) | 90 |
| β (°) | 112.268(6) | 85.553(2) | 105.3420(6) | 100.387(1) |
| γ (°) | 90 | 83.264(3) | 98.8340(6) | 90 |
| Volume (Å3) | 1204.94(12) | 1497.46(8) | 1528.942(19) | 6274.45(12) |
| Z | 2 | 2 | 2 | 8 |
| Density (calcd) (mg/m3) | 1.718 | 1.665 | 1.661 | 1.649 |
| Absorption coefficient (mm−1) | 11.641 | 5.018 | 9.964 | 9.723 |
| F (000) | 608 | 740 | 756 | 3088 |
| Crystal size (mm3) | 0.255 × 0.034 × 0.026 | 0.194 × 0.121 × 0.082 | 0.248 × 0.193 × 0.120 | 0.113 × 0.035 × 0.010 |
| θ (°) | 3.727 to 77.196 | 2.90 to 29.59 | 3.845 to 76.876 | 3.44 to 76.91 |
| Index ranges | −16 ≤ h ≤ 15 | −12 ≤ h ≤ 12 | −11 ≤ h ≤ 11 | −32 ≤ h ≤ 32 |
| −8 ≤ k ≤ 8 | −13 ≤ k ≤ 13 | −15 ≤ k ≤ 15 | −12 ≤ k ≤ 12 | |
| −19 ≤ l ≤ 19 | −21 ≤ l ≤ 21 | −19 ≤ l ≤ 19 | −30 ≤ l ≤ 28 | |
| Reflections collected | 24,420 | 20,276 | 34,268 | 39,269 |
| Independent reflections Rint | 2760(0.1335) | 7306(0.0341) | 6409(0.0268) | 6584(0.0339) |
| Completeness to θmax (%) | 99.8 | 99.2 | 100.0 | 99.4 |
| Absorption correction | Gaussian | Gaussian | Gaussian | Gaussian |
| Max and min transmission | 0.788 and 0.291 | 0.714 and 0.472 | 0.455 and 0.227 | 0.913 and 0.516 |
| Refinement method | Full-matrix Least-squares on F2 | Full-matrix Least-squares on F2 | Full-matrix Least-squares on F2 | Full-matrix Least-squares on F2 |
| Data/restraints/parameters | 2760/0/147 | 7306/0/380 | 6409/0/389 | 6584/0/398 |
| Goodness-of-fit on F2 | 1.139 | 1.046 | 1.085 | 1.041 |
| Final R indices [I > 2σ(I)] | R1 = 0.0442, wR2 = 0.0983 | R1 = 0.0255, wR2 = 0.0437 | R1 = 0.0178, wR2 = 0.0439 | R1 = 0.0266, wR2 = 0.0674 |
| R indices (all data) | R1 = 0.0464, wR2 = 0.0998 | R1 = 0.0324, wR2 = 0.0465 | R1 = 0.0187, wR2 = 0.0442 | R1 = 0.0305, wR2 = 0.0701 |
| Largest diff. peak and hole | 1.982 and −1.411 | 1.009 and −0.762 | 0.639 and −0.859 | 1.508 and −1.500 |
| 2 | 12 | 13 | 14 | |
|---|---|---|---|---|
| Au–C(8) | 2.031(8) | 2.012(3) | 2.008(2) | 2.008(3) |
| Au–C(30) | 2.026(9) | |||
| C(30)–N(3) | 1.113(12) | |||
| Au–S(1) | 2.2856(7) | 2.2851(6) | 2.3012(8) | |
| S(1)–C(30) | 1.751(3) | 1.755(2) | 1.735(3) | |
| C(36)–O(1) | 1.249(3) | 1.211(3) | 1.215(5) | |
| C(36)–O(2) | 1.304(3) | 1.349(3) | 1.337(5) | |
| O(2)–C(37) | 1.442(3) | 1.450(5) |
| 2 | 12 | 13 | 14 | |
|---|---|---|---|---|
| C(8)–Au–S | 177.48(8) | 175.20(6) | 173.45(9) | |
| C(8)–Au–C(30) | 179.6(4) | |||
| Au–S–C(30) | 108.40(10) | 109.44(8) | 108.83(12) | |
| Au–C(30)–N(3) | 177.0(8) | |||
| S(1)–C(30)–S(2) | ||||
| O(1)–C(36)–O(2) | 122.8(3) | 122.9(2) | 123.4(3) | |
| O(1)–C(36)–C(33) | 120.8(3) | 125.2(2) | 124.2(4) | |
| O(2)–C(36)–C(33) | 116.4(3) | 111.9(2) | 112.4(3) | |
| C(36)–O(2)–C(37) | 115.4(2) | 117.0(3) |
| HCT-116wt | HCT-116 p53−/− | MCF-7topo | |
|---|---|---|---|
| 2 | 14.8 ± 1.9 | - | 10.8 ± 0.9 |
| 3 | 1.5 ± 0.1 | - | 0.28 ± 0.03 |
| 4 | 8.0 ± 0.1 | 3.8 ± 0.4 | 0.36 ± 0.03 |
| 5 | 6.2 ± 0.3 | 2.0 ± 0.6 | 1.5 ± 0.3 |
| 12 | 5.5 ± 0.1 | 2.7 ± 0.2 | 5.4 ± 0.5 |
| 13 | 18.1 ± 6.5 | 9.5 ± 0.6 | 21.3 ± 3.4 |
| 14 | 6.8 ± 0.2 | 7.9 ± 0.2 | 13.2 ± 3.7 |
| 15 | 4.5 ± 1.2 | 6.6 ± 0.3 | 7.1 ± 0.3 |
| 16 | 2.8 ± 0.1 | 4.5 ± 0.6 | 6.3 ± 0.5 |
| 17 | 2.9 ± 0.1 | 3.7 ± 0.2 | 5.4 ± 0.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curran, D.; Dada, O.; Müller-Bunz, H.; Rothemund, M.; Sánchez-Sanz, G.; Schobert, R.; Zhu, X.; Tacke, M. Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A. Molecules 2018, 23, 2031. https://doi.org/10.3390/molecules23082031
Curran D, Dada O, Müller-Bunz H, Rothemund M, Sánchez-Sanz G, Schobert R, Zhu X, Tacke M. Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A. Molecules. 2018; 23(8):2031. https://doi.org/10.3390/molecules23082031
Chicago/Turabian StyleCurran, Danielle, Oyinlola Dada, Helge Müller-Bunz, Matthias Rothemund, Goar Sánchez-Sanz, Rainer Schobert, Xiangming Zhu, and Matthias Tacke. 2018. "Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A" Molecules 23, no. 8: 2031. https://doi.org/10.3390/molecules23082031
APA StyleCurran, D., Dada, O., Müller-Bunz, H., Rothemund, M., Sánchez-Sanz, G., Schobert, R., Zhu, X., & Tacke, M. (2018). Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A. Molecules, 23(8), 2031. https://doi.org/10.3390/molecules23082031