3. Materials and Methods
3.1. Synthetic Methods and Molecular Characterization
All starting materials were purchased from Sigma-Aldrich (St. Louis, MO, USA), Alfa-Aesar (Ward Hill, MA, USA), and TCI (Nihonbashi, Japan) and were used without further purification. Reactions were performed under an atmosphere of dry nitrogen. An Initiator microwave system (Biotage, Uppsala, Sweden) was used for microwave-assisted reaction. LC-MS was performed on a system consisting of an electrospray ionization (ESI) source in a LCMS-2020 liquid chromatography-mass spectrometer system (Shimadzu, Kyoto, Japan; column: Shim-pack GIS, 100 × 3.0 mm, 3 μm ODS). A Teledyne ISCO flash purification system (Lincoln, NE, USA) with various prepacked silica gel cartridges was used for flash column chromatography. 1H- and 13C-NMR spectra were recorded in the indicated solvent on an AVANCE III HD (400 and 100 MHz for 1H and 13C, respectively) spectrometer (Bruker, Billerica, MA, USA). Chemical shifts are reported as δ values in parts per million downfield from TMS (δ 0.0) as the internal standard in CD3OD, DMSO-d6 or CDCl3. The purity of the compounds was evaluated on a Shimadzu reverse-phase analytical HPLC system (column: Ace C18, 150 × 4.6 mm, 3 μm). Purities of all compounds that were subjected to biological assay were >95%.
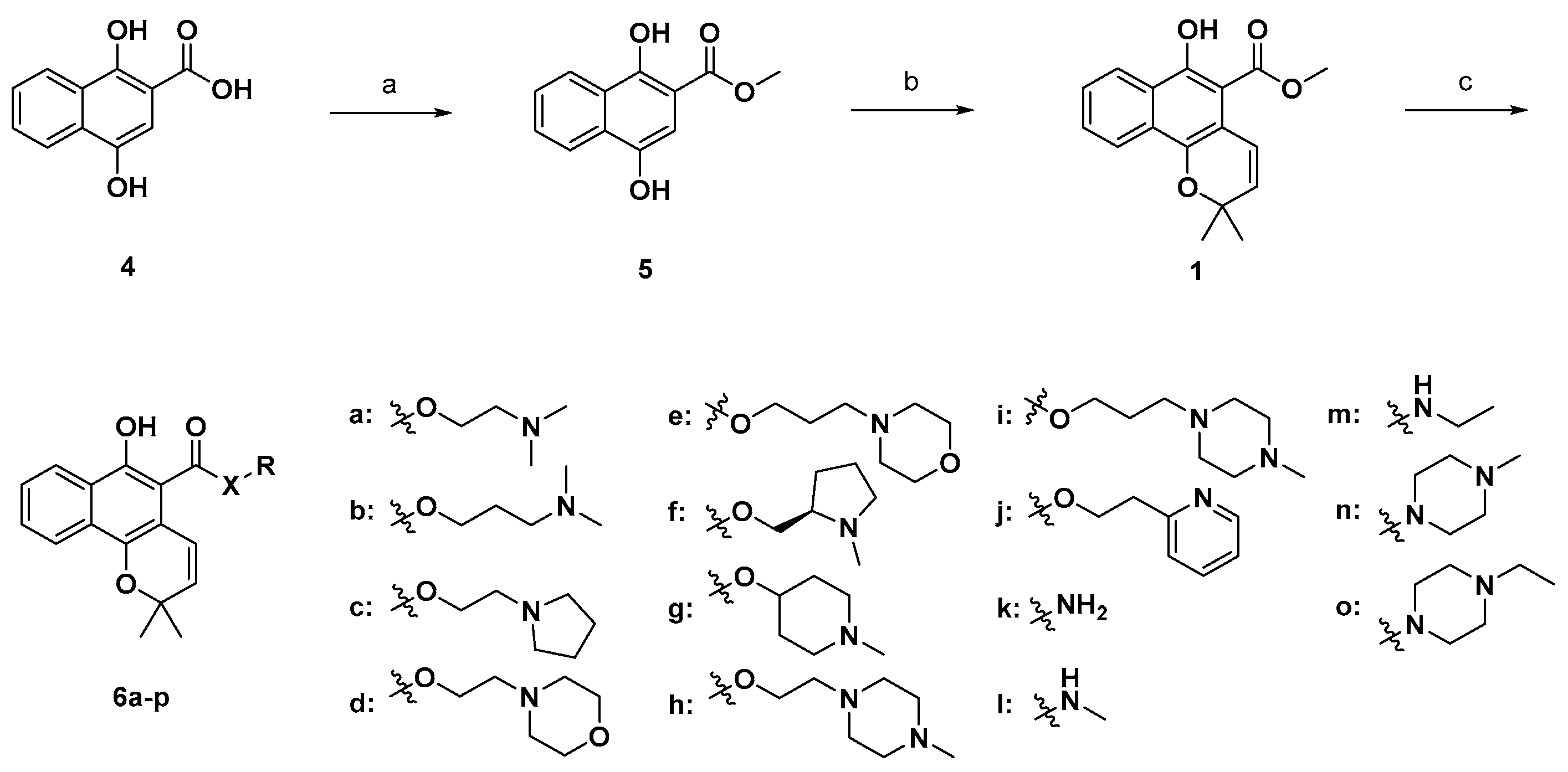
3.2. General Method A (6a–o)
The mixture of mollugin (0.35 mmol), alcohol (3.52 mmol), and catalytic p-TsOH (0.035 mmol) in 2 mL microwave vial was placed in the cavity of microwave reactor, and then stirred for 3 h at 160 °C. The produced brown mixture was dried under vacuum and subjected to purification (20 g silica gel cartridge, dichloromethane-MeOH) to give the title product.
2-(Dimethylamino)ethyl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6a). Yield: 43%; yellow oil; 1H-NMR (CDCl3) δ 11.76 (bs, 1H), 8.36 (d, J = 8.4 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.61 (t, J = 7.2 Hz, 1H), 7.51 (t, J = 7.2 Hz, 1H), 7.10 (d, J = 10.0 Hz, 1H), 5.67 (d, J = 10.0 Hz, 1H), 4.54 (d, J = 6.0 Hz, 2H), 2.76 (d, J = 6.0 Hz, 2H), 2.35 (s, 6H), 1.48 (s, 6H); 13C-NMR (CDCl3) δ 170.42, 154.37, 141.43, 128.99, 128.89, 128.68, 126.12, 125.27, 123.98, 122.00, 121.84, 112.84, 103.81, 74.84, 62.66, 57.17, 45.43, 26.96; MS (ESI, m/z): calcd for C20H24NO4 [M + H]+ 342, found 342.
3-(Dimethylamino)propyl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6b): Yield: 31%; yellow gel; 1H-NMR (CDCl3) δ 12.18 (bs, 1H), 8.36 (dd, J = 8.3, 0.4 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.62–7.59 (m, 1H), 7.52–7.48 (m, 1H), 7.13 (d, J = 10.0 Hz, 1H), 5.66 (d, J = 10.0 Hz, 1H), 4.49 (dd, J = 6.4 Hz, 2H), 2.47 (dd, J = 7.1 Hz, 2H), 2.28 (s, 6H), 2.04–1.97 (m, 2H), 1.49 (s, 6H); MS (ESI, m/z) calcd for C21H26NO4 [M + H]+ 356, found 356.
2-(Pyrrolidin-1-yl)ethyl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6c): Yield: 9%; brown oil; 1H-NMR (CDCl3) δ 11.81 (bs, 1H), 8.35 (d, J = 8.3 Hz, 1H), 8.16 (d, J = 8.1 Hz, 1H), 7.60–7.56 (m, 1H), 7.52–7.47 (m, 1H), 7.09 (d, J = 10.0 Hz, 1H), 5.66 (d, J = 10.0 Hz, 1H), 4.57 (t, J = 5.9 Hz, 2H), 2.91 (t, J = 5.9 Hz, 2H), 2.66–7.64 (m, 4H), 1.85–1.82 (m, 4H), 1.49 (s, 6H); MS (ESI, m/z) calcd for C22H26NO4 [M + H]+ 368, found 368.
2-Morpholinoethyl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6d): Yield: 30%; yellow gel; 1H-NMR (CDCl3) δ 11.61 (s, 1H), 8.36 (d, J = 8.0 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.62–7.58 (m, 1H), 7.52–7.48 (m, 1H), 7.18 (d, J = 10.0 Hz, 1H), 5.66 (d, J = 10.0 Hz, 1H), 4.55 (t, J = 5.6 Hz, 2H), 3.76 (t, J = 4.6 Hz, 4H), 2.80 (t, J = 5.7 Hz, 2H), 2.59 (t, J = 4.3 Hz, 4H), 1.49 (s, 6H); 13C-NMR (DMSO-d6) δ 168.3, 149.8, 140.8, 129.9, 128.7, 127.1, 126.5, 124.9, 123.4, 121.4, 121.1, 112.4, 107.2, 75.1, 63.1, 59.9, 54.2, 51.5, 26.5; MS (ESI, m/z) calcd for C22H26NO5 [M + H]+ 384, found 384.
3-Morpholinopropyl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6e): Yield: 23%; yellow gel; 1H-NMR (CDCl3) δ 11.99 (bs, 1H), 8.36 (d, J = 8.3 Hz, 1H), 8.18 (d, J = 8.3 Hz, 1H), 7.65–7.61 (m, 1H), 7.55–7.51 (m, 1H), 6.99 (d, J = 10.0 Hz, 1H), 5.69 (d, J = 10.0 Hz, 1H), 4.53 (t, J = 6.1 Hz, 2H), 3.99 (t, J = 4.7 Hz, 4H), 3.17–3.13 (m, 4H), 2.37–2.30 (m, 2H), 1.85–1.60 (m, 2H), 1.50 (s, 6H); MS (ESI, m/z) calcd for C23H28NO5 [M + H]+ 398, found 398.
(R)-(1-Methylpyrrolidin-2-yl)methyl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6f): Yield: 16%; yellow oil; 1H-NMR (CDCl3) δ 11.92 (s, 1H), 8.35 (d, J = 8.3 Hz, 1H), 8.16 (d, J = 8.3 Hz, 1H), 7.62–7.57 (m, 1H), 7.51–7.47 (m, 1H), 7.17 (d, J = 10.0 Hz, 1H), 5.67 (d, J = 10.0 Hz, 1H), 5.28–5.24 (m, 1H), 3.67 (d, J = 6.0 Hz, 2H), 2.80 (m, 1H), 2.51 (m, 2H), 2.37–2.32 (m, 4H), 2.00–1.90 (m, 3H), 1.50 (s, 6H); MS (ESI, m/z) calcd for C22H26NO4 [M + H]+ 368, found 368.
1-Methylpiperidin-4-yl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6g): Yield: 17%; pale green oil; 1H-NMR (CDCl3) δ 11.97 (s, 1H), 8.36 (d, J = 8.2 Hz, 1H), 8.17 (d, J = 8.3 Hz, 1H), 7.63–7.59 (m, 1H), 7.53–7.49 (m, 1H), 7.23 (d, J = 10.0 Hz, 1H), 5.66 (d, J = 10.0 Hz, 1H), 4.59 (m, 2H), 3.89 (m, 2H), 3.72 (m, 2H), 3.59 (m, 2H), 3.41 (s, 3H), 3.38 (m, 1H), 1.49 (s, 6H); MS (ESI, m/z) calcd for C22H26NO4 [M + H]+ 368, found 368.
2-(4-Methylpiperazin-1-yl)ethyl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6h): Yield: 34%; brown oil; 1H-NMR (MeOD) δ 8.36 (d, J = 8.2 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.62–7.58 (m, 1H), 7.52–7.48 (m, 1H), 7.16 (d, J = 10.0 Hz, 1H), 5.66 (d, J = 10.0 Hz, 1H), 4.55 (t, J = 5.6 Hz, 2H), 2.80 (t, J = 5.7 Hz, 2H), 2.63–2.47 (m, 8H), 2.31 (s, 3H), 1.49 (s, 6H); MS (ESI, m/z) calcd for C23H29N2O4 [M + H]+ 397, found 397.
3-(4-Methylpiperazin-1-yl)propyl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6i): Yield: 13%; yellow gel; 1H-NMR (MeOD) δ 8.34 (d, J = 8.3 Hz, 1H), 8.16 (d, J = 8.2 Hz, 1H), 7.66 (m, 1H), 7.56 (m, 1H), 7.14 (d, J = 10.0 Hz, 1H), 5.83 (d, J = 10.0 Hz, 1H), 4.61 (t, J = 6.2 Hz, 2H), 3.76 (m, 2H), 3.37 (s, 8H), 3.02 (s, 3H), 2.36 (m, 2H), 1.51 (s, 6H); MS (ESI, m/z) calcd for C24H31N2O4 [M + H]+ 411, found 411.
2-(Pyridin-2-yl)ethyl 6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (6j): Yield: 11%; yellow oil; 1H-NMR (CDCl3) δ 12.00 (bs, 1H), 8.95 (d, J = 5.3 Hz, 1H), 8.33 (d, J = 8.3 Hz, 1H), 8.22 (m, 1H), 8.15 (d, J = 8.3 Hz, 1H), 7.78–7.72 (m, 2H), 7.64–7.60 (m, 1H), 7.53–7.50 (m, 1H), 6.78 (d, J = 10.0 Hz, 1H), 5.56 (d, J = 10.0 Hz, 1H), 4.88 (d, J = 6.0 Hz, 2H), 3.67 (d, J = 6.0 Hz, 2H), 1.46 (s, 6H); MS (ESI, m/z) calcd for C23H22NO4 [M + H]+ 376, found 376.
6-Hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxamide (6k): Yield: 35%; brown gel; 1H-NMR (CDCl3) δ 12.00 (s, 1H), 8.39 (d, J = 8.2 Hz, 1H), 8.20 (d, J = 8.2 Hz, 1H), 7.67–7.64 (m, 1H), 7.56–7.52 (m, 1H), 7.35–7.26 (m, 3H), 5.73 (d, J = 9.9 Hz, 1H), 1.51 (s, 6H); MS (ESI, m/z) calcd for C16H16NO3 [M + H]+ 270, found 270.
6-Hydroxy-N,2,2-trimethyl-2H-benzo[h]chromene-5-carboxamide (6l): Yield: 52%; yellow solid; HPLC purity: 4.5 min, 96.3%; 1H-NMR (CDCl3) δ 12.23 (s, 1H), 8.34–8.32 (m, 1H), 8.17–8.14 (m, 1H), 7.60–7.55 (m, 1H), 7.53–7.49 (m, 1H), 6.68 (d, J = 10.0 Hz, 1H), 5.90 (s, 1H), 5.74 (d, J = 10.0 Hz, 1H), 3.08 (d, J = 4.9 Hz, 3H), 1.53 (s, 6H); MS (ESI, m/z) calcd for C17H18NO3 [M + H]+ 284, found 284.
N-Ethyl-6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxamide (6m): Yield: 34%; orange solid; 1H-NMR (CDCl3) δ 12.20 (s, 1H), 8.32 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 8.2 Hz, 1H), 7.59–7.55 (m, 1H), 7.53–7.49 (m, 1H), 6.69 (d, J = 9.7 Hz, 1H), 5.90 (m, 1H), 5.75 (d, J = 9.7 Hz, 1H), 3.60–3.53 (m, 2H), 1.54 (s, 6H), 1.30 (t, J = 7.2 Hz, 3H); MS (ESI, m/z) calcd for C18H20NO3 [M + H]+ 298, found 298.
(6-Hydroxy-2,2-dimethyl-2H-benzo[h]chromen-5-yl)(4-methylpiperazin-1-yl)methanone (6n): Yield: 32%; brown oil; 1H-NMR (CDCl3) δ 8.19 (d, J = 8.4 Hz, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.51 (m, 2H), 6.37 (d, J = 9.8 Hz, 1H), 5.69 (d, J = 9.8 Hz, 1H), 3.72 (m, 2H), 3.69 (bs, 1H), 3.61 (m, 2H), 2.53 (m, 2H), 2.33 (m, 2H), 2.30 (s, 3H), 1.49 (s, 6H); 13C-NMR (CDCl3) δ 169.02, 146.63, 141.53, 129.99, 127.28, 126.55, 126.14, 125.69, 122.97, 121.84, 120.63, 111.71, 110.64, 77.24, 75.61, 54.97, 45.89, 27.19 ; MS (ESI, m/z): calcd for C21H25N2O3 [M + H]+ 353, found 353.
4-Ethylpiperazin-1-yl)(6-hydroxy-2,2-dimethyl-2H-benzo[h]chromen-5-yl)methanone (6o): Yield: 16%; pale yellow gel; 1H-NMR (CDCl3) δ 12.20 (s, 1H), 8.22 (d, J = 7.8 Hz, 1H), 8.14 (d, J = 7.8 Hz, 1H), 7.51 (m, 2H), 6.38 (d, J = 9.8 Hz, 1H), 5.69 (d, J = 9.8 Hz, 1H), 3.76 (m, 2H), 3.51 (m, 2H), 2.58 (m, 2H), 2.30 (m, 2H), 2.42 (q, J = 7.2 Hz, 2H), 1.50 (s, 6H), 1.08 (t, J = 7.2 Hz, 3H); MS (ESI, m/z): calcd for C22H27N2O3 [M + H]+ 367, found 368.
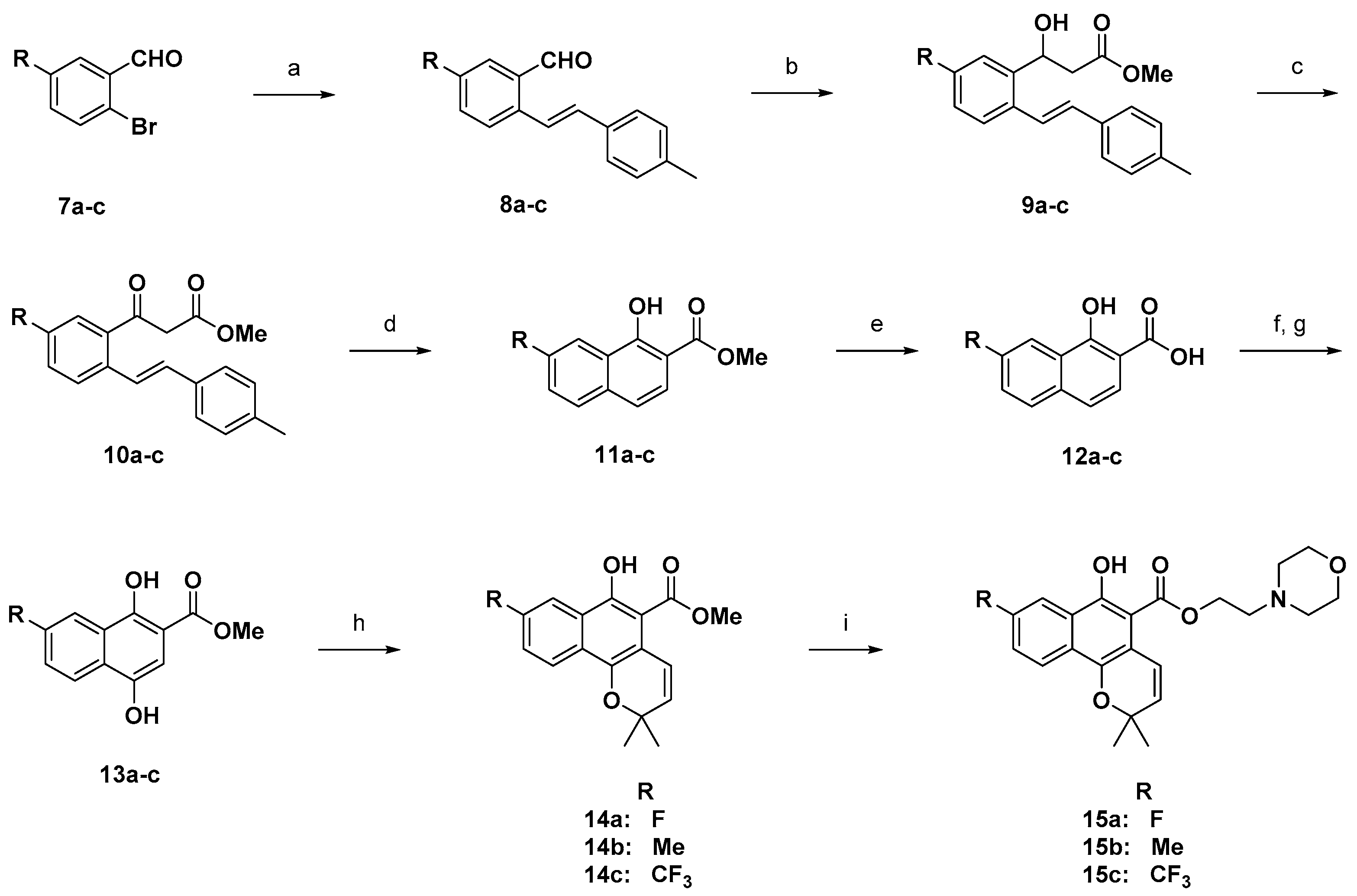
3.3. General Method B (Compounds 8a–c)
Various 2-bormobenzaldehyde (25 mmol), 4-methylstyrene (1.5 eq.), palladium (II) acetate (0.1 eq.) and tris(o-tolyl)phosphine (0.2 eq.) were dissolved in triethylamine (0.8 M) and added in a pressure tube. The pressure tube was tightly capped and heated for 15 h at 130 °C. The reaction mixture was quenched with saturated aqueous ammonium chloride and then organic materials were extracted with dichloromethane (two times). The combined organic layer was dried over magnesium sulfate, filtered, and concentrated under vacuum. The produced mixture was subjected to flash chromatography on silica gel using hexane/ethyl acetate (0 to 2% for 60 min) to give the wanted products. Yield 65–75%.
(E)-5-Fluoro-2-(4-methylstyryl)benzaldehyde (8a): Yield: 71%; Yellow oil; 1H-NMR (CDCl3) δ 10.32 (s, 1H), 7.82 (d, J = 16.1 Hz, 1H), 7.65–7.70 (m, 1H), 7.50–7.85 (m, 1H), 7.42–7.48 (m, 2H), 7.28–7.35 (m, 1H), 7.16–7.22 (m, 2H), 6.95 (d, J = 16.1 Hz, 1H), 2.38 (s, 3H); MS (ESI, m/z): calcd for C16H14FO [M + H]+ 241, found 241.
(E)-5-Methyl-2-(4-methylstyryl)benzaldehyde (8b): Yield: 73%; Yellow oil; 1H-NMR (CDCl3) δ 10.31 (s, 1H), 7.94 (d, J = 16.1 Hz, 1H), 7.58–7.65 (m, 2H), 7.42–7.49 (m, 2H), 7.35–7.41 (m, 1H), 7.18 (m, J = 7.8 Hz, 2H), 6.99 (d, J = 16.1 Hz, 1H), 2.42 (s, 3H), 2.37 (s, 3H); MS (ESI, m/z): calcd for C17H17O [M + H]+ 237, found 237.
(E)-5-Trifluoromethyl-2-(4-methylstyryl)benzaldehyde (8c): Yield: 75%; Yellow oil; 1H-NMR (CDCl3) δ 10.32 (s, 1H), 7.81 (d, J = 16.1 Hz, 1H), 7.62–7.68 (m, 1H), 7.51–7.55 (m, 1H), 7.42–7.45 (m, 2H), 7.26–7.31 (m, 1H), 7.13–7.24 (m, 2H), 6.97 (d, J = 16.1 Hz, 1H), 2.38 (s, 3H); MS (ESI, m/z): calcd for C17H14F3O [M + H]+ 291, found 291.
3.4. General Method C (Compounds 9a–c)
(4-Methylstyryl)benzaldehyde 8a–c (15 mmol) was dissolved in anhydrous benzene (50 mL). To this solution, Zinc powder (1.5 eq.) and methyl 2-bromoacetate (2.0 eq.) were added to the reaction mixture, which was then refluxed for 5 h. The reaction was quenched with saturated aqueous ammonium chloride and the organic materials were extracted with dichloromethane (two times). The combined organic layer was dried over magnesium sulfate, filtered, and concentrated under vacuum. The mixture was purified by flash chromatography on silica gel using hexane/ethyl acetate (5 to 20% for 60 min) to give the target products; Yield 79–82%.
Methyl (E)-3-hydroxy-3-(5-fluoro-2-(4-methylstyryl)phenyl)propionate (9a): Yield: 79%; yellow oil; 1H-NMR (CDCl3) δ 7.50 (dd, J = 8.7, 5.75 Hz, 1H), 7.36–7.47 (m, 2H), 7.30 (dd, J = 10.0, 2.7 Hz, 1H), 7.12 (d, J = 16.1 Hz, 1H), 6.98 (td, J = 8.4, 2.81 Hz, 1H), 6.78–6.94 (m, 3H), 5.50 (dt, J = 9.2, 3.1 Hz, 1H), 3.83 (s, 3H), 3.73 (s, 3H), 3.37 (d, J = 3.2 Hz, 1H), 2.55–2.77 (m, 2H); MS (ESI, m/z): calcd for C19H20FO3 [M + H]+ 315, found 315.
Methyl (E)-3-hydroxy-3-(5-methyl-2-(4-methylstyryl)phenyl)propanoate (9b): Yield: 80%; yellow oil; 1H-NMR (CDCl3) δ 7.47 (d, J = 7.8 Hz, 1H); 7.36–7.43 (m, 3H), 7.32 (d, J = 15.9 Hz, 1H), 7.17 (d, J = 7.8 Hz, 2H), 7.08–7.13 (m, 1H), 6.91 (d, J = 15.9 Hz, 1H), 5.52 (ddd, J = 7.8, 4.9, 3.2 Hz, 1H), 3.72 (s, 3H), 3.21 (d, J = 2.9 Hz, 1H), 2.67–2.74 (m, 2H), 2.36 (ss, 6H); MS (ESI, m/z): calcd for C20H23O3 [M + H]+ 311, found 311.
Methyl (E)-3-hydroxy-3-(5-(trifluoromethyl)-2-(4-methylstyryl)phenyl)propanoate (9c): Yield: 82%; yellow oil; 1H-NMR (CDCl3) δ 7.86 (s, 1H) 7.66 (d, J = 8.3 Hz, 1H), 7.51–7.59 (m, 1H), 7.42 (m, J = 8.1 Hz, 2H), 7.30 (d, J = 15.9 Hz, 1H), 7.19 (m, J = 7.8 Hz, 2H), 7.02 (d, J = 15.9 Hz, 1H), 5.56 (dt, J = 8.1, 3.9 Hz, 1H), 3.73 (s, 3H), 3.47 (d, J = 3.2 Hz, 1H), 2.66–2.73 (m, 2H), 2.37 (s, 3H); MS (ESI, m/z): calcd for C20H20F3O3 [M + H]+ 365, found 365.
3.5. General Method D (Compounds 10a–c)
Various methyl (4-methylstyryl) phenyl propanoate 9a–c (12 mmol) and Dess-Martin periodinane (1.3 equiv) were dissolved in anhydrous dichloromethane, and stirred at room temperature for 2 h. The reaction mixture was quenched with saturated sodium bicarbonate water solution and extracted with dichloromethane (two times). The combined organic layer was washed with brine and dried over magnesium sulfate, filtered and concentrated under vacuum. The product was purified by flash chromatography on silica gel using hexane/ethyl acetate (1 to 5% for 60 min). Product obtained as a pale yellow oil. Yield 86–90%.
Methyl (E)-3-(5-Fluoro-2-(4-methylstyryl)phenyl)-3-oxopropanoate (10a): Yield: 87%; yellow oil; 1H-NMR (CDCl3) δ 7.61–7.71 (m, 1H), 7.50 (d, J = 16.4 Hz, 1H), 7.40 (t, J = 8.4 Hz, 2H), 7.28–7.36 (m, 1H), 7.09–7.24 (m, 3H), 6.85–6.98 (m, 1H), 3.94 (s, 2H), 3.77 (m, 3H), 2.36 (s, 3H); MS (ESI, m/z): calcd for C19H18FO3 [M + H]+ 313, found 313.
Methyl (E)-3-(5-methyl-2-(4-methylstyryl)phenyl)-3-oxopropanoate (10b): Yield: 86%; yellow oil; 1H-NMR (CDCl3) δ 7.50–7.67 (m, 2H), 7.27–7.44 (m, 4H), 7.16 (d, J = 8.1 Hz, 2H), 6.83–7.05 (m, 1H), 3.97 (s, 2H), 3.73–3.80 (m, 3H), 2.40 (s, 3 H), 2.35 (s, 3 H); MS (ESI, m/z): calcd for C20H21O3 [M + H]+ 309, found 309.
Methyl (E)-3-(5-(trifluoromethyl)-2-(4-methylstyryl)phenyl)-3-oxopropanoate (10c): Yield: 90%; yellow oil; 1H-NMR (CDCl3) δ 7.70–7.89 (m, 2H), 7.66 (dd, J = 8.3, 2.0 Hz, 1H), 7.57 (d, J = 16.1 Hz, 1H), 7.35–7.48 (m, 2H), 7.19 (d, J = 7.8 Hz, 2H), 7.08 (dd, J = 16.1, 13. 5 Hz, 1H), 3.99 (s, 2H), 3.74–3.82 (m, 3H), 2.38 (s, 3H); MS (ESI, m/z): calcd for C20H18F3O3 [M + H]+ 363, found 363.
3.6. General Method E (Compounds 11a–c)
Various methyl (4-methylstyryl) phenyl) oxopropanoate 10a–c (10 mmol) was dissolved in anhydrous dichloroethane (50 mL). Palladium (II) trifluoroacetate (0.2 equiv), copper (II) acetate (1.0 equiv) and methyl acrylate (3.0 equiv) were added in reaction mixture and stirred at 100 °C for 10 h. The reaction mixture was quenched with saturated ammonium chloride water solution and extracted with dichloromethane (two times). The combined organic layer was washed with brine and dried over magnesium sulfate, filtered and concentrated under vacuum. The product was purified by flash chromatography on silica gel using hexane/ethyl acetate (0 to 2% for 60 min). Product obtained as a white solid. Yield 90–95%.
Methyl 7-fluoro-1-hydroxy-2-naphthoate (11a): Yield: 90%; white solid; 1H-NMR (CDCl3) δ 11.91 (s, 1H), 8.01 (dd, J = 10.0, 2.7 Hz, 1H), 7.65–7.83 (m, 2H) 7.37 (td, J = 8.7, 2.7 Hz, 1H), 7.28 (d, J = 8.8 Hz, 1H), 4.01 (s, 3H); MS (ESI, m/z): calcd for C12H10FO3 [M + H]+ 221, found 221.
Methyl 7-methyl-1-hydroxy-2-naphthoate (11b): Yield: 92%; white solid; 1H-NMR (CDCl3) δ 11.95 (s, 1H), 8.19 (s, 1H), 7.67 (d, J = 8.3 Hz, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.44 (dd, J = 8.3, 1.7 Hz, 1H), 7.24 (d, J = 9.1 Hz, 1H), 4.00 (s, 3H), 2.54 (s, 3H); MS (ESI, m/z): calcd for C13H13O3 [M + H]+ 217, found 217.
Methyl 7-(trifluoromethyl)-1-hydroxy-2-naphthoate (11c): Yield: 95%; white solid; 1H-NMR (CDCl3) δ 12.07 (s, 1H), 8.72 (s, 1H), 7.82–7.95 (m, 2H), 7.76 (dd, J = 8. 6, 1.7 Hz, 1H), 7.33 (d, J = 8.8 Hz, 1H), 4.02 (s, 3 H); MS (ESI, m/z): calcd for C13H9F3O3 [M + H]+ 271, found 271.
3.7. General Method F (Compounds 12a–c)
Various methyl hydroxy-2-naphthoate 11a–c (9 mmol) was dissolved in tetrahydrofuran and stirred at 25 °C for 10 min. The excess amount of potassium hydroxide solution was added in the reaction mixture and stirred at 90 °C for 20 h. The reaction mixture was cooled, acidified with 6N hydrogen chloride solution and extracted with ethyl acetate (two times). The combined organic layer was washed with brine and dried over magnesium sulfate, filtered and concentrated under vacuum. The product was purified by flash chromatography on silica gel using dichloromethane/methanol (10 to 30% for 60 min). Product obtained as a yellow solid. Yield 95–98%.
7-Fluoro-1-hydroxy-2-naphthoic acid (12a): Yield: 95%; yellow solid; 1H-NMR (CD3OD) δ 8.14 (s, 1H), 7.76 (d, J = 8.7, 1H), 7.71 (d, J = 8.4, 1H), 7.45 (dd, J = 8.4, 1.8 Hz, 1H), 7.25 (d, J = 8.7, 1H); MS (ESI, m/z): calcd for C11H8FO3 [M + H]+ 207, found 207.
7-Methyl-1-hydroxy-2-naphthoic acid (12b): Yield: 97%; yellow solid; 1H-NMR (CD3OD) δ 7.94 (dd, J = 10.3, 2.7 Hz, 1H), 7.89–7.81 (m, 2H), 7.40 (td, J = 8.7, 2.7 Hz, 1H), 7.32 (d, J = 8.7, 1H); MS (ESI, m/z): calcd for C12H11O3 [M + H]+ 203, found 203.
7-(Trifluoromethyl)-1-hydroxy-2-naphthoic acid (12c): Yield: 98%; yellow solid; 1H-NMR (MeOD) δ 7.91 (dd, J = 10.3, 2. 5 Hz, 1H), 7.87–7.75 (m, 2H), 7.38 (td, J = 8.7, 2.7 Hz, 1H), 7.33–7.24 (m, 1 H); MS (ESI, m/z): calcd for C12H8F3O3 [M + H]+ 257, found 257.
3.8. General Method G (Compounds 13a–c)
Various 1-hydroxy-2-naphthoic acids 12a–c (6 mmol) was dissolved in a mixed solution of 10% sodium hydroxide and 1,4-dioxane and stirred at 0 °C for 1 h. A saturated aqueous solution of potassium persulfate (1.5 equiv) was slowly added to the reaction mixture during 4–5 h. The reaction mixture was continuously stirred at 20 °C for overnight. The reaction mixture was acidified with 6N HCl solution and extracted with ethyl acetate (two times). The combined organic layer was washed with brine and dried over magnesium sulfate, filtered and concentrated under vacuum. The dried mixture was dissolved in N,N-dimethyl-formamide, 3.0 equivalent of potassium hydrogen carbonate was added to reaction mixture. Which was stirred at 40 °C for 0.5 h 3.0 equivalent of Iodomethane was added in reaction mixture, which was stirred at 40 °C for overnight. The reaction mixture was quenched with saturated sodium bicarbonate water solution and extracted with dichloromethane (two times). The combined organic layer was washed with brine and dried over magnesium sulfate, filtered and concentrated under vacuum. The product was purified by flash chromatography on silica gel using hexane/ethyl acetate (5 to 10% for 60 min). Product obtained as a pale yellow solid. Yield 80–82%.
Methyl 7-fluoro-1,4-dihydroxy-2-naphthoate (13a): Yield: 81%; yellow solid; 1H-NMR (CDCl3) δ 9.80 (s, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 4.4 Hz, 2H), 7.40 (m, 1H), 7.14 (s, 1H), 3.96 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 170.5, 158.8, 152.0, 145.3, 127.9, 127.3, 120.4, 119.9, 119.0, 115.0, 106.4, 53.2; MS (ESI, m/z): calcd for C12H10FO4 [M + H]+ 237, found 237.
Methyl 7-methyl-1,4-dihydroxy-2-naphthoate (13b): Yield: 80%; yellow solid; 1H-NMR (CDCl3) δ 11.54 (s, 1H), 8.18 (s, 1H), 8.02 (d, J = 8. 6 Hz, 1H), 7.49 (dd, J = 8.4, 1.59 Hz, 1H), 7.04 (s, 1H), 4.84 (s, 1H), 3.98 (s, 3H), 2.55 (s, 3H); MS (ESI, m/z): calcd for C13H13O4 [M + H]+ 233, found 233.
Methyl 7-(trifluoromethyl)-1,4-dihydroxy-2-2-naphthoate (13c): Yield: 82%; yellow solid; 1H-NMR (CDCl3) δ 11.60 (s, 1H), 8.70 (s, 1H), 8.26 (d, J = 8. 6 Hz, 1H), 7.72–7.84 (d, J = 8. 6 Hz, 1H), 7.22 (s, 1H), 5.24 (s, 1H), 4.00 (s, 3H); MS (ESI, m/z): calcd for C13H9F3O4 [M + H]+ 286, found 286.
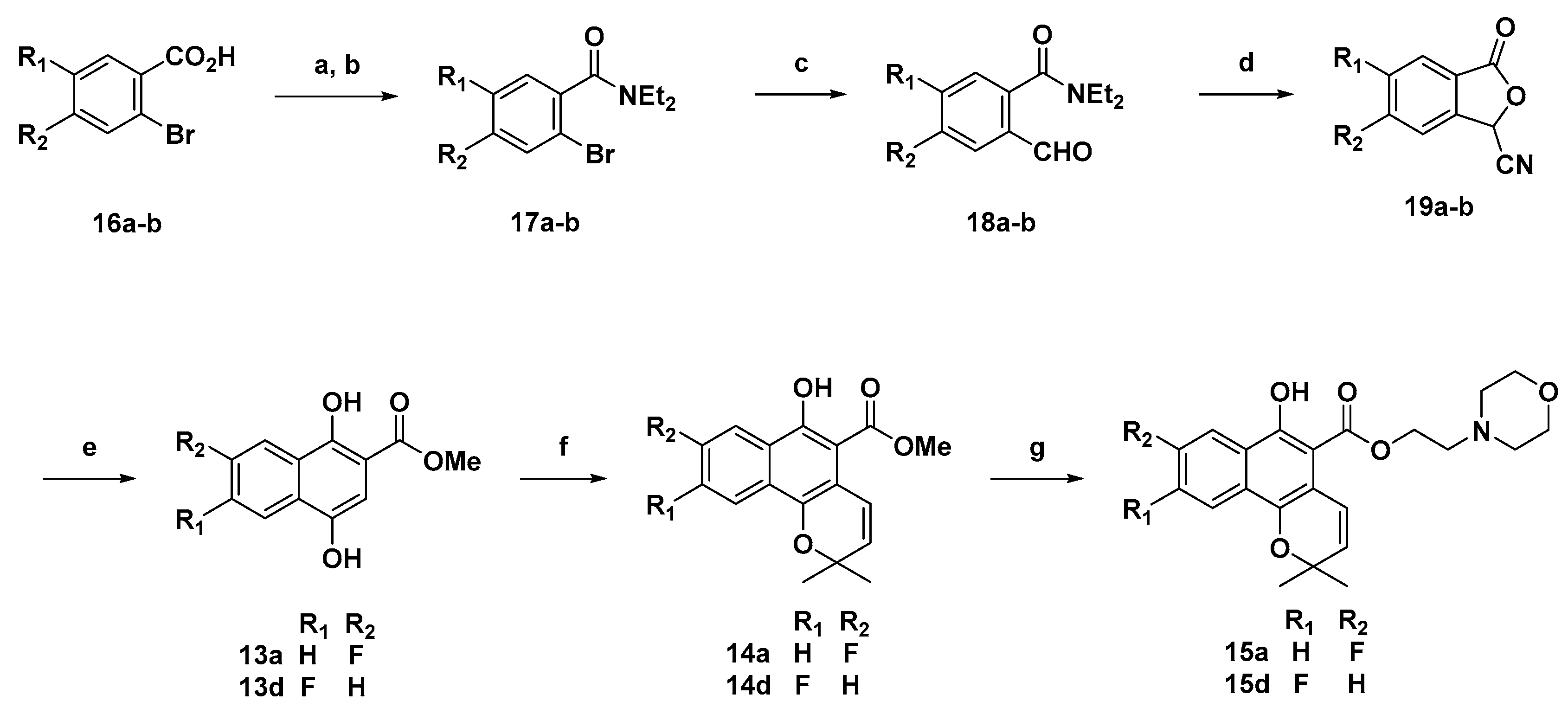
3.9. Methyl fluoro-1,4-dihydroxy-2-naphthoates 13a,d from 19a–b
LiOtBu (6 mL. 1 M in THF, 6.0 mmol) was added to stirred solution of the phthalide (0.35 g, 2.0 mmol) in dry THF (10 mL) at −60 °C under an inert atmosphere. The resulting solution was stirred at the same temperature for 30 min after which a solution of methyl acrylate (0.362 mL, 4.0 mmol) in dry THF (10 mL) was added. The reaction mixture was stirred for another 30 min at −60 °C followed by 8 h stirring at room temperature. The reaction was then quenched with saturated ammonium chloride solution (20 mL) and THF was evaporated under reduced pressure. The residue was then extracted with ethyl acetate (2 × 20 mL). The combined extracts were washed with brine, dried (Na2SO4), filtered, and concentrated to afford the crude product which was purified by column chromatography to obtain the pure compound.
Methyl 7-fluoro-1,4-dihydroxy-2-naphthoate (13a): Yield = 95%.
Methyl 6-fluoro-1,4-dihydroxy-2-naphthoate (13d): Yield = 56%; 1H-NMR (CDCl3) δ 11.59 (s, 1H), 8.40 (dd, J = 9.2, 5.7 Hz, 1H), 7.31 (dd, J = 10.2, 2.6 Hz, 1H), 7.31 (td, J = 8.7, 2.6 Hz, 1H), 7.13 (s, 1H), 4.97 (s, 1H), 3.98 (s, 3H); MS (ESI, m/z): calcd for C12H10FO4 [M + H]+ 237, found 237.
3.10. General Method H (Compounds 14a–d)
Various methyl 1,4-dihydroxy-2-naphthoate derivatives 13a–d (5 mmol), phenylboronic acid (2.0 equiv), glacial acetic acid (5.0 equiv), and 3-methylbut-2-enal (3.0 equiv) were dissolved in anhydrous toluene and refluxed for 6 h under nitrogen gas in an apparatus fitted with a Dean–Stark trap. The reaction mixture was cooled, quenched with saturated sodium bicarbonate water solution, and extracted with dichloromethane (two times). The combined organic layer was washed with brine and dried over magnesium sulfate, filtered and concentrated under vacuum. The product was purified by flash chromatography on silica gel using hexane/ethyl acetate (5 to 20% for 60 min). Product obtained as a pale yellow solid. Yield 63–66%;
Methyl 8-fluoro-6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (14a): Yield: 68%; yellow solid; 1H-NMR (CDCl3) δ 12.10 (s, 1H), 8.16 (d, J = 8. 6 Hz, 1H), 7.39 (td, J = 8.1, 4.7 Hz, 1H), 7.22 (ddd, J = 12.7, 7.7, 1.1 Hz, 1H), 7.05 (d, J = 10.0 Hz, 1H), 5.67 (d, J = 10.0 Hz, 1H), 4.03 (s, 3H), 1.51 (s, 6H); 13C-NMR (100 MHz, CDCl3) δ 172.1, 158.8, 155.5, 141.3, 129.1, 127.5, 126.2, 122.0, 120.0, 119.1, 115.3, 114.1, 103.2, 74.5, 52.5, 26.6; MS (ESI, m/z): calcd for C17H16FO4 [M + H]+ 303, found 303.
Methyl 6-hydroxy-2,2,8-trimethyl-2H-benzo[h]chromene-5-carboxylate (14b): Yield: 65%; yellow solid; 1H-NMR (CDCl3) δ 12.13 (s, 1H), 8.14 (s, 1H), 8.07 (d, J = 8. 6 Hz, 1H), 7.39–7.52 (d, J = 8. 6 Hz, 1H), 7.10 (d, J = 10.0 Hz, 1H), 5.65 (d, J = 10.0 Hz, 1H), 4.01 (s, 3H), 2.53 (s, 3H), 1.48 (m, 6H); MS (ESI, m/z): calcd for C18H19O4 [M + H]+ 299, found 299.
Methyl 6-hydroxy-2,2-dimethyl-8-(trifluoromethyl)-2H-benzo[h]chromene-5-carboxylate (14c): Yield: 66%; yellow solid; 1H-NMR (CDCl3) δ 12.21 (s, 1H), 8.67 (s, 1H), 8.27 (d, J = 8.8 Hz, 1H), 7.75 (dd, J = 8.8, 1.7 Hz, 1H), 7.13 (d, J = 10.0 Hz, 1H), 5.74 (d, J = 10.0 Hz, 1H), 4.04 (s, 3H), 1.50 (s, 6H); MS (ESI, m/z): calcd for C18H16F3O4 [M + H]+ 353, found 353.
Methyl 9-fluoro-6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (14d): Yield = 71%; yellow solid; 1H-NMR (CDCl3) δ 12.23 (s, 1H), 8.37 (dd, J = 9.1, 5.6 Hz, 1H), 7.75 (dd, J = 10.5, 2.5 Hz, 1H), 7.18–7.32 (m, 1H), 7.11 (d, J = 10.0 Hz, 1H), 5.70 (d, J = 10.0 Hz, 1H), 4.02 (s, 3H), 1.48 (s, 6H); MS (ESI, m/z): calcd for C17H16FO4 [M + H]+ 303, found 303.
3.11. General Method I (Compounds 15a–d)
Various methyl 6-hydroxy-2H-benzo[h]chromene-5-carboxylates 14a–c (3 mmol), sodium methoxide (2.0 equiv) and an alkyl alcohol (7.0 equiv) were dissolved in anhydrous toluene and added to a microwave vial, which was placed in the microwave cavity, and stirred at 150 °C for 1.5 h. After cooling and being neutralized via addition of 1.0 equivalent of the 6N hydrogen chloride solution, the mixture was concentrated under vacuum. The product was purified by flash chromatography on silica gel using hexane/ethyl acetate (30 to 100% 20 min). Product obtained as a pale yellow oil. Yield 40–59%.
2-Morpholinoethyl 8-fluoro-6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (15a): Yield: 59%; yellow oil; 1H-NMR (CDCl3) δ 11.64 (s, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.38 (m, 1H), 7.20 (m, 1H), 7.08 (d, J = 10.0 Hz, 1H), 5.66 (d, J = 10.0 Hz, 1H), 4.56 (t, J = 5.6 Hz, 2H), 3.76 (t, J = 4.4 Hz, 4H), 2.79 (t, J = 5.6 Hz, 2H), 2.59 (m, 4H), 1.50 (s, 6H); 13C-NMR (CDCl3) δ 169.8, 158.9, 153.1, 141.3, 129.3, 127.6, 126.1, 121.7, 119.9, 118.8, 114.8, 114.3, 104.9, 74.8, 66.8, 61.6, 56.5, 53.5, 26.7; MS (ESI, m/z): calcd for C22H25FO5 [M + H]+ 402, found 402.
2-Morpholinoethyl 6-hydroxy-2,2,8-trimethyl-2H-benzo[h]chromene-5-carboxylate (15b): Yield: 41%; yellow oil; 1H-NMR (CDCl3) δ 8.14 (s, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.48 (dd, J = 8.5, 1.8 Hz, 1H), 6.93 (dd, J = 10.0, 3.2 Hz, 1H), 5.65 (d, J = 10.0 Hz, 1H), 4.87 (m, 2H), 3.97 (m, 4H), 3.68 (m, 2H), 3.53 (m, 2H), 2.99 (m, 2H), 2.54 (s, 3H), 1.47 (s, 6H); MS (ESI, m/z): calcd for C23H28NO5 [M + H]+ 398, found 399.
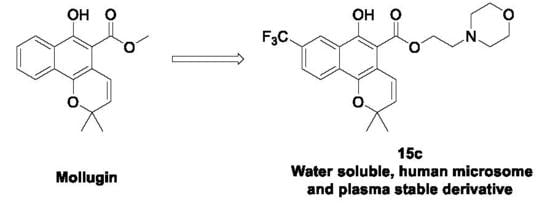
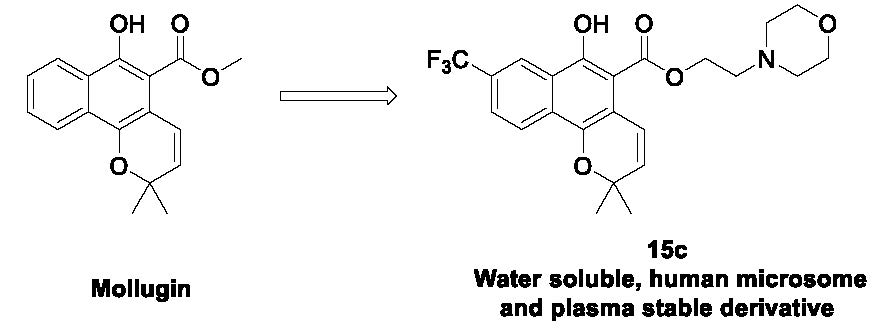
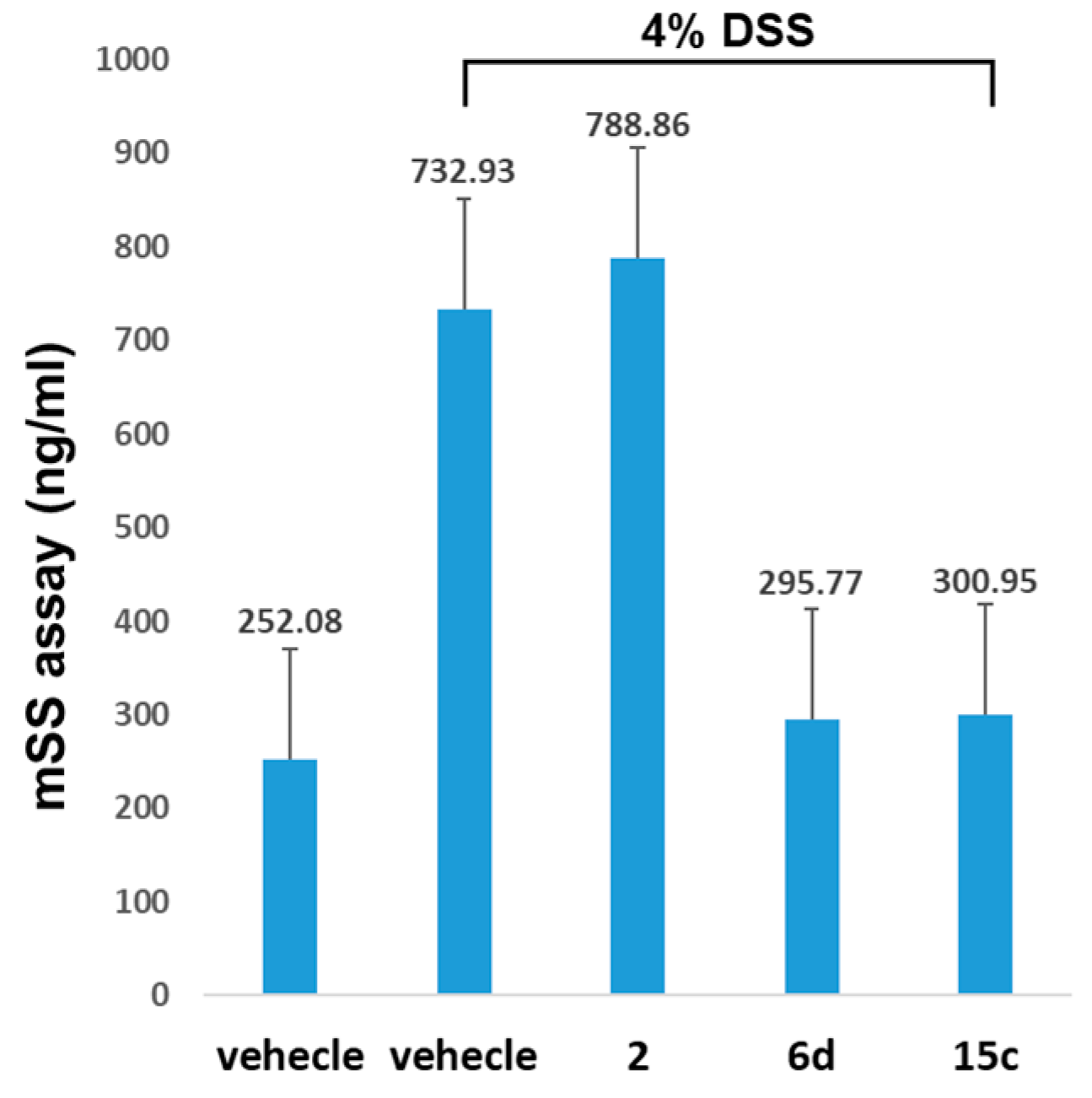
2-Morpholinoethyl 6-hydroxy-2,2-dimethyl-8-(trifluoromethyl)-2H-benzo[h]chromene-5-carboxylate (15c): Yield: 45%; yellow oil; 1H-NMR (CDCl3) δ 11.57 (s, 1H), 8.65 (s, 1H), 8.26 (d, J = 8.8 Hz, 1H), 7.73 (dd, J = 8.7, 1.6 Hz, 1H), 7.17 (d, J = 10.0 Hz, 1H), 5.73 (d, J = 10.0 Hz, 1H), 4.58 (t, J = 5.6 Hz, 2H), 3.71–3.84 (m, 4H), 2.80 (t, J = 5.6 Hz, 2H), 2.53–2.66 (m, 4H), 1.50 (s, 6H); 13C-NMR (CDCl3) δ 170.6, 156.8, 141.7, 130.5, 130.3, 128.4, 125.4, 124.4, 124.1, 123.2, 122.1, 121.9, 114.0, 102.7, 75.1, 63.7, 59.6, 56.3, 52.8, 26.7; MS (ESI, m/z): calcd for C23H25F3NO5 [M + H]+ 452, found 452.
2-Morpholinoethyl 9-fluoro-6-hydroxy-2,2-dimethyl-2H-benzo[h]chromene-5-carboxylate (15d): Yield = 55%; yellow oil; 1H-NMR (CDCl3) δ 8.37 (dd, J = 9.1, 5.6 Hz, 1H), 7.75 (dd, J = 10.3, 2.5 Hz, 1H), 7.26 (m, 1H), 6.95 (d, J = 10.0 Hz, 1H), 5.71 (d, J = 10.0 Hz, 1H), 4.88 (m, 2H), 3.98 (m, 4H), 3.64 (m, 2H), 3.51 (m, 2H), 2.99 (m, 2H), 1.48 (s, 6H); MS (ESI, m/z): calcd for C22H25FO5 [M + H]+ 402, found 402.
3.12. Synthesis of 17a–b
To a benzoic acid (10 mmol) solution in CH2Cl2 (20 mL), SOCl2 (5 mL) was added via syringe. After addition of DMF (3 drops), the reaction mixture was stirred for 5 h at room temperature, and then concentrated to dryness under reduced pressure. The produced acid chloride was dissolved in CH2Cl2 (30 mL) and cooled under ice bath. Diethylamine (25 mmol) was added dropwise and the resulting colorless suspension was stirred for 3 h at room temperature. After addition of brine (80 mL) and CH2Cl2 (50 mL), the organic layer was partitioned, washed with brine, dried with MgSO4, filtered, and concentrated under reduced pressure to yield the diethylamide which was purified by flash column chromatography to provide the target compound.
2-Bromo-N,N-diethyl-5-fluorobenzamide (17a): Yield: 97%; colorless oil; 1H-NMR (CDCl3) δ 7.53 (m, 1H), 7.00–6.95 (m, 2H), 3.82 (q, J = 6.8 Hz, 1H), 3.33 (q, J = 7.2 Hz, 1H), 3.20–3.12 (m, 2H), 1.27 (t, J = 6.8 Hz, 3H), 1.09 (t, J = 7.2 Hz, 3H); 13C-NMR (CDCl3) δ 167.0, 161.7, 140.3, 134.3, 117.2, 114.7, 113.5, 42.7, 39.0, 13.8, 13.6; MS (ESI, m/z): calcd for C11H14BrFNO [M + H]+ 274, found 274.
2-Bromo-N,N-diethyl-4-fluorobenzamide (17b): Yield: 97%; colorless oil; 1H-NMR (CDCl3) δ 6.94 (m, 1H), 6.41–6.29 (m, 4H), 5.74 (s, 3H), 2.53 (q, J = 7.5 Hz, 4H), 1.16 (m J = 7.5 Hz, 6H).
3.13. Synthesis of 18a–b from 17a–b
To a solution of amide 17a–b (9.0 mmol) in dry THF (30 mL), BuLi (1.6 M, 1.3 eq) was added dropwise at −78 °C. The resulting pale yellow solution was stirred for 15 min at the same temperature. DMF (2.5 eq) was added dropwise, and the reaction mixture was stirred for 30 min before slowly warming to ambient temperature over 2 h. The reaction mixture was quenched with a saturated solution of NH4Cl (50 mL) and extracted with CH2Cl2 (3 × 35 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford a crude orange oil. Purification of the crude oil by flash chromatography afforded the target products 18a–b.
N,N-Diethyl-5-fluoro-2-formylbenzamide (18a): Yield: 42%; colorless oil; 1H-NMR (CDCl3) δ 10.37 (s, 1H), 7.62 (dd, J = 5.2, 3.2 Hz, 1H), 7.20 (dd, J = 8.8, 5.6 Hz, 1H), 7.10 (d, J = 7.6 Hz, 1H), 3.60 (q, J = 7.2 Hz, 2H), 3.08 (q, J = 7.2 Hz, 2H), 1.33 (t, J = 7.2 Hz, 3H), 1.03 (t, J = 7.2 Hz, 3H); 13C-NMR (CDCl3) δ 186.6, 168.2, 164.7, 139.6, 136.1, 122.9, 120.9, 116.7, 42.6, 38.9, 13.6, 12.2; MS (ESI, m/z): calcd for C12H15FNO2 [M + H]+ 224, found 224.
N,N-Diethyl-4-fluoro-2-formylbenzamide (18b): Yield: 55%; colorless oil; 1H-NMR (CDCl3) δ 10.02 (s, 1H), 7.64 (m, 1H), 7.37 (m, 2H), 3.62 (q, J = 7.1 Hz, 2H), 3.14 (q, J = 7.1 Hz, 2H), 1.31 (t, J = 7.1 Hz, 3H), 1.07 (t, J = 7.1 Hz, 3H); MS (ESI, m/z): calcd for C12H15FNO2 [M + H]+ 224, found 224.
3.14. Synthesis of 19a–b from 18a–b
To a solution of the formylarylamide (0.85 g, 4.80 mmol) in CH2Cl2 (20 mL) at 0–5 °C was added 18-crown-6 (127 mg, 0.48 mmol) and KCN (31 mg, 0.48 mmol). After 10 min, TMSCN (667 mg, 6.7 mmol) was added dropwise and the mixture was stirred for 30 min. The solvent was removed under reduced pressure and AcOH (5 mL) was added to the residue. After stirring for 12 h at room temperature, the reaction mixture was slowly poured into a saturated solution of NaHCO3 (100 mL) with stirring. After 30 min (no more gas evolution), organic materials were extracted using ethyl acetate (50 mL), washed with brine, dried with MgSO4, filtered, and dried under reduced pressure. Purification of the crude product by flash chromatography afforded the target compounds.
5-Fluoro-3-oxo-1,3-dihydroisobenzofuran-1-carbonitrile (19a): Yield: 42%; white gel; 1H-NMR (CDCl3) δ 7.83 (d, J = 7.7 Hz, 1H), 7.76 (m, 1H), 7.5 (m, 1H), 6.17 (s, 1H); 13C-NMR (CDCl3) δ 166.2, 156.8, 134.2, 127.8, 127.3, 122.6, 122.4, 112.5, 63.1; MS (ESI, m/z): calcd for C9H5FNO2 [M + H]+ 178, found 178.
4-Fluoro-3-oxo-1,3-dihydroisobenzofuran-1-carbonitrile (19b): Yield: 51%; white gel; 1H-NMR (CDCl3) δ 8.02 (m, 1H), 7.43 (m, 2H), 6.08 (s, 1H); MS (ESI, m/z): calcd for C9H5FNO2 [M + H]+ 178, found 178.4. Biological methods.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}