
Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery

 , and
, and 
Abstract
1. Introduction
2. Relevance of PPIs in the Drug Discovery Paradigm
2.1. Structural Features of PPIs
2.2. Characteristics of PPI Modulators
3. Emerging In Silico Approaches for PPI Drug Discovery
3.1. Discerning the PPI Network Topology
3.2. Harnessing the Power of Machine Learning Algorithms for PPIs
3.3. Elucidation of Interface Characteristics and Hot Spot Contribution in PPIs
3.4. Exploring the PPI Interface through Macromolecular Docking and Virtual Screening
3.5. Exploiting Hot Spot Regions for Fragment-Based Design
3.6. Unraveling the Structural and Functional Aspects of PPIs Using MD Simulations
4. Pitfalls of CADD in the Discovery of PPI Inhibitors
Author Contributions
Funding
Conflicts of Interest
References
- Armour, D.; de Groot, M.J.; Edwards, M.; Perros, M.; Price, D.A.; Stammen, B.L.; Wood, A. The discovery of ccr5 receptor antagonists for the treatment of hiv infection: Hit-to-lead studies. ChemMedChem 2006, 1, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Filikov, A.V.; James, T.L. Structure-based design of ligands for protein basic domains: Application to the hiv-1 tat protein. J. Comput. Aid. Mol. Des. 1998, 12, 229–240. [Google Scholar] [CrossRef]
- Kim, C.U.; Lew, W.; Williams, M.A.; Liu, H.; Zhang, L.; Swaminathan, S.; Bischofberger, N.; Chen, M.S.; Mendel, D.B.; Tai, C.Y.; et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997, 119, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, P.; Kokkola, T.; Suuronen, T.; Poso, A.; Jarho, E.; Lahtela-Kakkonen, M. Virtual screening approach of sirtuin inhibitors results in two new scaffolds. Eur. J. Pharm. Sci. 2015, 76, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Tiwari, S.; Srivastava, K.K.; Siddiqi, M.I. Identification of novel inhibitors of mycobacterium tuberculosis pkng using pharmacophore based virtual screening, docking, molecular dynamics simulation, and their biological evaluation. J. Chem. Inf. Model. 2015, 55, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.N.; Mutyala, R.; Aparoy, P.; Reddanna, P.; Reddy, M.R. Computer aided drug design approaches to develop cyclooxygenase based novel anti-inflammatory and anti-cancer drugs. Curr. Pharm. Des. 2007, 13, 3505–3517. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Jee, J.G.; Yin, C.H.; Sandoval, C.; Jayabose, S.; Kitamura, D.; Bach, E.A.; Baeg, G.H. Nsc114792, a novel small molecule identified through structure-based computational database screening, selectively inhibits jak3. Mol. Cancer 2010, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, M.N.D.S.; Speck-Planche, A. Computer-aided drug design, synthesis and evaluation of new anti-cancer drugs. Curr. Top. Med. Chem. 2012, 12, 2703–2704. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.Q.; Zhou, R.; Lian, F.L.; Liu, Y.; Chen, L.M.; Shi, Z.; Zhang, N.X.; Zheng, M.Y.; Shen, B.R.; Jiang, H.L.; et al. Virtual screening and biological evaluation of novel small molecular inhibitors against protein arginine methyltransferase 1 (prmt1). Org. Biomol. Chem. 2014, 12, 9665–9673. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Li, G.H.; Zhao, D.Y.; Yu, H.Y.; Zheng, X.L.; Peng, X.D.; Zhang, X.; Fu, T.; Hu, X.Q.; Niu, M.S.; et al. Computational discovery and experimental verification of tyrosine kinase inhibitor pazopanib for the reversal of memory and cognitive deficits in rat model neurodegeneration. Chem. Sci. 2015, 6, 2812–2821. [Google Scholar] [CrossRef] [PubMed]
- Hoang, V.H.; Tran, P.T.; Cui, M.; Ngo, V.T.H.; Ann, J.; Park, J.; Lee, J.; Choi, K.; Cho, H.; Kim, H.; et al. Discovery of potent human glutaminyl cyclase inhibitors as anti-alzheimer’s agents based on rational design. J. Med. Chem. 2017, 60, 2573–2590. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Zhao, Z.M. A comparative study of cancer proteins in the human protein-protein interaction network. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Aguilar, A.; Bernard, D.; Wang, S.M. Small-molecule inhibitors of the mdm2-p53 protein-protein interaction (mdm2 inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Laraia, L.; McKenzie, G.; Spring, D.R.; Venkitaraman, A.R.; Huggins, D.J. Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein-protein interactions. Chem. Biol. 2015, 22, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed]
- Sable, R.; Jois, S. Surfing the protein-protein interaction surface using docking methods: Application to the design of ppi inhibitors. Molecules 2015, 20, 11569–11603. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, L.; Chothia, C.; Janin, J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999, 285, 2177–2198. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Unraveling hot spots in binding interfaces: Progress and challenges. Curr. Opin. Struct. Biol. 2002, 12, 14–20. [Google Scholar] [CrossRef]
- Moza, B.; Buonpane, R.A.; Zhu, P.; Herfst, C.A.; Rahman, A.K.M.N.U.; McCormick, J.K.; Kranz, D.M.; Sundberg, E.J. Long-range cooperative binding effects in a t cell receptor variable domain. Proc. Natl. Acad. Sci. USA 2006, 103, 9867–9872. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Janin, J. Principles of protein-protein recognition. Nature 1975, 256, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Janin, J. Principles of protein-protein recognition from structure to thermodynamics. Biochimie 1995, 77, 497–505. [Google Scholar] [CrossRef]
- Miller, S.; Lesk, A.M.; Janin, J.; Chothia, C. The accessible surface area and stability of oligomeric proteins. Nature 1987, 328, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Argos, P. An investigation of protein subunit and domain interfaces. Protein Eng. 1988, 2, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Miller, S.; Chothia, C. Surface, subunit interfaces and interior of oligomeric proteins. J. Mol. Biol. 1988, 204, 155–164. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Protein-protein interactions: A review of protein dimer structures. Prog. Biophys. Mol. Biol. 1995, 63, 31–65. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Janin, J. Dissecting protein-protein recognition sites. Proteins 2002, 47, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, R.P.; Chakrabarti, P.; Rodier, F.; Janin, J. Dissecting subunit interfaces in homodimeric proteins. Proteins 2003, 53, 708–719. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Sternberg, M.J. The contribution of missense mutations in core and rim residues of protein-protein interfaces to human disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Wu, F.; Jernigan, R.L.; Dobbs, D.; Honavar, V. Characterization of protein-protein interfaces. Protein J. 2008, 27, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. A bh3 mimetic for killing cancer cells. Cell 2016, 165, 1560. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; de Oliveira, M.R.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase ii study of single-agent navitoclax (abt-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Eradat, H.; Grosicki, S.; Catalano, J.; Cosolo, W.; Dyagil, I.S.; Yalamanchili, S.; Chai, A.; Sahasranaman, S.; Punnoose, E.; et al. A phase 2 study of the bh3 mimetic bcl2 inhibitor navitoclax (abt-263) with or without rituximab, in previously untreated b-cell chronic lymphocytic leukemia. Leukemia Lymphoma 2015, 56, 2826–2833. [Google Scholar] [CrossRef] [PubMed]
- Brachet, P.E.; Fabbro, M.; Leary, A.; Medioni, J.; Follana, P.; Lesoin, A.; Frenel, J.S.; Lacourtoisie, S.A.; Floquet, A.; Gladieff, L.; et al. A gineco phase ii study of navitoclax (abt 263) in women with platinum resistant/refractory recurrent ovarian cancer (roc). Ann. Oncol. 2017, 28. [Google Scholar] [CrossRef]
- Tolcher, A.W.; LoRusso, P.; Arzt, J.; Busman, T.A.; Lian, G.N.; Rudersdorf, N.S.; Vanderwal, C.A.; Kirschbrown, W.; Holen, K.D.; Rosen, L.S. Safety, efficacy, and pharmacokinetics of navitoclax (abt-263) in combination with erlotinib in patients with advanced solid tumors. Cancer Chemoth. Pharm. 2015, 76, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of amg 232, a potent, selective, and orally bioavailable mdm2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The bet bromodomain inhibitor otx015 affects pathogenetic pathways in preclinical b-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Zucca, E.; Bekradda, M.; Gomez-Roca, C.; Delord, J.P.; de La Motte Rouge, T.; Uro-Coste, E.; de Braud, F.; Pelosi, G.; French, C.A. Clinical response of carcinomas harboring the brd4-nut oncoprotein to the targeted bromodomain inhibitor otx015/mk-8628. Cancer Discov. 2016, 6, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, C.A.; Mitsuuchi, Y.; Burns, J.M.; Neiman, E.M.; Condon, S.M.; Yu, G.Y.; Seipel, M.E.; Kapoor, G.S.; LaPorte, M.G.; Rippin, S.R.; et al. Birinapant (tl32711), a bivalent smac mimetic, targets traf2-associated ciaps, abrogates tnf-induced nf-kb activation, and is active in patient-derived xenograft models. Mol. Cancer Ther. 2014, 13, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Vetma, V.; Rozanc, J.; Charles, E.M.; Hellwig, C.T.; Alexopoulos, L.G.; Rehm, M. Examining the in vitro efficacy of the iap antagonist birinapant as a single agent or in combination with dacarbazine to induce melanoma cell death. Oncol. Res. 2017, 25, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Schilder, R.J.; Martin, L.P.; Levin, M.; Graham, M.A.; Weng, D.E.; Adjei, A.A. A phase i study of the smac-mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol. Cancer Ther. 2015, 14, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Noonan, A.M.; Bunch, K.P.; Chen, J.Q.; Herrmann, M.A.; Lee, J.M.; Kohn, E.C.; O’Sullivan, C.C.; Jordan, E.; Houston, N.; Takebe, N.; et al. Pharmacodynamic markers and clinical results from the phase 2 study of the smac mimetic birinapant in women with relapsed platinum-resistant or -refractory epithelial ovarian cancer. Cancer 2016, 122, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Paller, C.J.; Antonarakis, E.S. Cabazitaxel: A novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des. Dev. Ther. 2011, 5, 117–124. [Google Scholar]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a dihydroisoquinolinone derivative (nvp-cgm097): A highly potent and selective mdm2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anti-Cancer Agent Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Stupp, R.; Van den Bent, M.J.; Erridge, S.G.; Reardon, D.A.; Hong, Y.; Wheeler, H.; Hegi, M.; Perry, J.R.; Picard, M.; Weller, M. Cilengitide in newly diagnosed glioblastoma with mgmt promoter methylation: Protocol of a multicenter, randomized, open-label, controlled phase iii trial (centric). J. Clin. Oncol. 2010, 28. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Barlesi, F.; Waller, C.F.; Bennouna, J.; Gridelli, C.; Goekkurt, E.; Verhoeven, D.; Szczesna, A.; Feurer, M.; Milanowski, J.; et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (nsclc) patients: Results of an open-label, randomized, controlled phase ii study (certo). Ann. Oncol. 2015, 26, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.P. End of the road: Confounding results of the core trial terminate the arduous journey of cilengitide for glioblastoma. Neuro-Oncology 2015, 17, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Leblanc, Y.; Cote, A.; Nasveschuk, C.G.; Taylor, A.M.; Harmange, J.C.; Audia, J.E.; Pardo, E.; et al. Discovery, design, and optimization of isoxazole azepine bet inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Cote, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a benzoisoxazoloazepine inhibitor (cpi-0610) of the bromodomain and extra-terminal (bet) family as a candidate for human clinical trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.A.; Abramson, J.; Maris, M.; Flinn, I.; Goy, A.; Mertz, J.; Sims, R.; Garner, F.; Senderowicz, A.; Younes, A. A phase i study of cpi-0610, a bromodomain and extra terminal protein (bet) inhibitor in patients with relapsed or refractory lymphoma. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Belani, C.P.; Eckardt, J. Development of docetaxel in advanced non-small-cell lung cancer. Lung Cancer 2004, 46, S3–S11. [Google Scholar] [CrossRef]
- Ford, H.E.R.; Marshall, A.; Bridgewater, J.A.; Janowitz, T.; Coxon, F.Y.; Wadsley, J.; Mansoor, W.; Fyfe, D.; Madhusudan, S.; Middleton, G.W.; et al. Docetaxel versus active symptom control for refractory oesophagogastric adenocarcinoma (cougar-02): An open-label, phase 3 randomised controlled trial. Lancet Oncol. 2014, 15, 78–86. [Google Scholar] [CrossRef]
- Feinman, H.E.; Price, D.K.; Figg, W.D. Piecing the puzzle together: Docetaxel cycles and current considerations in the treatment of metastatic castration-resistant prostate cancer. Cancer Biol. Ther. 2017, 18, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.W.; Kwon, O.K.; Chung, H.Y.; Yu, W.; Kim, J.G. Taxanes in the treatment of advanced gastric cancer. Molecules 2016, 21, 651. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Rosenthal, J.; Andreeff, M.; Zernovak, O.; Kumar, P.; Gajee, R.; Chen, S.Q.; Rosen, M.; Song, S.; Kochan, J.; et al. Phase 1 dose escalation study of mdm2 inhibitor ds-3032b in patients with hematological malignancies - preliminary results. Blood 2016, 128, 593. [Google Scholar]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Tcheng, J.E.; Talley, J.D.; O’Shea, J.C.; Gilchrist, I.C.; Kleiman, N.S.; Grines, C.L.; Davidson, C.J.; Lincoff, A.M.; Califf, R.M.; Jennings, L.K.; et al. Clinical pharmacology of higher dose eptifibatide in percutaneous coronary intervention (the pride study). Am. J. Cardiol. 2001, 88, 1097–1102. [Google Scholar] [CrossRef]
- Fung, J.J. Tacrolimus and transplantation: A decade in review. Transplantation 2004, 77, S41–S43. [Google Scholar] [CrossRef] [PubMed]
- Mirguet, O.; Gosmini, R.; Toum, J.; Clement, C.A.; Barnathan, M.; Brusq, J.M.; Mordaunt, J.E.; Grimes, R.M.; Crowe, M.; Pineau, O.; et al. Discovery of epigenetic regulator i-bet762: Lead optimization to afford a clinical candidate inhibitor of the bet bromodomains. J. Med. Chem. 2013, 56, 7501–7515. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Kang, M.H.; Reynolds, C.P.; Morton, C.L.; Kolb, E.A.; Gorlick, R.; Keir, S.T.; Carol, H.; Lock, R.; Maris, J.M.; et al. Initial testing (stage 1) of lcl161, a smac mimetic, by the pediatric preclinical testing program. Pediatr. Blood Cancer 2012, 58, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase i dose-escalation study of lcl161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Zuo, Y.; Yang, X.; Lu, J.; Zhan, L.L.; Xu, L.P.; Zhang, C.; Zhu, H.C.; Liu, J.; Liu, Z.M.; et al. Smac mimetic compound lcl161 sensitizes esophageal carcinoma cells to radiotherapy by inhibiting the expression of inhibitor of apoptosis protein. Tumor Biol. 2014, 35, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Martin, B.P.; Andrews, D.A.; Hogg, S.J.; Banerjee, A.; Grigoriadis, G.; Johnstone, R.W.; Shortt, J. The smac mimetic, lcl-161, reduces survival in aggressive myc-driven lymphoma while promoting susceptibility to endotoxic shock. Oncogenesis 2016, 5, e216. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.L.; Pflugfelder, S.C.; Zhang, S.; Shojaei, A.; Haque, R. Lifitegrast, a novel integrin antagonist for treatment of dry eye disease. Ocul. Surf. 2016, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.F.; McEachern, D.; Li, S.Q.; Ellis, M.J.; Wang, S.M. Reactivation of p53 by mdm2 inhibitor mi-77301 for the treatment of endocrine-resistant breast cancer. Mol. Cancer. Ther. 2016, 15, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Sun, W.; Zhao, Y.J.; McEachern, D.; Meaux, I.; Barriere, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.C.; Liu, L.; et al. Sar405838: An optimized inhibitor of mdm2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Blum, S.S.; Fung, H.B.; Bandres, J.C. Maraviroc: A ccr5-receptor antagonist for the treatment of hiv-1 infection. Clin. Ther. 2008, 30, 1228–1250. [Google Scholar] [CrossRef]
- Van Der Ryst, E. Maraviroc—A ccr5 antagonist for the treatment of hiv-1 infection. Front. Immunol. 2015, 6, 1228–1250. [Google Scholar] [CrossRef] [PubMed]
- Woodhead, A.J.; Angove, H.; Carr, M.G.; Chessari, G.; Congreve, M.; Coyle, J.E.; Cosme, J.; Graham, B.; Day, P.J.; Downham, R.; et al. Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydrois oindol-2-yl]methanone (at13387), a novel inhibitor of the molecular chaperone hsp90 by fragment based drug design. J. Med. Chem. 2010, 53, 5956–5969. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.J.; Jiang, N.; Liu, J.J.; Zhao, C.L.; Glenn, K.; Wen, Y.; et al. Discovery of rg7112: A small-molecule mdm2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.J.; Zhang, Z.M.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of rg7388, a potent and selective p53-mdm2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- King, S.; Short, M.; Harmon, C. Glycoprotein iib/iiia inhibitors: The resurgence of tirofiban. Vasc. Pharmacol. 2016, 78, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Milla, M.E.; Sauer, R.T. P22 arc repressor: Folding kinetics of a single-domain, dimeric protein. Biochemistry 1994, 33, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, K.; Krebs, M. The v-atpase: Small cargo, large effects. Curr. Opin. Plant Biol. 2010, 13, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Nooren, I.M.; Thornton, J.M. Diversity of protein-protein interactions. EMBO J. 2003, 22, 3486–3492. [Google Scholar] [CrossRef] [PubMed]
- Hayer-Hartl, M.; Bracher, A.; Hartl, F.U. The groel-groes chaperonin machine: A nano-cage for protein folding. Trends Biochem. Sci. 2016, 41, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.R.; Diboun, I.; Dessailly, B.H.; Lees, J.G.; Orengo, C. Transient protein-protein interactions: Structural, functional, and network properties. Structure 2010, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Nooren, I.M.; Thornton, J.M. Structural characterisation and functional significance of transient protein-protein interactions. J. Mol. Biol. 2003, 325, 991–1018. [Google Scholar] [CrossRef]
- Reichmann, D.; Rahat, O.; Cohen, M.; Neuvirth, H.; Schreiber, G. The molecular architecture of protein-protein binding sites. Curr. Opin. Struct. Biol. 2007, 17, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Janin, J. The kinetics of protein-protein recognition. Proteins 1997, 28, 153–161. [Google Scholar] [CrossRef]
- Li, W.; Keeble, A.H.; Giffard, C.; James, R.; Moore, G.R.; Kleanthous, C. Highly discriminating protein-protein interaction specificities in the context of a conserved binding energy hotspot. J. Mol. Biol. 2004, 337, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, D.; Thiel, S.; Vajda, S.; Camacho, C.J. Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 11287–11292. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.R.; Brower, R.C.; Vajda, S.; Camacho, C.J. Dynamical view of the positions of key side chains in protein-protein recognition. Biophys. J. 2001, 80, 635–642. [Google Scholar] [CrossRef]
- Kraich, M.; Klein, M.; Patino, E.; Harrer, H.; Nickel, J.; Sebald, W.; Mueller, T.D. A modular interface of il-4 allows for scalable affinity without affecting specificity for the il-4 receptor. BMC Biol. 2006, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.T.; Huang, K.Y.; Su, M.G.; Lee, T.Y.; Bretana, N.A.; Chang, W.C.; Chen, Y.J.; Chen, Y.J.; Huang, H.D. Dbptm 3.0: An informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 2013, 41, D295–D305. [Google Scholar] [CrossRef] [PubMed]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.Y.; Walther, D. The roles of post-translational modifications in the context of protein interaction networks. PLoS. Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef] [PubMed]
- Minguez, P.; Parca, L.; Diella, F.; Mende, D.R.; Kumar, R.; Helmer-Citterich, M.; Gavin, A.C.; van Noort, V.; Bork, P. Deciphering a global network of functionally associated post-translational modifications. Mol. Syst. Biol. 2012, 8, 599. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, S.G.; Raghunathan, D.; Nicholson, J.; Hupp, T.R.; Lane, D.P.; Verma, C.S. Chemical states of the n-terminal “lid” of mdm2 regulate p53 binding: Simulations reveal complexities of modulation. Cell Cycle 2011, 10, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ballester, G.; Blanes-Mira, C.; Serrano, L. The tryptophan switch: Changing ligand-binding specificity from type i to type ii in sh3 domains. J. Mol. Biol. 2004, 335, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Saksela, K.; Permi, P. Sh3 domain ligand binding: What’s the consensus and where’s the specificity? FEBS Lett. 2012, 586, 2609–2614. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Ma, B.; Nussinov, R. Folding and binding cascades: Shifts in energy landscapes. Proc. Natl. Acad. Sci. USA 1999, 96, 9970–9972. [Google Scholar] [CrossRef] [PubMed]
- Maity, A.; Majumdar, S.; Priya, P.; De, P.; Saha, S.; Ghosh Dastidar, S. Adaptability in protein structures: Structural dynamics and implications in ligand design. J. Biomol. Struct. Dyn. 2015, 33, 298–321. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. 2015, 589, 2498–2506. [Google Scholar] [CrossRef] [PubMed]
- Bourgeas, R.; Basse, M.J.; Morelli, X.; Roche, P. Atomic analysis of protein-protein interfaces with known inhibitors: The 2p2i database. PLoS ONE 2010, 5, e9598. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, O.; Reynes, C.H.; Camproux, A.C.; Villoutreix, B.O. Rationalizing the chemical space of protein-protein interaction inhibitors. Drug Discov. Today 2010, 15, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Kuenemann, M.A.; Bourbon, L.M.L.; Labbé, C.M.; Villoutreix, B.O.; Sperandio, O. Which three-dimensional characteristics make efficient inhibitors of protein–protein interactions? J. Chem. Inf. Model. 2014, 54, 3067–3079. [Google Scholar] [CrossRef] [PubMed]
- Basse, M.J.; Betzi, S.; Bourgeas, R.; Bouzidi, S.; Chetrit, B.; Hamon, V.; Morelli, X.; Roche, P. 2p2idb: A structural database dedicated to orthosteric modulation of protein-protein interactions. Nucleic Acids Res. 2013, 41, D824–D827. [Google Scholar] [CrossRef] [PubMed]
- Labbe, C.M.; Laconde, G.; Kuenemann, M.A.; Villoutreix, B.O.; Sperandio, O. Ippi-db: A manually curated and interactive database of small non-peptide inhibitors of protein-protein interactions. Drug Discov. Today 2013, 18, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Morelli, X.; Bourgeas, R.; Roche, P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2p2i). Curr. Opin. Chem. Biol. 2011, 15, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Villoutreix, B.O.; Labbe, C.M.; Lagorce, D.; Laconde, G.; Sperandio, O. A leap into the chemical space of protein-protein interaction inhibitors. Curr. Pharm. Des. 2012, 18, 4648–4667. [Google Scholar] [CrossRef] [PubMed]
- Petsalaki, E.; Russell, R.B. Peptide-mediated interactions in biological systems: New discoveries and applications. Curr. Opin. Biotechnol. 2008, 19, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Wanner, J.; Fry, D.C.; Peng, Z.; Roberts, J. Druggability assessment of protein-protein interfaces. Future Med. Chem. 2011, 3, 2021–2038. [Google Scholar] [CrossRef] [PubMed]
- Garner, J.; Harding, M.M. Design and synthesis of alpha-helical peptides and mimetics. Org. Biomol. Chem. 2007, 5, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.A.; Wilson, A.J. Helix-mediated protein--protein interactions as targets for intervention using foldamers. Amino Acids 2011, 41, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Nevola, L.; Giralt, E. Modulating protein-protein interactions: The potential of peptides. Chem. Commun. 2015, 51, 3302–3315. [Google Scholar] [CrossRef] [PubMed]
- Zoller, F.; Markert, A.; Barthe, P.; Zhao, W.; Weichert, W.; Askoxylakis, V.; Altmann, A.; Mier, W.; Haberkorn, U. Combination of phage display and molecular grafting generates highly specific tumor-targeting miniproteins. Angew. Chem. Int. Ed. Engl. 2012, 51, 13136–13139. [Google Scholar] [CrossRef] [PubMed]
- Seoane, M.D.; Petkau-Milroy, K.; Vaz, B.; Mocklinghoff, S.; Folkertsma, S.; Milroy, L.-G.; Brunsveld, L. Structure-activity relationship studies of miniproteins targeting the androgen receptor-coactivator interaction. MedChemComm 2013, 4, 187–192. [Google Scholar] [CrossRef]
- Winter, A.; Higueruelo, A.P.; Marsh, M.; Sigurdardottir, A.; Pitt, W.R.; Blundell, T.L. Biophysical and computational fragment-based approaches to targeting protein-protein interactions: Applications in structure-guided drug discovery. Q. Rev. Biophys. 2012, 45, 383–426. [Google Scholar] [CrossRef] [PubMed]
- Valkov, E.; Sharpe, T.; Marsh, M.; Greive, S.; Hyvoenen, M. Targeting protein-protein interactions and fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 145–179. [Google Scholar] [PubMed]
- Gleeson, M.P.; Hersey, A.; Montanari, D.; Overington, J. Probing the links between in vitro potency, admet and physicochemical parameters. Nat. Rev. Drug Discov. 2011, 10, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Davis, A.M. Time-related differences in the physical property profiles of oral drugs. J. Med. Chem. 2004, 47, 6338–6348. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, J.R. The evolution of synthetic oral drug properties. Bioorg. Med. Chem. Lett. 2005, 15, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Keseru, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Mason, J.S.; Overington, J.P. Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol. 2006, 16, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, J.; Fontanillo, C. Protein-protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.H.; Lopes, T.J.; Mah, N.; Shoemaker, J.E.; Matsuoka, Y.; Fontaine, J.F.; Louis-Jeune, C.; Eisfeld, A.J.; Neumann, G.; Perez-Iratxeta, C.; et al. Adding protein context to the human protein-protein interaction network to reveal meaningful interactions. PLoS Comput. Biol. 2013, 9, e1002860. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Betel, D.; Hogue, C.W.V. Bind: The biomolecular interaction network database. Nucleic Acids Res. 2003, 31, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. Biogrid: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Chatr-aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The biogrid interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Prasad, T.S.K.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human protein reference database-2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Hermjakob, H.; Montecchi-Palazzi, L.; Lewington, C.; Mudali, S.; Kerrien, S.; Orchard, S.; Vingron, M.; Roechert, B.; Roepstorff, P.; Valencia, A.; et al. Intact: An open source molecular interaction database. Nucleic Acids Res. 2004, 32, D452–D455. [Google Scholar] [CrossRef] [PubMed]
- Aranda, B.; Achuthan, P.; Alam-Faruque, Y.; Armean, I.; Bridge, A.; Derow, C.; Feuermann, M.; Ghanbarian, A.T.; Kerrien, S.; Khadake, J.; et al. The intact molecular interaction database in 2010. Nucleic Acids Res. 2010, 38, D525–D531. [Google Scholar] [CrossRef] [PubMed]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The intact molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. Mint, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Pagel, P.; Kovac, S.; Oesterheld, M.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Mark, P.; Stumpflen, V.; Mewes, H.W.; et al. The mips mammalian protein-protein interaction database. Bioinformatics 2005, 21, 832–834. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Stransky, M.; Waegele, B.; Schmidt, T.; Doudieu, O.N.; Mpflen, V.S.; et al. Corum: The comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008, 36, D646–D650. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Waegele, B.; Lechner, M.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Mewes, H.W. Corum: The comprehensive resource of mammalian protein complexes-2009. Nucleic Acids Res. 2010, 38, D497–D501. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.K.; Pacifico, S.; Liu, G.Z.; Finley, R.L. Droid: The drosophila interactions database, a comprehensive resource for annotated gene and protein interactions. BMC Genom. 2008, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Murali, T.; Pacifico, S.; Yu, J.K.; Guest, S.; Roberts, G.G.; Finley, R.L. Droid 2011: A comprehensive, integrated resource for protein, transcription factor, rna and gene interactions for drosophila. Nucleic Acids Res. 2011, 39, D736–D743. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Lopez, D.; Gutierrez, M.A.; Lopes, K.P.; Prieto, C.; Santamaria, R.; De Las Rivas, J. Apid interactomes: Providing proteome-based interactomes with controlled quality for multiple species and derived networks. Nucleic Acids Res. 2016, 44, W529–W535. [Google Scholar] [CrossRef] [PubMed]
- Ptak, R.G.; Fu, W.; Sanders-Beer, B.E.; Dickerson, J.E.; Pinney, J.W.; Robertson, D.L.; Rozanov, M.N.; Katz, K.S.; Maglott, D.R.; Pruitt, K.D.; et al. Cataloguing the hiv type 1 human protein interaction network. Aids Res. Hum. Retrov. 2008, 24, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Sanders-Beer, B.E.; Katz, K.S.; Maglott, D.R.; Pruitt, K.D.; Ptak, R.G. Human immunodeficiency virus type 1, human protein interaction database at ncbi. Nucleic Acids Res. 2009, 37, D417–D422. [Google Scholar] [CrossRef] [PubMed]
- Pinney, J.W.; Dickerson, J.E.; Fu, W.; Sanders-Beer, B.E.; Ptak, R.G.; Robertson, D.L. Hiv-host interactions: A map of viral perturbation of the host system. Aids 2009, 23, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Ako-Adjei, D.; Fu, W.; Wallin, C.; Katz, K.S.; Song, G.; Darji, D.; Brister, J.R.; Ptak, R.G.; Pruitt, K.D. Hiv-1, human interaction database: Current status and new features. Nucleic Acids Res. 2015, 43, D566–D570. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Park, B.; Kim, H.; Hong, J.; Park, J. Hpid: The human protein interaction database. Bioinformatics 2004, 20, 2466–2470. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Nanduri, B. Hpidb—A unified resource for host-pathogen interactions. BMC Bioinform. 2010, 11, S16. [Google Scholar] [CrossRef] [PubMed]
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. Hpidb 2.0: A curated database for host-pathogen interactions. Database 2016. [Google Scholar] [CrossRef] [PubMed]
- Razick, S.; Magklaras, G.; Donaldson, I.M. Irefindex: A consolidated protein interaction database with provenance. BMC Bioinform. 2008, 9, 405. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.; Razick, S.; Turinsky, A.L.; Vlasblom, J.; Crowdy, E.K.; Cho, E.; Morrison, K.; Donaldson, I.M.; Wodak, S.J. Irefweb: Interactive analysis of consolidated protein interaction data and their supporting evidence. Database 2010. [Google Scholar] [CrossRef] [PubMed]
- Chautard, E.; Ballut, L.; Thierry-Mieg, N.; Ricard-Blum, S. Matrixdb, a database focused on extracellular protein-protein and protein-carbohydrate interactions. Bioinformatics 2009, 25, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Chautard, E.; Fatoux-Ardore, M.; Ballut, L.; Thierry-Mieg, N.; Ricard-Blum, S. Matrixdb, the extracellular matrix interaction database. Nucleic Acids Res. 2011, 39, D235–D240. [Google Scholar] [CrossRef] [PubMed]
- Launay, G.; Salza, R.; Multedo, D.; Thierry-Mieg, N.; Ricard-Blum, S. Matrixdb, the extracellular matrix interaction database: Updated content, a new navigator and expanded functionalities. Nucleic Acids Res. 2015, 43, D321–D327. [Google Scholar] [CrossRef] [PubMed]
- Calderone, A.; Castagnoli, L.; Cesareni, G. Mentha: A resource for browsing integrated protein-interaction networks. Nat. Methods 2013, 10, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Beuming, T.; Skrabanek, L.; Niv, M.Y.; Mukherjee, P.; Weinstein, H. Pdzbase: A protein-protein interaction database for pdz-domains. Bioinformatics 2005, 21, 827–828. [Google Scholar] [CrossRef] [PubMed]
- Klapa, M.I.; Tsafou, K.; Theodoridis, E.; Tsakalidis, A.; Moschonas, N.K. Reconstruction of the experimentally supported human protein interactome: What can we learn? BMC Syst. Biol. 2013, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Gioutlakis, A.; Klapa, M.I.; Moschonas, N.K. Pickle 2.0: A human protein-protein interaction meta-database employing data integration via genetic information ontology. PLoS ONE 2017, 12, e0186039. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.M.; Vallenius, T.; Ovaska, K.; Westermarck, J.; Makela, T.P.; Hautaniemi, S. Integrated network analysis platform for protein-protein interactions. Nat. Methods 2009, 6, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.J.; Pinese, M.; Kassahn, K.S.; Waddell, N.; Pearson, J.V.; Grimmond, S.M.; Biankin, A.V.; Hautaniemi, S.; Wu, J.M. Pina v2.0: Mining interactome modules. Nucleic Acids Res. 2012, 40, D862–D865. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Tripathi, L.P.; Prathipati, P.; Mizuguchi, K. Network analysis and in silico prediction of protein-protein interactions with applications in drug discovery. Curr. Opin. Struct. Biol. 2017, 44, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Kinoshita, K.; Nakamura, H. Hub promiscuity in protein-protein interaction networks. Int. J. Mol. Sci. 2010, 11, 1930–1943. [Google Scholar] [CrossRef] [PubMed]
- Raman, K. Construction and analysis of protein-protein interaction networks. Autom. Exp. 2010, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Choi, S.; Hyeon, C. Mapping the intramolecular signal transduction of g-protein coupled receptors. Proteins 2014, 82, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Lee, Y.; Choi, S. Understanding g protein-coupled receptor allostery via molecular dynamics simulations: Implications for drug discovery. Methods Mol. Biol. 2018, 1762, 455–472. [Google Scholar] [PubMed]
- Keskin, O.; Tuncbag, N.; Gursoy, A. Predicting protein-protein interactions from the molecular to the proteome level. Chem. Rev. 2016, 116, 4884–4909. [Google Scholar] [CrossRef] [PubMed]
- Aragues, R.; Sali, A.; Bonet, J.; Marti-Renom, M.A.; Oliva, B. Characterization of protein hubs by inferring interacting motifs from protein interactions. PLoS Comput. Biol. 2007, 3, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.M.; Lu, L.J.; Xia, Y.; Gerstein, M.B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science 2006, 314, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Mason, S.P.; Barabasi, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wang, J.; Wang, J.; Wu, F.X.; Pan, Y. Rechecking the centrality-lethality rule in the scope of protein subcellular localization interaction networks. PLoS ONE 2015, 10, e0130743. [Google Scholar] [CrossRef] [PubMed]
- Wuchty, S. Controllability in protein interaction networks. Proc. Natl. Acad. Sci. USA 2014, 111, 7156–7160. [Google Scholar] [CrossRef] [PubMed]
- Wuchty, S.; Boltz, T.; Kucuk-McGinty, H. Links between critical proteins drive the controllability of protein interaction networks. Proteomics 2017. [Google Scholar] [CrossRef] [PubMed]
- Feldman, I.; Rzhetsky, A.; Vitkup, D. Network properties of genes harboring inherited disease mutations. Proc. Natl. Acad. Sci. USA 2008, 105, 4323–4328. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.D.; Murali, T.M.; Sobral, B.W. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008, 4, e32. [Google Scholar] [CrossRef] [PubMed]
- Vinayagam, A.; Gibson, T.E.; Lee, H.J.; Yilmazel, B.; Roesel, C.; Hu, Y.; Kwon, Y.; Sharma, A.; Liu, Y.Y.; Perrimon, N.; et al. Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets. Proc. Natl. Acad. Sci. USA 2016, 113, 4976–4981. [Google Scholar] [CrossRef] [PubMed]
- Kanhaiya, K.; Czeizler, E.; Gratie, C.; Petre, I. Controlling directed protein interaction networks in cancer. Sci. Rep. 2017, 7, 10327. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, V.; Nacher, J.C.; Akutsu, T.; Ishitsuka, M.; Osadcenco, A.; Sunitha, V.; Bagler, G.; Schwartz, J.-M.; Robertson, D.L. Network controllability: Viruses are driver agents in dynamic molecular systems. bioRxiv 2018. [Google Scholar] [CrossRef]
- Pazos, F.; Valencia, A. In silico two-hybrid system for the selection of physically interacting protein pairs. Proteins 2002, 47, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hopf, T.A.; Scharfe, C.P.; Rodrigues, J.P.; Green, A.G.; Kohlbacher, O.; Sander, C.; Bonvin, A.M.; Marks, D.S. Sequence co-evolution gives 3d contacts and structures of protein complexes. eLife 2014, 3, e03430. [Google Scholar] [CrossRef] [PubMed]
- Walhout, A.J.M.; Sordella, R.; Lu, X.W.; Hartley, J.L.; Temple, G.F.; Brasch, M.A.; Thierry-Mieg, N.; Vidal, M. Protein interaction mapping in c-elegans using proteins involved in vulval development. Science 2000, 287, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Gerstein, M. Analyzing protein function on a genomic scale: The importance of gold-standard positives and negatives for network prediction. Curr. Opin. Microbiol. 2004, 7, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Wu, B.; Jansen, R.; Gerstein, M.; Zhao, H. Information assessment on predicting protein-protein interactions. BMC Bioinform. 2004, 5, 154. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.J.; Xia, Y.; Paccanaro, A.; Yu, H.; Gerstein, M. Assessing the limits of genomic data integration for predicting protein networks. Genome Res. 2005, 15, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Yu, H.; Greenbaum, D.; Kluger, Y.; Krogan, N.J.; Chung, S.; Emili, A.; Snyder, M.; Greenblatt, J.F.; Gerstein, M. A bayesian networks approach for predicting protein-protein interactions from genomic data. Science 2003, 302, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Nakamura, H. Filtering high-throughput protein-protein interaction data using a combination of genomic features. BMC Bioinform. 2005, 6, 100. [Google Scholar]
- Ofran, Y.; Rost, B. Isis: Interaction sites identified from sequence. Bioinformatics 2007, 23, e13–e16. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Dobbs, D.; Bonvin, A.M.; Honavar, V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015, 589, 3516–3526. [Google Scholar] [CrossRef] [PubMed]
- Sikic, M.; Tomic, S.; Vlahovicek, K. Prediction of protein-protein interaction sites in sequences and 3d structures by random forests. PLoS Comput. Biol. 2009, 5, e1000278. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, P.; Huang, D.S.; Li, J.J.; Lok, T.M.; Lyu, M.R. Predicting protein interaction sites from residue spatial sequence profile and evolution rate. FEBS Lett. 2006, 580, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Prediction of protein-protein interaction sites using patch analysis. J. Mol. Biol. 1997, 272, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Analysis of protein-protein interaction sites using surface patches. J. Mol. Biol. 1997, 272, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.R.; Westhead, D.R. Improved prediction of protein-protein binding sites using a support vector machines approach. Bioinformatics 2005, 21, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.R.; Needham, C.J.; Bulpitt, A.J.; Westhead, D.R. Insights into protein-protein interfaces using a bayesian network prediction method. J. Mol. Biol. 2006, 362, 365–386. [Google Scholar] [CrossRef] [PubMed]
- Porollo, A.; Meller, J. Prediction-based fingerprints of protein-protein interactions. Proteins 2007, 66, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Mizuguchi, K. Applying the naive bayes classifier with kernel density estimation to the prediction of protein-protein interaction sites. Bioinformatics 2010, 26, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Dhole, K.; Singh, G.; Pai, P.P.; Mondal, S. Sequence-based prediction of protein-protein interaction sites with l1-logreg classifier. J. Theor. Biol. 2014, 348, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Dhole, K.; Pai, P.P.; Mondal, S. Springs: Prediction of protein-protein interaction sites using artificial neural networks. Peer. J. PrePrints. 2014, 2, e266v2. [Google Scholar]
- Du, T.; Liao, L.; Wu, C.H.; Sun, B. Prediction of residue-residue contact matrix for protein-protein interaction with fisher score features and deep learning. Methods 2016, 110, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.L.; Zhou, B.; Lai, L.H.; Pei, J.F. Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinform. 2017, 18, 277. [Google Scholar] [CrossRef] [PubMed]
- Du, X.Q.; Sun, S.W.; Hu, C.L.; Yao, Y.; Yan, Y.T.; Zhang, Y.P. Deepppi: Boosting prediction of protein-protein interactions with deep neural networks. J. Chem. Inf. Model. 2017, 57, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.R.; Gough, D.A. Predicting protein--protein interactions from primary structure. Bioinformatics 2001, 17, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.W.; Liu, M. Prediction of protein-protein interactions using random decision forest framework. Bioinformatics 2005, 21, 4394–4400. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, H.X. Prediction of interface residues in protein-protein complexes by a consensus neural network method: Test against nmr data. Proteins 2005, 61, 21–35. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Bonvin, A.M. Cport: A consensus interface predictor and its performance in prediction-driven docking with haddock. PLoS ONE 2011, 6, e17695. [Google Scholar] [CrossRef] [PubMed]
- Dohkan, S.; Koike, A.; Takagi, T. Improving the performance of an svm-based method for predicting protein-protein interactions. In Silico Biol. 2006, 6, 515–529. [Google Scholar] [PubMed]
- Negi, S.S.; Schein, C.H.; Oezguen, N.; Power, T.D.; Braun, W. Interprosurf: A web server for predicting interacting sites on protein surfaces. Bioinformatics 2007, 23, 3397–3399. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Zhou, H.X. Meta-ppisp: A meta web server for protein-protein interaction site prediction. Bioinformatics 2007, 23, 3386–3387. [Google Scholar] [CrossRef] [PubMed]
- Minhas, F.; Geiss, B.J.; Ben-Hur, A. Pairpred: Partner-specific prediction of interacting residues from sequence and structure. Proteins 2014, 82, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Budagyan, L.; Raush, E.; Totrov, M.; Abagyan, R. Pier: Protein interface recognition for structural proteomics. Proteins 2007, 67, 400–417. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.D.; Zhang, C.; Liu, S.; Zhou, Y.Q. Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 2006, 34, 3698–3707. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Mizuguchi, K. Partner-aware prediction of interacting residues in protein-protein complexes from sequence data. PLoS ONE 2011, 6, e29104. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Basu, S.; Kundu, M.; Nasipuri, M.; Plewczynski, D. Ppi_svm: Prediction of protein-protein interactions using machine learning, domain-domain affinities and frequency tables. Cell Mol. Biol. Lett. 2011, 16, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Z.; Yu, L.Z.; Wen, Z.N.; Li, M.L. Using support vector machine combined with auto covariance to predict proteinprotein interactions from protein sequences. Nucleic Acids Res 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.H.; Li, K.B. Predicting protein-protein interaction sites using sequence descriptors and site propensity of neighboring amino acids. Int. J. Mol. Sci. 2016, 17, 1788. [Google Scholar] [CrossRef] [PubMed]
- Zellner, H.; Staudigel, M.; Trenner, T.; Bittkowski, M.; Wolowski, V.; Icking, C.; Merkl, R. Prescont: Predicting protein-protein interfaces utilizing four residue properties. Proteins 2012, 80, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Deng, L.; Fisher, M.; Guan, J.H.; Honig, B.; Petrey, D. Predus: A web server for predicting protein interfaces using structural neighbors. Nucleic Acids Res. 2011, 39, W283–W287. [Google Scholar] [CrossRef] [PubMed]
- Baspinar, A.; Cukuroglu, E.; Nussinov, R.; Keskin, O.; Gursoy, A. Prism: A web server and repository for prediction of protein-protein interactions and modeling their 3d complexes. Nucleic Acids Res. 2014, 42, W285–W289. [Google Scholar] [CrossRef] [PubMed]
- Neuvirth, H.; Raz, R.; Schreiber, G. Promate: A structure based prediction program to identify the location of protein-protein binding sites. J. Mol. Biol. 2004, 338, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Ramasamy, S.; Raghava, G.P.S. A simple approach for predicting protein-protein interactions. Curr. Protein Pept. Sci. 2010, 11, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Jones, S. Sharp2: Protein-protein interaction predictions using patch analysis. Bioinformatics 2006, 22, 1794–1795. [Google Scholar] [CrossRef] [PubMed]
- Valente, G.T.; Acencio, M.L.; Martins, C.; Lemke, N. The development of a universal in silico predictor of protein-protein interactions. PLoS ONE 2013, 8, e65587. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; van Dijk, A.D.J.; Bonvin, A.M.J.J. Whiscy: What information does surface conservation yield? Application to data-driven docking. Proteins 2006, 63, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Dobbs, D.; Honavar, V. A two-stage classifier for identification of protein-protein interface residues. Bioinformatics 2004, 20 (Suppl. 1), i371–i378. [Google Scholar] [CrossRef]
- Basse, M.J.; Betzi, S.; Morelli, X.; Roche, P. 2p2idb v2: Update of a structural database dedicated to orthosteric modulation of protein-protein interactions. Database 2016. [Google Scholar] [CrossRef] [PubMed]
- Higueruelo, A.P.; Jubb, H.; Blundell, T.L. Timbal v2: Update of a database holding small molecules modulating protein-protein interactions. Database 2013. [Google Scholar] [CrossRef] [PubMed]
- Higueruelo, A.P.; Schreyer, A.; Bickerton, G.R.; Pitt, W.R.; Groom, C.R.; Blundell, T.L. Atomic interactions and profile of small molecules disrupting protein-protein interfaces: The timbal database. Chem. Biol. Drug Des. 2009, 74, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Labbe, C.M.; Kuenemann, M.A.; Zarzycka, B.; Vriend, G.; Nicolaes, G.A.; Lagorce, D.; Miteva, M.A.; Villoutreix, B.O.; Sperandio, O. Ippi-db: An online database of modulators of protein-protein interactions. Nucleic Acids Res. 2016, 44, D542–D547. [Google Scholar] [CrossRef] [PubMed]
- Reynes, C.; Host, H.; Camproux, A.C.; Laconde, G.; Leroux, F.; Mazars, A.; Deprez, B.; Fahraeus, R.; Villoutreix, B.O.; Sperandio, O. Designing focused chemical libraries enriched in protein-protein interaction inhibitors using machine-learning methods. PLoS Comput. Biol. 2010, 6, e1000695. [Google Scholar] [CrossRef] [PubMed]
- Miteva, M.A.; Violas, S.; Montes, M.; Gomez, D.; Tuffery, P.; Villoutreix, B.O. Faf-drugs: Free adme/tox filtering of compound collections. Nucleic Acids Res. 2006, 34, W738–W744. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Galons, H.; Miteva, M.A.; Villoutreix, B.O. Faf-drugs2: Free adme/tox filtering tool to assist drug discovery and chemical biology projects. BMC Bioinform. 2008, 9, 396. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Maupetit, J.; Baell, J.; Sperandio, O.; Tuffery, P.; Miteva, M.A.; Galons, H.; Villoutreix, B.O. The faf-drugs2 server: A multistep engine to prepare electronic chemical compound collections. Bioinformatics 2011, 27, 2018–2020. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. Faf-drugs3: A web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, W200–W207. [Google Scholar] [CrossRef] [PubMed]
- Hamon, V.; Bourgeas, R.; Ducrot, P.; Theret, I.; Xuereb, L.; Basse, M.J.; Brunel, J.M.; Combes, S.; Morelli, X.; Roche, P. 2p2i hunter: A tool for filtering orthosteric protein-protein interaction modulators via a dedicated support vector machine. J. R. Soc. Interface 2014, 11, 20130860. [Google Scholar] [CrossRef] [PubMed]
- Bordner, A.J.; Abagyan, R. Statistical analysis and prediction of protein-protein interfaces. Proteins 2005, 60, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Acuner Ozbabacan, S.E.; Engin, H.B.; Gursoy, A.; Keskin, O. Transient protein–protein interactions. Protein Eng. Des. Sel. 2011, 24, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Gursoy, A.; Keskin, O. Prediction of protein-protein interactions: Unifying evolution and structure at protein interfaces. Phys. Biol. 2011, 8, 035006. [Google Scholar] [CrossRef] [PubMed]
- Acuner Ozbabacan, S.E.; Gursoy, A.; Keskin, O.; Nussinov, R. Conformational ensembles, signal transduction and residue hot spots: Application to drug discovery. Curr. Opin. Drug Discov. Dev. 2010, 13, 527–537. [Google Scholar]
- Bradshaw, R.T.; Patel, B.H.; Tate, E.W.; Leatherbarrow, R.J.; Gould, I.R. Comparing experimental and computational alanine scanning techniques for probing a prototypical protein-protein interaction. Protein Eng. Des. Sel. 2011, 24, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Zhang, S.X. Computational prediction of protein hot spot residues. Curr. Pharm. Des. 2012, 18, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Computational alanine scanning to probe protein-protein interactions: A novel approach to evaluate binding free energies. J. Am. Chem. Soc. 1999, 121, 8133–8143. [Google Scholar] [CrossRef]
- Kruger, D.M.; Gohlke, H. Drugscore(ppi) webserver: Fast and accurate in silico alanine scanning for scoring protein-protein interactions. Nucleic Acids Res. 2010, 38, W480–W486. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.Q.; Yan, Y.N.; Sun, Z.X.; Song, J.N.; Zhang, J.Z.H. Interaction entropy for computational alanine scanning in protein-protein binding. WIREs Comput. Mol. Sci. 2018, 8, e1342. [Google Scholar] [CrossRef]
- Fernandez-Recio, J.; Totrov, M.; Skorodumov, C.; Abagyan, R. Optimal docking area: A new method for predicting protein-protein interaction sites. Proteins 2005, 58, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, S.; Fernandez-Recio, J. Identification of hot-spot residues in protein-protein interactions by computational docking. BMC Bioinform. 2008, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.M.; Blundell, T.L.; Fernandez-Recio, J. Pydock: Electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins 2007, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Vendrell, J.; Aviles, F.X.; Fernandez-Recio, J. Structural and functional characterization of binding sites in metallocarboxypeptidases based on optimal docking area analysis. Proteins 2007, 68, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Fratev, F.; Jonsdottir, S.O.; Pajeva, I. Structural insight into the unc-45-myosin complex. Proteins 2013, 81, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Aumentado-Armstrong, T.T.; Istrate, B.; Murgita, R.A. Algorithmic approaches to protein-protein interaction site prediction. Algorithms Mol. Biol. 2015, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Yang, J.S.; Choi, Y.; Ryu, S.H.; Kim, S. Evolutionary conservation in multiple faces of protein interaction. Proteins 2009, 77, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Weigt, M.; White, R.A.; Szurmant, H.; Hoch, J.A.; Hwa, T. Identification of direct residue contacts in protein-protein interaction by message passing. Proc. Natl. Acad. Sci. USA 2009, 106, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Ingles-Prieto, A.; Ibarra-Molero, B.; Delgado-Delgado, A.; Perez-Jimenez, R.; Fernandez, J.M.; Gaucher, E.A.; Sanchez-Ruiz, J.M.; Gavira, J.A. Conservation of protein structure over four billion years. Structure 2013, 21, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Norel, R.; Honig, B.H. Protein interface conservation across structure space. Proc. Natl. Acad. Sci. USA 2010, 107, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Q.; Feng, K.Y.; Chen, L.; Huang, T.; Cai, Y.D. Prediction of protein-protein interaction sites by random forest algorithm with mrmr and ifs. PLoS ONE 2012, 7, e43927. [Google Scholar] [CrossRef] [PubMed]
- Higa, R.H.; Tozzi, C.L. Prediction of binding hot spot residues by using structural and evolutionary parameters. Genet. Mol. Biol. 2009, 32, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Meireles, L.M.C.; Domling, A.S.; Camacho, C.J. Anchor: A web server and database for analysis of protein-protein interaction binding pockets for drug discovery. Nucleic Acids Res. 2010, 38, W407–W411. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Dobbs, D.; Honavar, V. Homppi: A class of sequence homology based protein-protein interface prediction methods. BMC Bioinform. 2011, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Darnell, S.J.; LeGault, L.; Mitchell, J.C. Kfc server: Interactive forecasting of protein interaction hot spots. Nucleic Acids Res. 2008, 36, W265–W269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Mitchell, J.C. Kfc2: A knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins 2011, 79, 2671–2683. [Google Scholar] [CrossRef] [PubMed]
- Darnell, S.J.; Page, D.; Mitchell, J.C. An automated decision-tree approach to predicting protein interaction hot spots. Proteins 2007, 68, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Keskin, O.; Gursoy, A. Hotpoint: Hot spot prediction server for protein interfaces. Nucleic Acids Res. 2010, 38, W402–W406. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The ftmap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.I.; Kim, D.; Lee, D. A feature-based approach to modeling protein-protein interaction hot spots. Nucleic Acids Res. 2009, 37, 2672–2687. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhang, Q.C.; Chen, Z.G.; Meng, Y.; Guan, J.H.; Zhou, S.G. Predhs: A web server for predicting protein-protein interaction hot spots by using structural neighborhood properties. Nucleic Acids Res. 2014, 42, W290–W295. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.K.; Karanicolas, J. Druggable protein interaction sites are more predisposed to surface pocket formation than the rest of the protein surface. PLoS Comput. Biol. 2013, 9, e1002951. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.P.; Hajduk, P.J. Effects of conformational dynamics on predicted protein druggability. ChemMedChem 2006, 1, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, K.; Rual, J.F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.; Goh, K.I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Rakers, C.; Bermudez, M.; Keller, B.G.; Mortier, J.; Wolber, G. Computational close up on protein-protein interactions: How to unravel the invisible using molecular dynamics simulations? WIREs Comput. Mol. Sci. 2015, 5, 345–359. [Google Scholar] [CrossRef]
- Vakser, I.A. Protein-protein docking: From interaction to interactome. Biophys. J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Janin, J. Protein-protein docking tested in blind predictions: The capri experiment. Mol. Biosyst. 2010, 6, 2351–2362. [Google Scholar] [CrossRef] [PubMed]
- Lesk, V.I.; Sternberg, M.J. 3d-garden: A system for modelling protein-protein complexes based on conformational refinement of ensembles generated with the marching cubes algorithm. Bioinformatics 2008, 24, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Schindler, C.E.; Chauvot de Beauchene, I.; Zacharias, M. A web interface for easy flexible protein-protein docking with attract. Biophys. J. 2015, 108, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guo, D.; Huang, Y.; Liu, S.; Xiao, Y. Aspdock: Protein-protein docking algorithm using atomic solvation parameters model. BMC Bioinform. 2011, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Palma, P.N.; Krippahl, L.; Wampler, J.E.; Moura, J.J. Bigger: A new (soft) docking algorithm for predicting protein interactions. Proteins 2000, 39, 372–384. [Google Scholar] [CrossRef]
- Pons, C.; Jimenez-Gonzalez, D.; Gonzalez-Alvarez, C.; Servat, H.; Cabrera-Benitez, D.; Aguilar, X.; Fernandez-Recio, J. Cell-dock: High-performance protein-protein docking. Bioinformatics 2012, 28, 2394–2396. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The cluspro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, S.; Ravikant, D.V.; Elber, R. Dock/pierr: Web server for structure prediction of protein-protein complexes. Methods Mol. Biol. 2014, 1137, 199–207. [Google Scholar] [PubMed]
- Roberts, V.A.; Thompson, E.E.; Pique, M.E.; Perez, M.S.; Ten Eyck, L.F. Dot2: Macromolecular docking with improved biophysical models. J. Comput. Chem. 2013, 34, 1743–1758. [Google Scholar] [CrossRef] [PubMed]
- Ausiello, G.; Cesareni, G.; Helmer-Citterich, M. Escher: A new docking procedure applied to the reconstruction of protein tertiary structure. Proteins 1997, 28, 556–567. [Google Scholar] [CrossRef]
- Bajaj, C.; Chowdhury, R.; Siddavanahalli, V. F2dock: Fast fourier protein-protein docking. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Nussinov, R.; Wolfson, H.J. Fiberdock: A web server for flexible induced-fit backbone refinement in molecular docking. Nucleic Acids Res. 2010, 38, W457–W461. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. Firedock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Aportela, E.; Lopez-Blanco, J.R.; Chacon, P. Frodock 2.0: Fast protein-protein docking server. Bioinformatics 2016, 32, 2386–2388. [Google Scholar] [CrossRef] [PubMed]
- Gabb, H.A.; Jackson, R.M.; Sternberg, M.J. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J. Mol. Biol. 1997, 272, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.J.; Willett, P.; Artymiuk, P.J. Protein docking using a genetic algorithm. Proteins 2001, 44, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Tovchigrechko, A.; Vakser, I.A. Gramm-x public web server for protein-protein docking. Nucleic. Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. Haddock: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. Hdock: A web server for protein-protein and protein-DNA/rna docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W.; Venkatraman, V. Ultra-fast fft protein docking on graphics processors. Bioinformatics 2010, 26, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Recio, J.; Totrov, M.; Abagyan, R. Icm-disco docking by global energy optimization with fully flexible side-chains. Proteins 2003, 52, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vavrusa, M.; Andreani, J.; Rey, J.; Tuffery, P.; Guerois, R. Interevdock: A docking server to predict the structure of protein-protein interactions using evolutionary information. Nucleic Acids Res. 2016, 44, W542–W549. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Garcia, B.; Roel-Touris, J.; Romero-Durana, M.; Vidal, M.; Jimenez-Gonzalez, D.; Fernandez-Recio, J. Lightdock: A new multi-scale approach to protein-protein docking. Bioinformatics 2018, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Kihara, D. Protein docking prediction using predicted protein-protein interface. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Ohue, M.; Shimoda, T.; Suzuki, S.; Matsuzaki, Y.; Ishida, T.; Akiyama, Y. Megadock 4.0: An ultra-high-performance protein-protein docking software for heterogeneous supercomputers. Bioinformatics 2014, 30, 3281–3283. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zeev, E.; Kowalsman, N.; Ben-Shimon, A.; Segal, D.; Atarot, T.; Noivirt, O.; Shay, T.; Eisenstein, M. Docking to single-domain and multiple-domain proteins: Old and new challenges. Proteins 2005, 60, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Patchdock and symmdock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Neveu, E.; Ritchie, D.W.; Popov, P.; Grudinin, S. Pepsi-dock: A detailed data-driven protein-protein interaction potential accelerated by polar fourier correlation. Bioinformatics 2016, 32, i693–i701. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. Piper: An fft-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Mitra, P.; Pal, D. Prune and probe-two modular web services for protein-protein docking. Nucleic Acids Res. 2011, 39, W229–W234. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Garcia, B.; Pons, C.; Fernandez-Recio, J. Pydockweb: A web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics 2013, 29, 1698–1699. [Google Scholar] [CrossRef] [PubMed]
- Lyskov, S.; Gray, J.J. The rosettadock server for local protein-protein docking. Nucleic Acids Res 2008, 36, W233–W238. [Google Scholar] [CrossRef] [PubMed]
- Terashi, G.; Takeda-Shitaka, M.; Kanou, K.; Iwadate, M.; Takaya, D.; Umeyama, H. The ske-dock server and human teams based on a combined method of shape complementarity and free energy estimation. Proteins 2007, 69, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.J.; Vajda, S. Protein docking along smooth association pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 10636–10641. [Google Scholar] [CrossRef] [PubMed]
- Torchala, M.; Moal, I.H.; Chaleil, R.A.; Fernandez-Recio, J.; Bates, P.A. Swarmdock: A server for flexible protein-protein docking. Bioinformatics 2013, 29, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Levieux, G.; Tiger, G.; Mader, S.; Zagury, J.F.; Natkin, S.; Montes, M. Udock, the interactive docking entertainment system. Faraday Discuss. 2014, 169, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. Zdock server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shimon, A.; Niv, M.Y. Anchordock: Blind and flexible anchor-driven peptide docking. Structure 2015, 23, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. Cabs-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Moll, M.; Devaurs, D.; Jackson, K.R.; Lizee, G.; Kavraki, L.E. Dinc 2.0: A new protein-peptide docking webserver using an incremental approach. Cancer Res. 2017, 77, e55–e57. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta flexpepdock web server--high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011, 39, W249–W253. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. Galaxypepdock: A protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43, W431–W435. [Google Scholar] [CrossRef] [PubMed]
- Trellet, M.; Melquiond, A.S.; Bonvin, A.M. A unified conformational selection and induced fit approach to protein-peptide docking. PLoS ONE 2013, 8, e58769. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. Hpepdock: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.F.; Xu, X.J.; Zou, X.Q. Fully blind docking at the atomic level for protein-peptide complex structure prediction. Structure 2016, 24, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Rey, J.; Schindler, C.E.M.; Zacharias, M.; Tuffery, P. The pepattract web server for blind, large-scale peptide-protein docking. Nucleic Acids Res. 2017, 45, W361–W364. [Google Scholar] [CrossRef] [PubMed]
- Donsky, E.; Wolfson, H.J. Pepcrawler: A fast rrt-based algorithm for high-resolution refinement and binding affinity estimation of peptide inhibitors. Bioinformatics 2011, 27, 2836–2842. [Google Scholar] [CrossRef] [PubMed]
- Trabuco, L.G.; Lise, S.; Petsalaki, E.; Russell, R.B. Pepsite: Prediction of peptide-binding sites from protein surfaces. Nucleic Acids Res. 2012, 40, W423–W427. [Google Scholar] [CrossRef] [PubMed]
- Saladin, A.; Rey, J.; Thevenet, P.; Zacharias, M.; Moroy, G.; Tuffery, P. Pep-sitefinder: A tool for the blind identification of peptide binding sites on protein surfaces. Nucleic Acids Res. 2014, 42, W221–W226. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Manavalan, B.; Govindaraj, R.G.; Choi, S. In silico approach to inhibition of signaling pathways of toll-like receptors 2 and 4 by st2l. PLoS ONE 2011, 6, e23989. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Choi, Y.M.; Lee, G.; Choi, S. Structure-function relationship of cytoplasmic and nuclear ikappab proteins: An in silico analysis. PLoS ONE 2010, 5, e15782. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Govindaraj, R.; Lee, G.; Choi, S. Molecular modeling-based evaluation of dual function of ikappabzeta ankyrin repeat domain in toll-like receptor signaling. J. Mol. Recognit. 2011, 24, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Galeazzi, R.; Laudadio, E.; Falconi, E.; Massaccesi, L.; Ercolani, L.; Mobbili, G.; Minnelli, C.; Scire, A.; Cianfruglia, L.; Armeni, T. Protein-protein interactions of human glyoxalase ii: Findings of a reliable docking protocol. Org. Biomol. Chem. 2018, 16, 5167–5177. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Schuster, D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Guner, O.; Clement, O.; Kurogi, Y. Pharmacophore modeling and three dimensional database searching for drug design using catalyst: Recent advances. Curr. Med. Chem. 2004, 11, 2991–3005. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for ligands and proteins (flap): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Barillari, C.; Marcou, G.; Rognan, D. Hot-spots-guided receptor-based pharmacophores (hs-pharm): A knowledge-based approach to identify ligand-anchoring atoms in protein cavities and prioritize structure-based pharmacophores. J. Chem. Inf. Model. 2008, 48, 1396–1410. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. Ligandscout: 3-d pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. Phase: A novel approach to pharmacophore modeling and 3d database searching. Chem. Biol. Drug. Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Domling, A.; Camacho, C.J. Anchorquery: Rapid online virtual screening for small-molecule protein-protein interaction inhibitors. Protein Sci. 2018, 27, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.H.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.M.; Mortenson, P.N.; Murray, C.W. Diverse, high-quality test set for the validation of protein-ligand docking performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. Icm—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Betzi, S.; Restouin, A.; Opi, S.; Arold, S.T.; Parrot, I.; Guerlesquin, F.; Morelli, X.; Collette, Y. Protein protein interaction inhibition (2p2i) combining high throughput and virtual screening: Application to the hiv-1 nef protein. Proc. Natl. Acad. Sci. USA 2007, 104, 19256–19261. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.B.; Chen, J.F.; Meagher, J.L.; Yang, C.Y.; Aguilar, A.; Liu, L.; Bai, L.C.; Gong, X.; Cai, Q.; Fang, X.L.; et al. Design of bcl-2 and bcl-xl inhibitors with subnanomolar binding affinities based upon a new scaffold. J. Med. Chem. 2012, 55, 4664–4682. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Bower, K.E.; Beuscher, A.E., 4th; Zhou, B.; Bobkov, A.A.; Olson, A.J.; Vogt, P.K. Stabilizers of the max homodimer identified in virtual ligand screening inhibit myc function. Mol. Pharmacol. 2009, 76, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Schuffenhauer, A.; Ruedisser, S.; Marzinzik, A.L.; Jahnke, W.; Blommers, M.; Selzer, P.; Jacoby, E. Library design for fragment based screening. Curr. Top. Med. Chem. 2005, 5, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.L.; Schneider, G. Scaffold architecture and pharmacophoric properties of natural products and trade drugs: Application in the design of natural product-based combinatorial libraries. J. Comb. Chem. 2001, 3, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Milroy, L.G.; Grossmann, T.N.; Hennig, S.; Brunsveld, L.; Ottmann, C. Modulators of protein-protein interactions. Chem. Rev. 2014, 114, 4695–4748. [Google Scholar] [CrossRef] [PubMed]
- Over, B.; Wetzel, S.; Grutter, C.; Nakai, Y.; Renner, S.; Rauh, D.; Waldmann, H. Natural-product-derived fragments for fragment-based ligand discovery. Nat. Chem. 2013, 5, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Camacho, C.J. Pocketquery: Protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res. 2012, 40, W387–392. [Google Scholar] [CrossRef] [PubMed]
- Geppert, T.; Bauer, S.; Hiss, J.A.; Conrad, E.; Reutlinger, M.; Schneider, P.; Weisel, M.; Pfeiffer, B.; Altmann, K.H.; Waibler, Z.; et al. Immunosuppressive small molecule discovered by structure-based virtual screening for inhibitors of protein-protein interactions. Angew. Chem. Int. Ed. 2012, 51, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Weisel, M.; Proschak, E.; Schneider, G. Pocketpicker: Analysis of ligand binding-sites with shape descriptors. Chem. Cent. J. 2007, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Geppert, T.; Hoy, B.; Wessler, S.; Schneider, G. Context-based identification of protein-protein interfaces and “hot-spot” residues. Chem. Biol. 2011, 18, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Basith, S.; Choi, S. Recent advances in structure-based drug design targeting class a g protein-coupled receptors utilizing crystal structures and computational simulations. J. Med. Chem. 2018, 61, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Verkhivker, G.M. Probing molecular mechanisms of the hsp90 chaperone: Biophysical modeling identifies key regulators of functional dynamics. PLoS ONE 2012, 7, e37605. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, E.S.; Jang, H.; Gursoy, A.; Keskin, O.; Li, Z.; Sacks, D.B.; Nussinov, R. Unraveling the molecular mechanism of interactions of the rho gtpases cdc42 and rac1 with the scaffolding protein iqgap2. J. Biol. Chem. 2018, 293, 3685–3699. [Google Scholar] [CrossRef] [PubMed]
- Sarvagalla, S.; Cheung, C.H.A.; Tsai, J.Y.; Hsieh, H.P.; Coumar, M.S. Disruption of protein-protein interactions: Hot spot detection, structure-based virtual screening and in vitro testing for the anti-cancer drug target survivin. RSC Adv. 2016, 6, 31947–31959. [Google Scholar] [CrossRef]
- Bastianelli, G.; Bouillon, A.; Nguyen, C.; Crublet, E.; Petres, S.; Gorgette, O.; Le-Nguyen, D.; Barale, J.C.; Nilges, M. Computational reverse-engineering of a spider-venom derived peptide active against plasmodium falciparum sub1. PLoS ONE 2011, 6, e21812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, H.; Bai, H.; Lai, L. Molecular dynamics simulations on the oligomer-formation process of the gnnqqny peptide from yeast prion protein sup35. Biophys. J. 2007, 93, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Baram, M.; Atsmon-Raz, Y.; Ma, B.; Nussinov, R.; Miller, Y. Amylin-abeta oligomers at atomic resolution using molecular dynamics simulations: A link between type 2 diabetes and alzheimer’s disease. Phys. Chem. Chem. Phys. 2016, 18, 2330–2338. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provaznik, I.; Choi, I. Computer aided drug design: Success and limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, S.G.; Lane, D.P.; Verma, C. Multiple peptide conformations give rise to similar binding affinities: Molecular simulations of p53-mdm2. J. Am. Chem. Soc. 2008, 130, 13514–13515. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Kay, L.E.; Forman-Kay, J.D. Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 2010, 23, 105–116. [Google Scholar] [CrossRef] [PubMed]




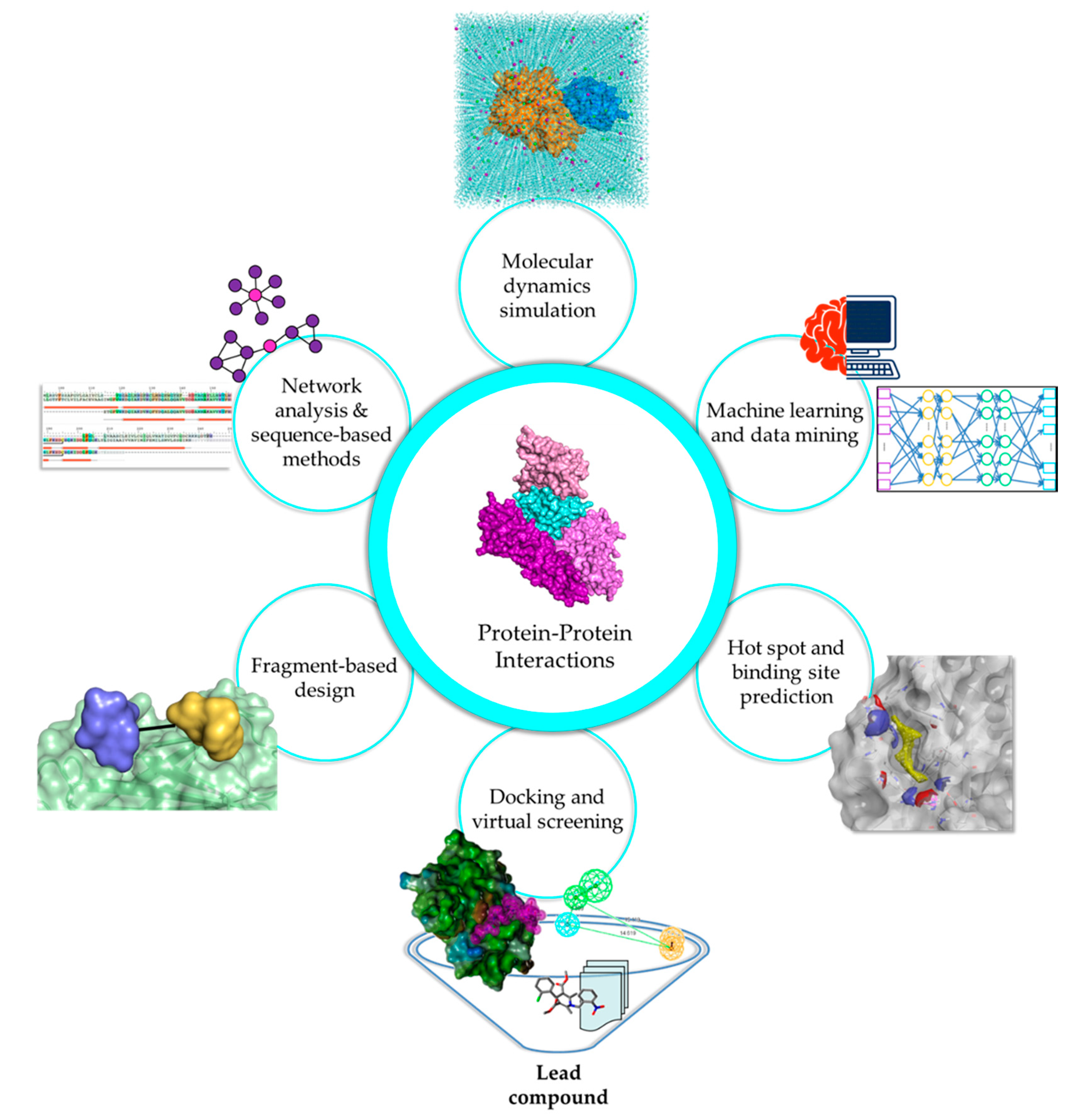{kind=link}
{kind=link}
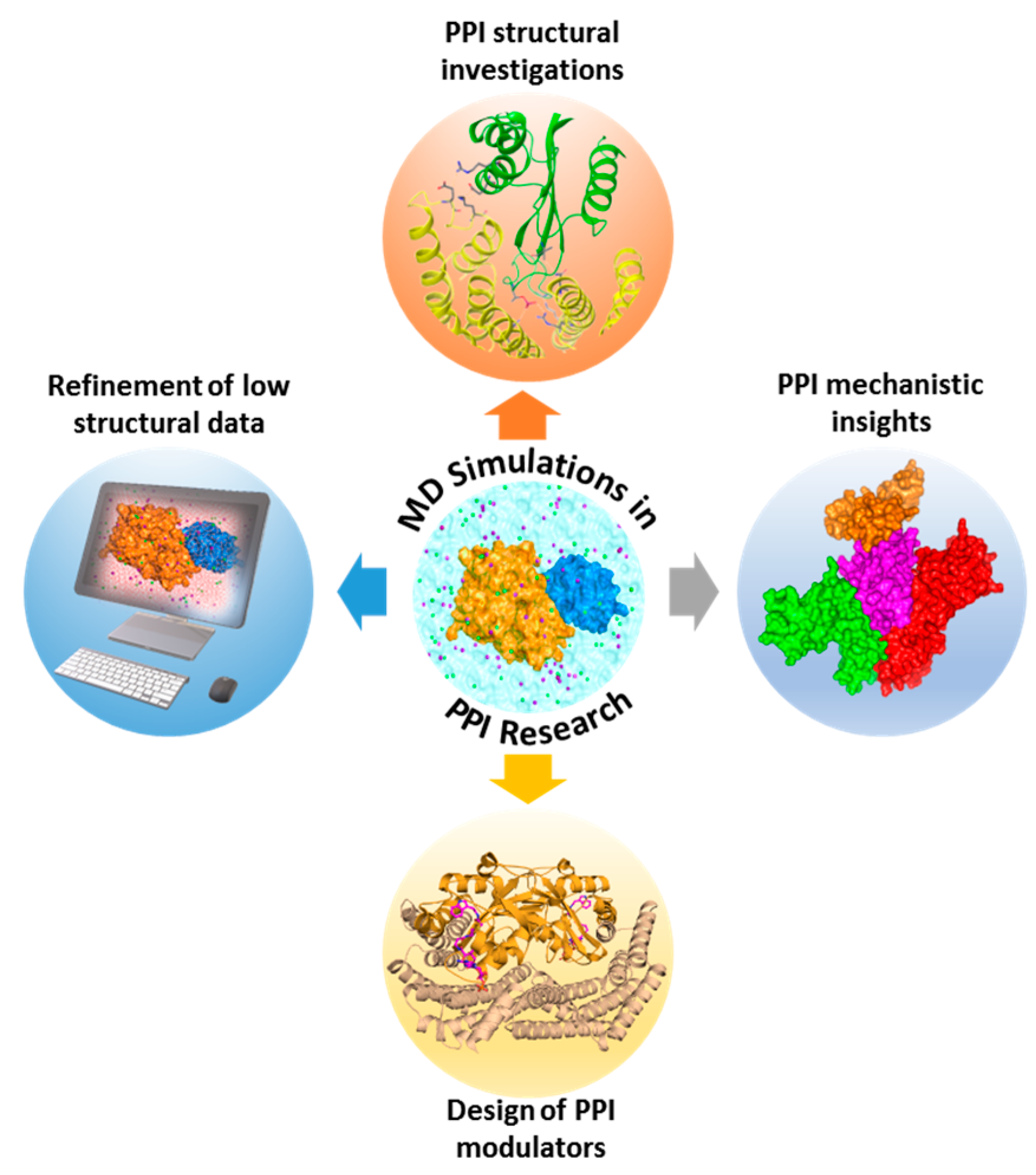{kind=link}
| Compound | Structure | Mode of Action | Identification Method | Clinical Status | Ref. |
|---|---|---|---|---|---|
| ABT-199 (Venetoclax) |  | Bcl-2/(BAX/BAK) inhibitor | Rational design for BCL-2 | Approved for chronic lymphocytic leukemia (CLL) with 17p deletion | [31] |
| ABT-263 (Navitoclax) |  | Bcl-2/(BAX/BAK) inhibitor | High-throughput screening (HTS) and fragment-based design | Phase 1/2 for various cancer types | [32,33,34,35] |
| AMG232 |  | p53/MDM2 inhibitor | Fragment-based design | Phase 1 for cancer | [36] |
| Birabresib (OTX015) |  | Bromodomain and extra-terminal (BET)/histone peptide inhibitor | Cell assays | Phase 1 for various cancer types | [37,38] |
| Birinapant (TL32711) |  | CIAP1-BIR3/Caspase-9 and XIAP-BIR3/second mitochondrial activator of caspases (SMAC) inhibitor | Dimerized SMAC mimetics | Phase 1/2 for various cancer types | [39,40,41,42] |
| Cabazitaxel |  | Microtubule inhibitor | Screening of semisynthetic taxane derivatives | Approved for prostate cancer | [43] |
| CGM097 |  | p53/MDM2 inhibitor | Virtual screening (VS), molecular modeling, and rational design based on crystal complex structure | Phase 1 for cancer | [44] |
| Cilengitide |  | Integrin αvβ3/αvβ5 inhibitor | Ligand-based design using Arg-Gly-Asp (RGD)-binding motif | Phase 1/2/3 for various cancer types | [45,46,47,48] |
| CPI-0610 |  | BET/histone peptide inhibitor | Structure-based drug design | Phase 1/2 for various cancer types | [49,50,51] |
| Docetaxel |  | Microtubule inhibitor | Semisynthetic taxane derivative | Approved for various cancer types | [52,53,54,55] |
| DS-3032b |  | p53/MDM2 inhibitor | Enzyme and cell assays | Phase 1 for leukemia | [56] |
| Eptifibatide |  | Glycoprotein IIb/IIIa inhibitor | Peptide-based (barbourin) design | Approved as platelet aggregation inhibitor | [57,58] |
| FK506 (Tacrolimus) |  | FK506-binding protein 12 (FKBP12)/Calcineurin inhibitor | In vitro and in vivo assays | Approved for immunosuppression/organ rejection | [59] |
| I-BET762 (Molibresib) |  | BET/histone peptide inhibitor | Cell-based HTS | Phase 2 for cancer | [60] |
| LCL161 |  | Inhibitor of apoptosis (IAP)/SMAC inhibitor | SMAC mimetics, cell assays | Phase 1/2 for various cancer types | [61,62,63,64]. |
| Lifitegrast (SAR1118) |  | Lymphocyte function-associated antigen-1 (LFA-1)/Intercellular adhesion molecule 1 ICAM-1 inhibitor | Structure-based rational design based on LFA-1 and ICAM-1 binding | Approved for dry eye | [65] |
| MI-77301 (SAR405838) |  | p53/MDM2 inhibitor | Structure-based rational design based on p53 peptide | Phase 1 for cancer | [66,67] |
| Maraviroc |  | CCR5/gp120 inhibitor | HTS | Approved for human immunodeficiency virus (HIV) | [68,69] |
| Onalespib (AT13387) |  | HSP90 inhibitor | Fragment-based design | Phase 1 for cancer | [70] |
| RG7112 |  | p53/MDM2 inhibitor | HTS and rational optimization of Nutlins | Phase 1 for cancer | [71] |
| RG7388 (Idasanutlin) |  | p53/MDM2 inhibitor | Rational optimization of RG7112, biochemical and cell assays | Phase 3 for acute myeloid leukemia, phase 1/2 for other various cancer types | [72] |
| Tirofiban |  | Glycoprotein IIb/IIIa inhibitor | Ligand-based design using RGD-binding motif | Approved as platelet aggregation inhibitor | [57,73] |
| Database | Website | Description | Number of Proteins | Number of Interactions | Ref. |
|---|---|---|---|---|---|
| BIND | http://download.baderlab.org/BINDTranslation/ | Biomolecular Interaction Network Database (Last update: 2004) | - | 198,905 | [132] |
| BioGRID | https://thebiogrid.org/ | Biological General Repository for Interaction Datasets (Last update: 2018) | - | 1,202,227 | [133,134] |
| DIP | http://dip.doe-mbi.ucla.edu/dip/ | Database of Interacting Proteins (Last update: 2017) | 28,823 | 81,762 | [135] |
| HPRD | http://www.hprd.org/ | Human Protein Reference Database (Last update: 2009) | 30,047 | 41,327 | [136] |
| IntAct | https://www.ebi.ac.uk/intact/ | IntAct Molecular Interaction Database (Last update: 2013) | 105,180 | 805,177 | [137,138,139] |
| MINT | https://mint.bio.uniroma2.it/ | Molecular INTeraction database (Last update: 2013) | 25,178 | 123,940 | [140] |
| MIPS | http://mips.helmholtz-muenchen.de/proj/ppi/ | Mammalian Protein-Protein Database (Last update: 2005) | 900 | 1800 | [141] |
| CORUM | http://mips.helmholtz-muenchen.de/corum/ | Comprehensive resource of mammalian protein complexes | - | 2837 | [142,143] |
| DroID | http://www.droidb.org/Index.jsp | Drosophila Interactions Database (Last update: 2017) | - | 262,631 | [144,145] |
| APID * | http://cicblade.dep.usal.es:8080/APID/init.action | Agile Protein Interactomes Dataserver | 90,379 | 678,441 | [146] |
| HIV Interaction DB * | https://www.ncbi.nlm.nih.gov/genome/viruses/retroviruses/hiv-1/interactions/ | Interactions between HIV-1 proteins with host cell proteins, other HIV-1 proteins, or proteins from HIV-associated disease organisms | - | 2589 | [147,148,149,150] |
| HPID * | http://wilab.inha.ac.kr/hpid/ | Human Protein Interaction Database | 4642 | 719,349 | [151] |
| HPIDB * | http://hpidb.igbb.msstate.edu/index.html | Database for host-pathogen interactions (Last update: 2016) | - | 45,238 | [152,153] |
| IRefWeb * | http://wodaklab.org/iRefWeb/ | Consolidated protein interaction database with provenance | 66,701 | 1,119,604 (distinct: 263,479) | [154,155] |
| MatrixDB * | http://matrixdb.univ-lyon1.fr/ | Extracellular Matrix Interaction Database | - | 9262 | [156,157,158] |
| Mentha * | http://mentha. uniroma2.it/ | Molecular interaction database (Last update: 2018) | 89,666 | 707,003 | [159] |
| PDZBase * | http://abc.med. cornell.edu/pdzbase | Database of PPIs which involve PDZ domains | - | ~300 | [160] |
| PICKLE * | http://www.pickle.gr/ | Protein InteraCtion KnowLedgebasE | - | 120,882 | [161,162] |
| PINA * | http://omics.bjcancer.org/pina/ | Protein Interaction Network Analysis | - | 365,930 | [163,164] |
| Tool/Server | Input Type | ML Algorithm | Features | Website URL | Ref. |
|---|---|---|---|---|---|
| Bock et al. | Structure | Support vector machine (SVM) | Primary structure and associated data | N/A | [205] |
| Chen et al. | Structure | Decision tree | Domain interaction data | Source code available upon request | [206] |
| Cons-PPISP | Structure | Neural network (NN) | Position-specific scoring matrix (PSSM), solvent accessibilities, and spatial neighbors of each residue | http://pipe.scs.fsu.edu/ppisp.html | [207] |
| CPORT * | Structure-based meta server | Scoring function | Combines six interface prediction methods: WHISCY, PIER, ProMate, cons-PPISP, SPPIDER, and PINUP into a consensus predictor | http://haddock.science.uu.nl/services/CPORT/ | [208] |
| DeepPPI | Sequence | Deep neural network (DNN) | Sequence features | http://ailab.ahu.edu.cn:8087/DeepPPI/index.html | [204] |
| Dohkan et al. | Structure | SVM | Domains and amino acid compositions | N/A | [209] |
| InterProSurf | Structure | Scoring function | Solvent accessible surface area (SASA), propensity of interface residues | http://curie.utmb.edu/prosurf.html | [210] |
| MetaPPI * | Structure | Scoring function | Raw scores from five prediction servers PPI−Pred, PPISP, PINUP, Promate, and SPPIDER | http://projects.biotec.tu-dresden.de/metappi/ | |
| Meta-PPISP * | Structure | Linear regression | Raw scores from three other servers: ProMate, PINUP, cons-PPISP | http://pipe.scs.fsu.edu/meta-ppisp.html | [211] |
| PAIRpred | Sequence or structure | Multiple pairwise kernel SVMs | Structural features: relative accessible surface area (rASA), residue depth, half sphere amino acid composition, protrusion index. Sequence features: PSSM and predicted rASA | Python code available at: http://combi.cs.colostate.edu/supplements/pairpred/ | [212] |
| PIER | Structure | Partial least square (PLS) regression | Solvent accessibility and evolutionary conservation | http://abagyan.ucsd.edu/PIER/ | [213] |
| PINUP | Structure | Empirical energy function | Side-chain energy score, residue interface propensity, and residue conservation score | http://sysbio.unl.edu/services/PINUP/ | [214] |
| PPiPP | Sequence | NN | Binary encoding of 20 amino acids and PSSM | http://mizuguchilab.org/netasa/ppipp/ | [215] |
| PPI_SVM | Structure | SVM | Physical interactions of constituent domains | N/A | [216] |
| Pred-PPI | Sequence | SVM | Conservation, electrostatic potential, hydrophobicity, propensity of interface residues, surface shape, and solvent accessible surface area | http://cic.scu.edu.cn/bioinformatics/predict_ppi/ | [217] |
| predPPIS | Sequence | SVM and Bayesian classifiers | Sequence features | http://bsaltools.ym.edu.tw/predppis/ | [218] |
| PresCont | Structure | SVM | SASA, hydrophobicity, conservation and the local environment of each amino acid on the protein surface | http://bioinf.ur.de/php/prescont.ph | [219] |
| PredUs | Structure | SVM | SASA, hydrophobicity, conservation and the local environment of each amino acid on the protein surface | http://bhapp.c2b2.columbia.edu/PredUs/ | [220] |
| PRISM | Structure | Scoring function | Geometric complementarity, conservation | http://cosbi.ku.edu.tr/prism/index.php | [221] |
| PROFisis | Sequence | NN | Sequence features | http://rostlab.org/owiki/index.php/PROFisis | [190] |
| ProMate | Structure | Composite probability | Multiple features like amino-acid propensities, pairwise amino-acid distribution, residue conservation, geometric properties, etc. | http://bioinfo41.weizmann.ac.il/promate/promate.html | [222] |
| ProPrInt | Sequence | SVM | Sequence features, PSSM | http://crdd.osdd.net/raghava/proprint/ | [223] |
| PSIVER | Sequence | Naïve Bayes classifier | PSSM, predicted solvent accessibility | http://mizuguchilab.org/PSIVER/ | [199] |
| SHARP2 | Structure | Scoring function | Solvation potential, hydrophobicity, accessible surface area, residue interface propensity, planarity and protrusion | N/A | [224] |
| SPPIDER | Sequence | SVM, NN | Fingerprints of protein interactions based on predicted relative solvent accessibility (experimental) | http://sppider.cchmc.org/ | [198] |
| Sun et al. | Sequence | DNN | Sequence features | N/A | [203] |
| UNISPPI | Sequence | Decision tree | Amino acid frequencies | N/A | [225] |
| WHISCY | Structure and multiple sequence alignment (MSA) | Scoring function | Residue conservation, interface propensity of residues | http://milou.science.uu.nl/services/WHISCY/ | [226] |
| Yan et al. | Sequence | SVM, Bayes | Interface residue neighborhoods | N/A | [227] |
| Tool/Server | Docking Algorithm | Website URL | Type | Ref. |
|---|---|---|---|---|
| AnchorDock | Global peptide docking | N/A | Standalone | [314] |
| CABS-dock | Global peptide docking | http://biocomp.chem.uw.edu.pl/CABSdock | Online | [315] |
| DINC | Global peptide docking | http://dinc.kavrakilab.org/ | Online | [316] |
| FlexPepDock | Local peptide docking | http://flexpepdock.furmanlab.cs.huji.ac.il/ | Online | [317] |
| GalaxyPepDock | Local peptide docking | http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=PEPDOCK | Online | [318] |
| HADDOCK peptide | Local peptide docking | http://www.bonvinlab.org/software/haddock2.2/ | Standalone, online | [319] |
| HPEPDOCK | Global peptide docking | http://huanglab.phys.hust.edu.cn/hpepdock/ | Online | [320] |
| MDockPep | Global peptide docking | N/A | Standalone | [321] |
| pepATTRACT | Global peptide docking | http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::pepATTRACT | Online, standalone | [322] |
| PepCrawler | Local peptide docking | http://bioinfo3d.cs.tau.ac.il/PepCrawler/ | Online, standalone | [323] |
| PepSite | Local peptide docking | http://pepsite2.russelllab.org/ | Online | [324] |
| PEP-SiteFinder | Local peptide docking | http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-SiteFinder/ | Online | [325] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macalino, S.J.Y.; Basith, S.; Clavio, N.A.B.; Chang, H.; Kang, S.; Choi, S. Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery. Molecules 2018, 23, 1963. https://doi.org/10.3390/molecules23081963
Macalino SJY, Basith S, Clavio NAB, Chang H, Kang S, Choi S. Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery. Molecules. 2018; 23(8):1963. https://doi.org/10.3390/molecules23081963
Chicago/Turabian StyleMacalino, Stephani Joy Y., Shaherin Basith, Nina Abigail B. Clavio, Hyerim Chang, Soosung Kang, and Sun Choi. 2018. "Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery" Molecules 23, no. 8: 1963. https://doi.org/10.3390/molecules23081963
APA StyleMacalino, S. J. Y., Basith, S., Clavio, N. A. B., Chang, H., Kang, S., & Choi, S. (2018). Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery. Molecules, 23(8), 1963. https://doi.org/10.3390/molecules23081963