Structural Revisions in Natural Ellagitannins

 , , , and
, , , and
Abstract
:
1. Introduction
2. Notice
3. Structural Revision in Ellagitannins
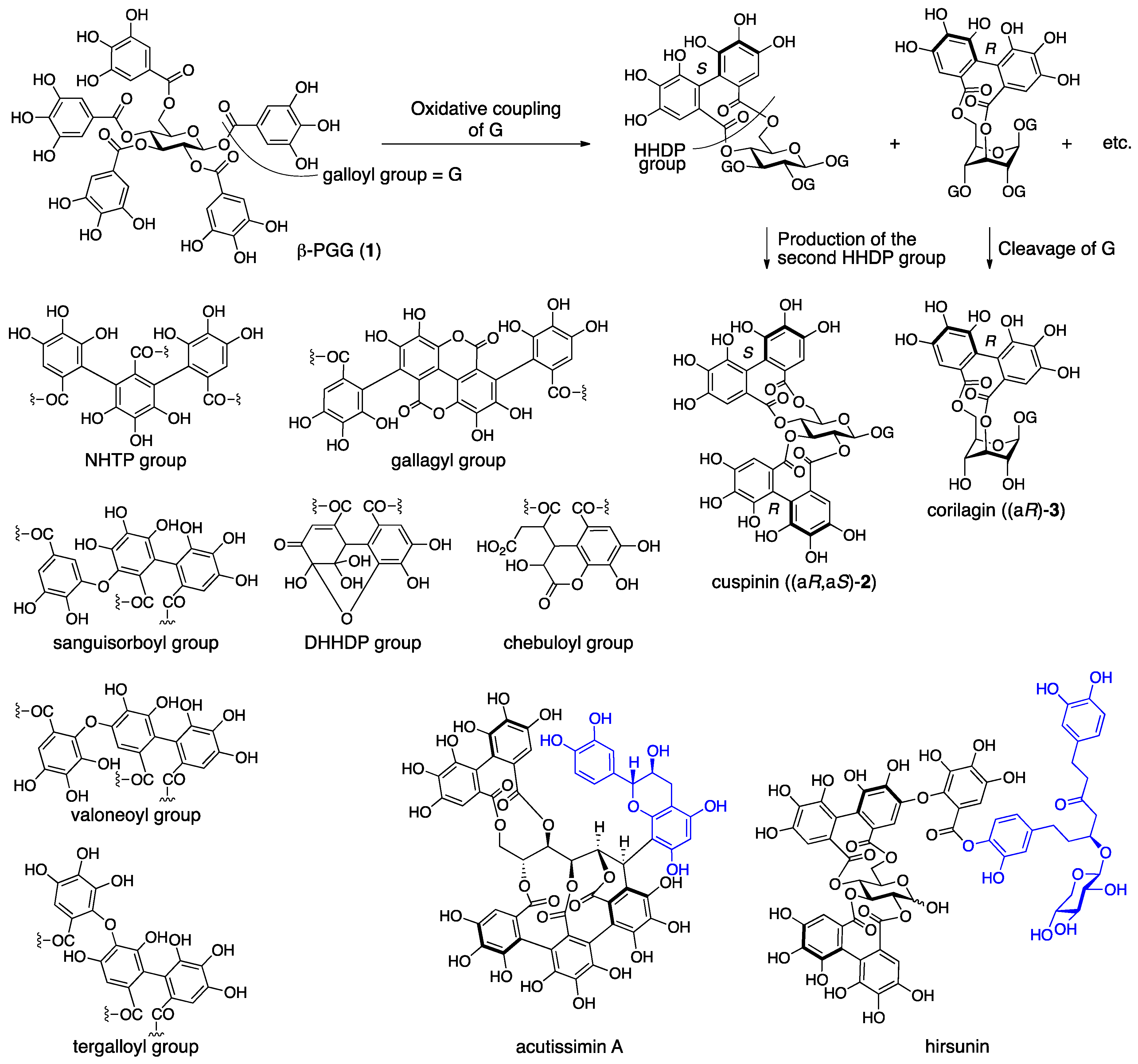
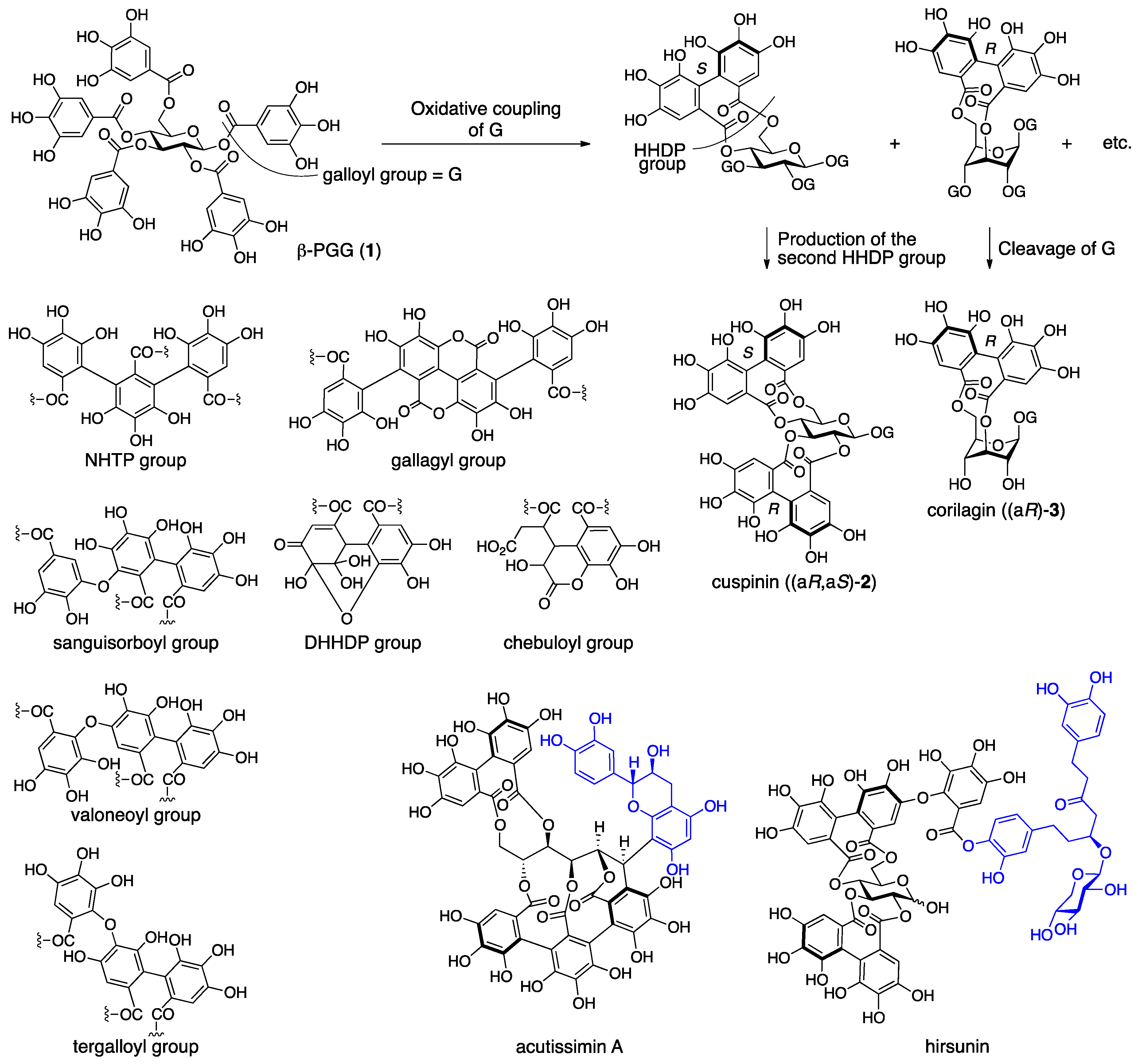
3.1. Correction of the Bonding Positions of the Galloyl and HHDP Groups and Correction of the Axial Chirality of the HHDP Group
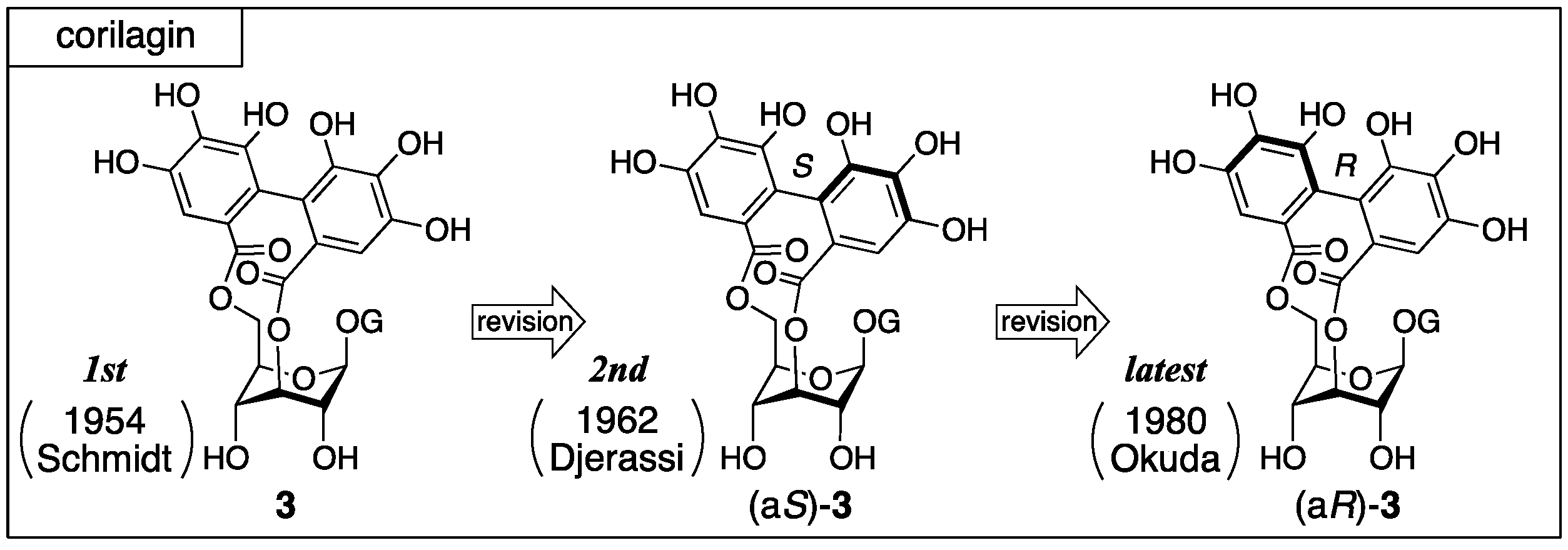
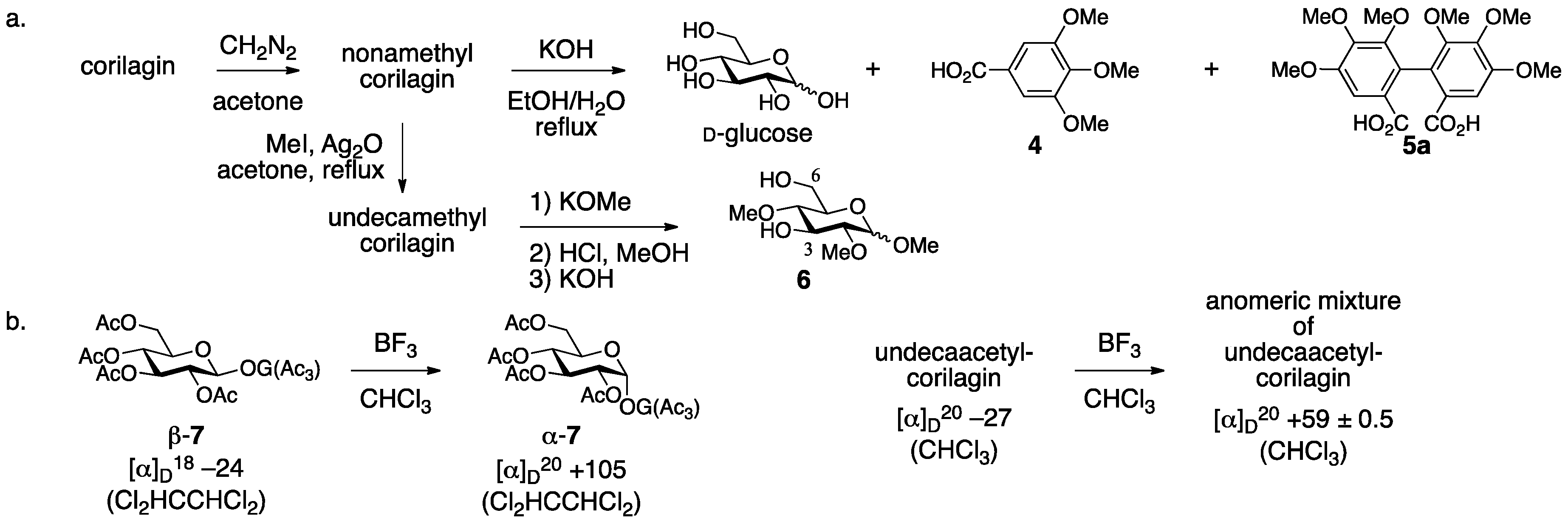
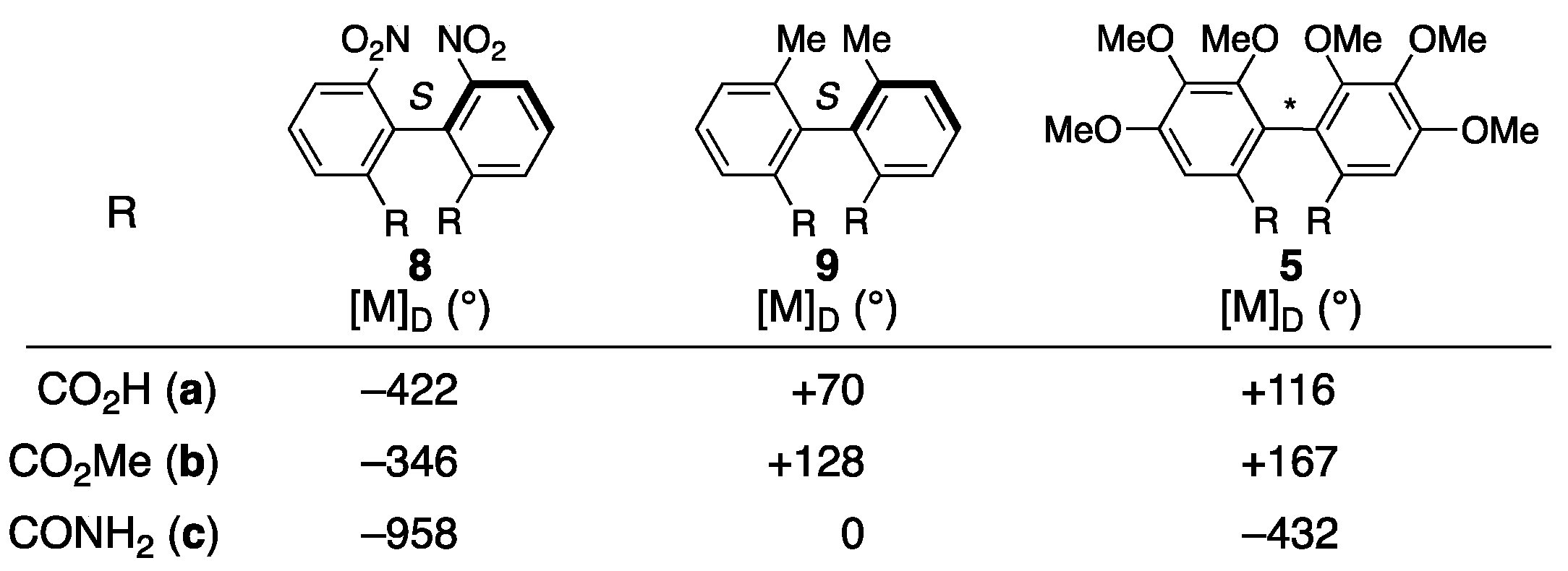
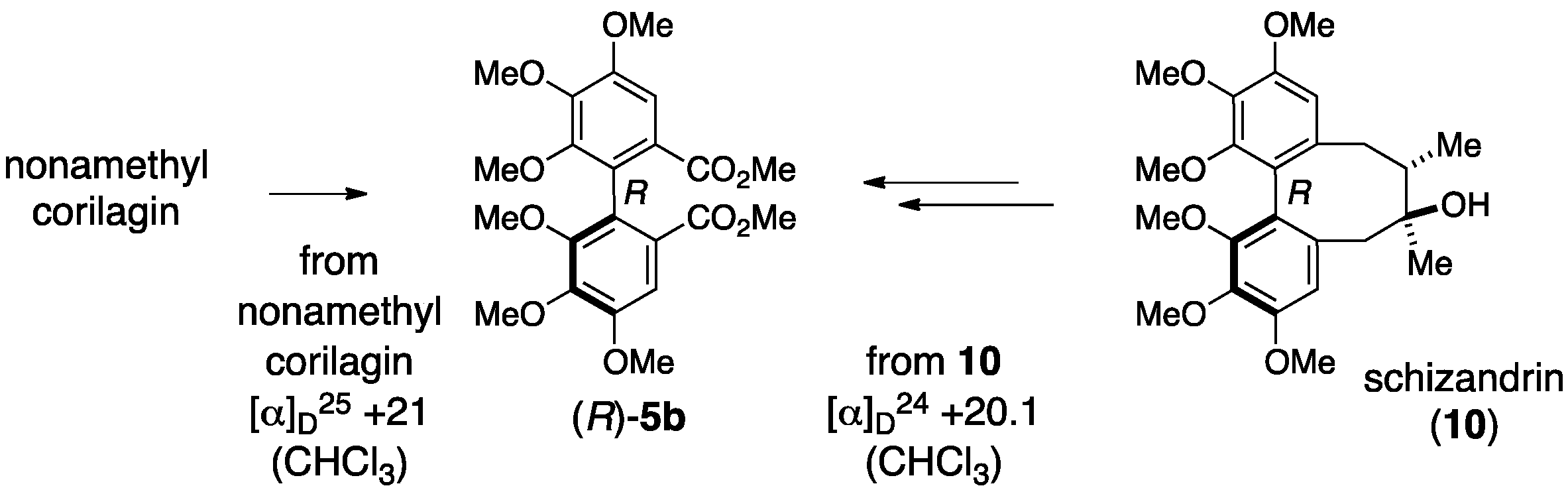
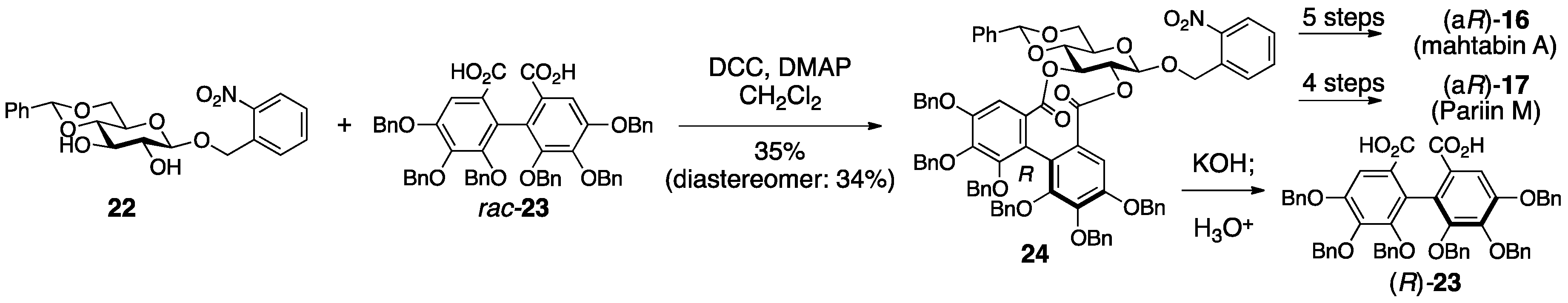
3.1.1. Corilagin
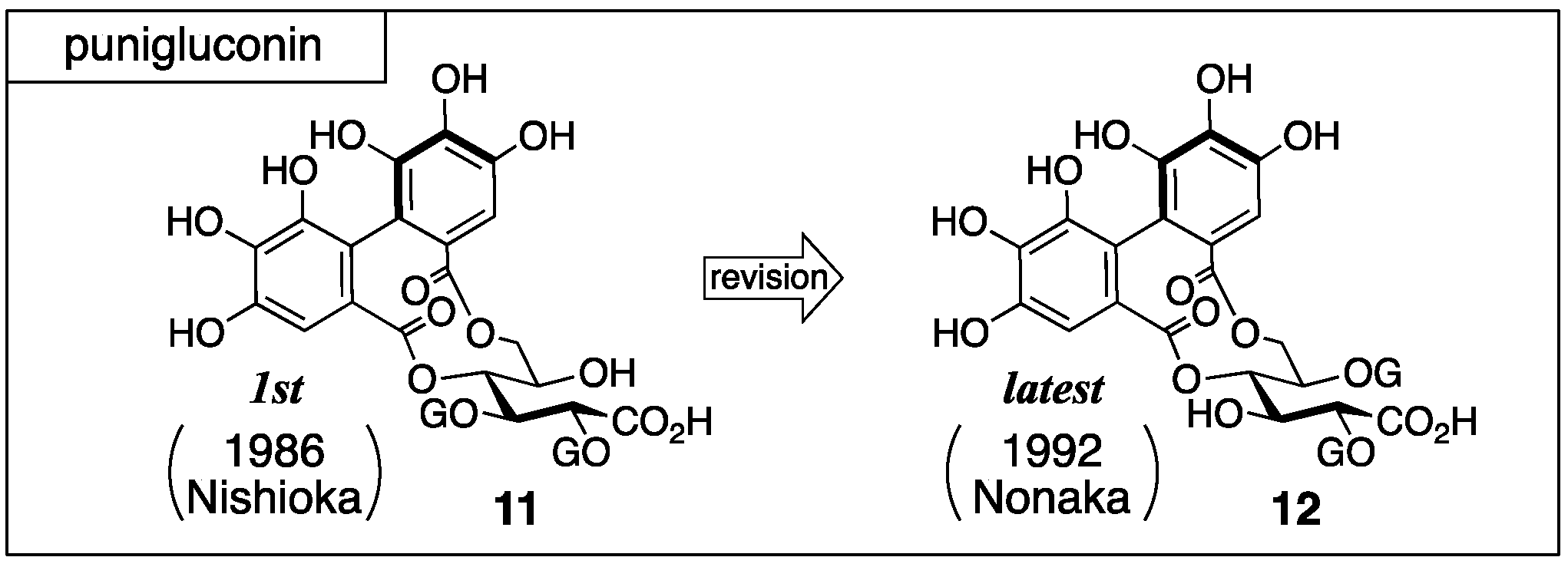
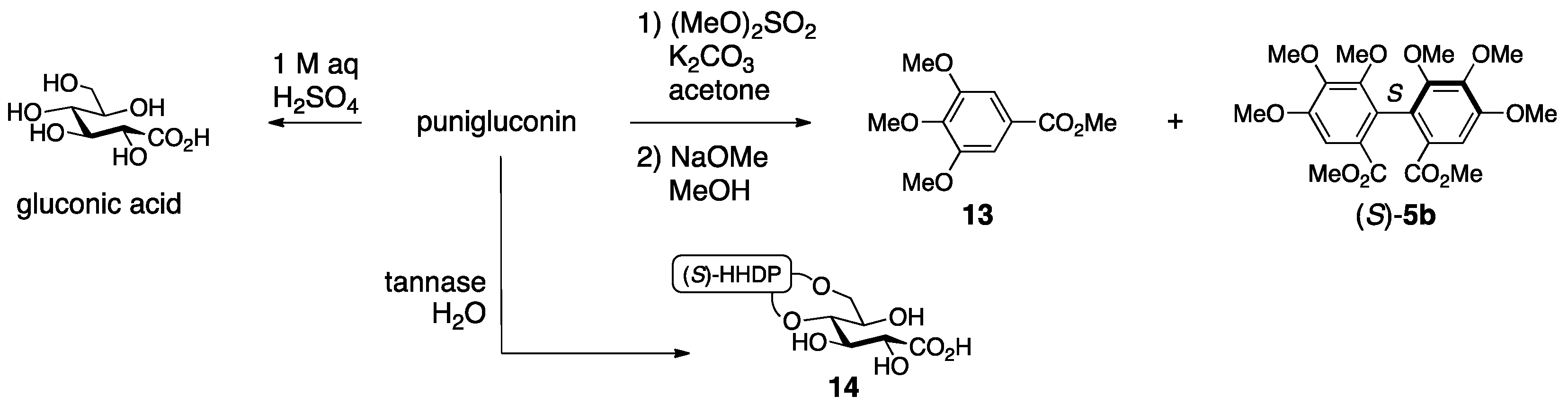
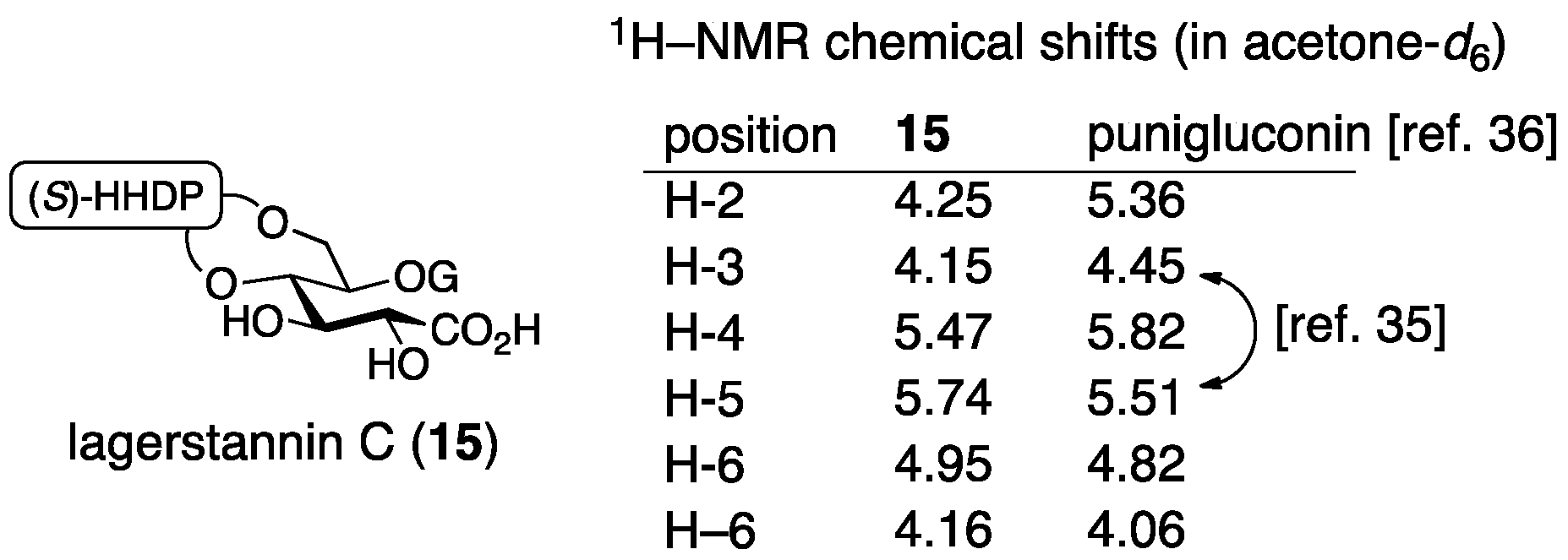
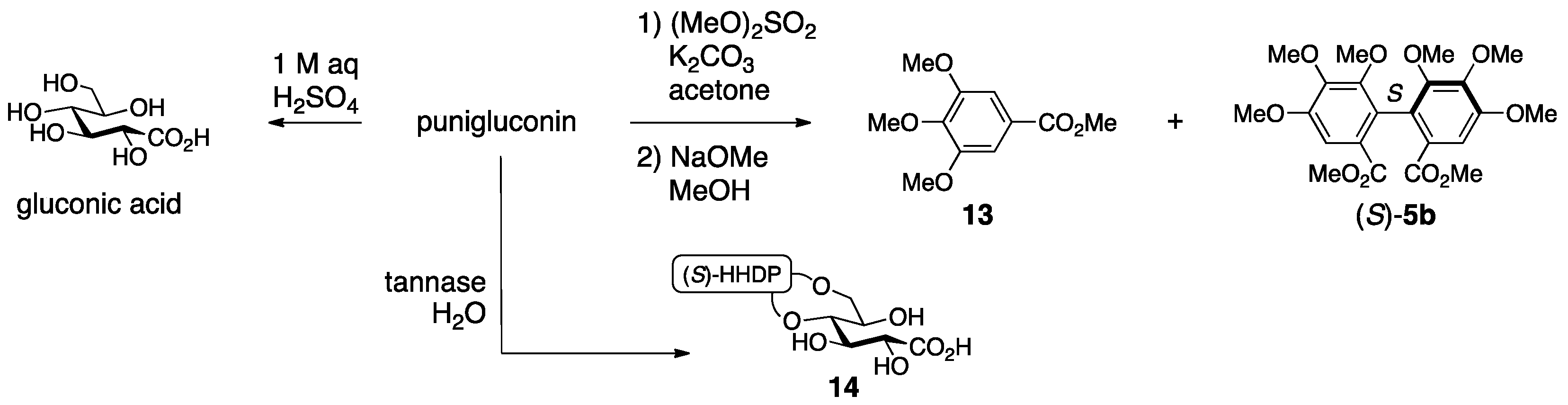
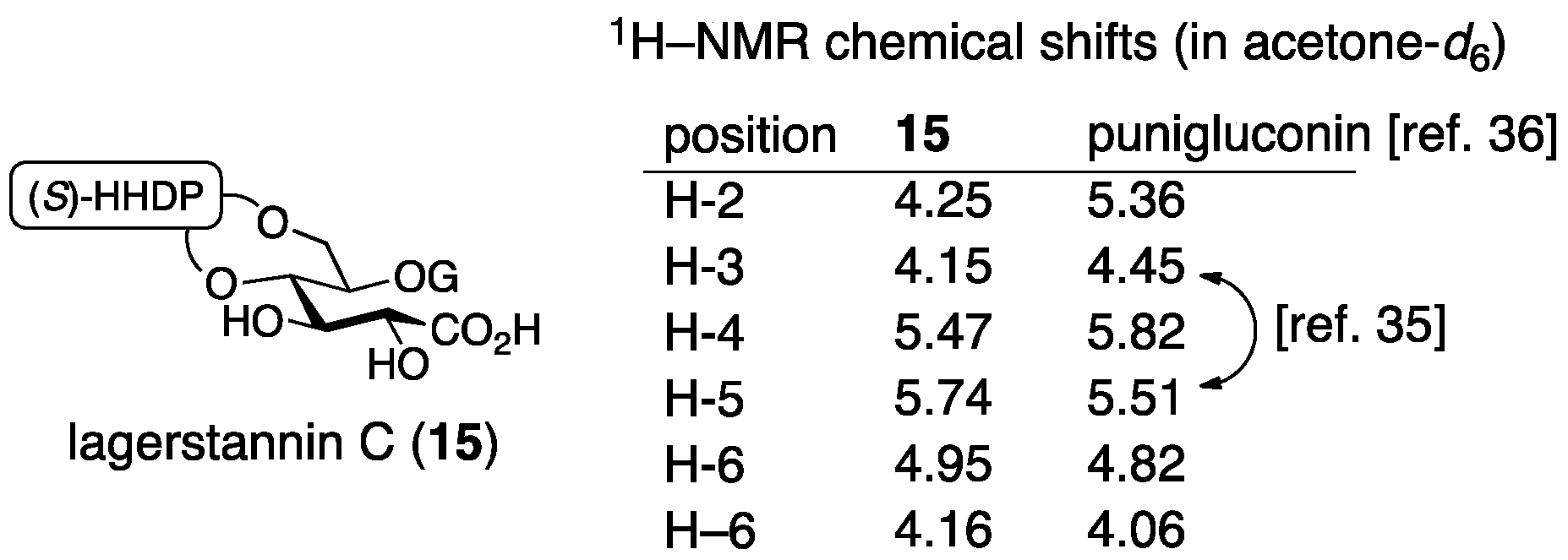
3.1.2. Punigluconin
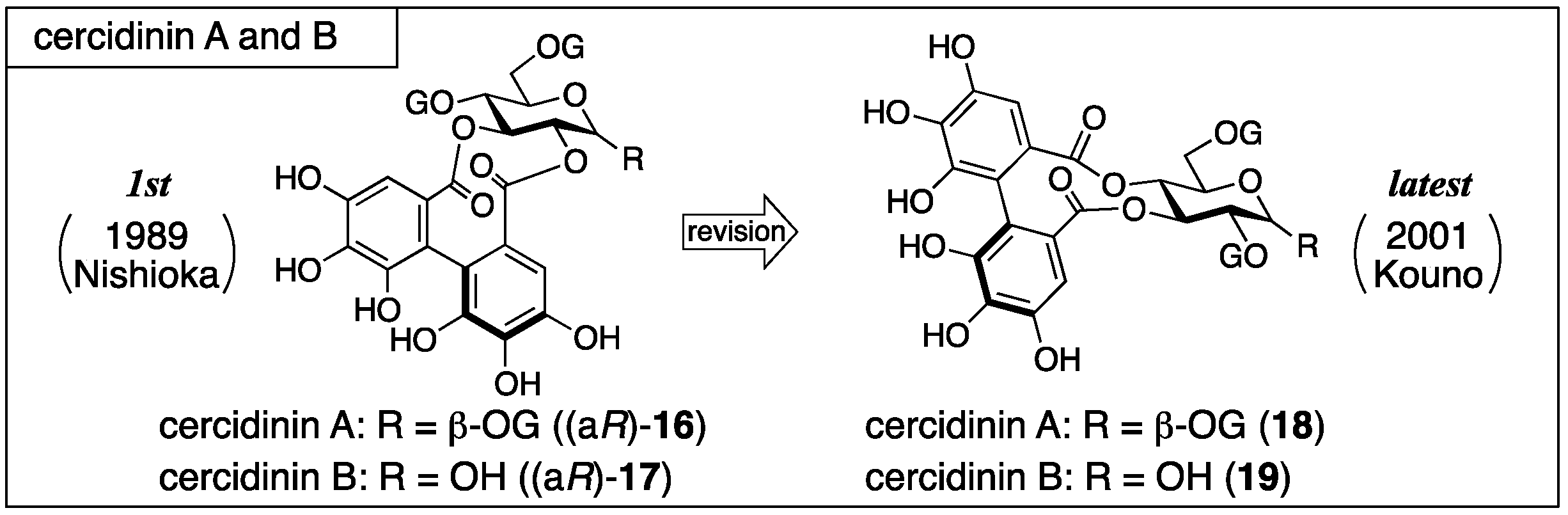
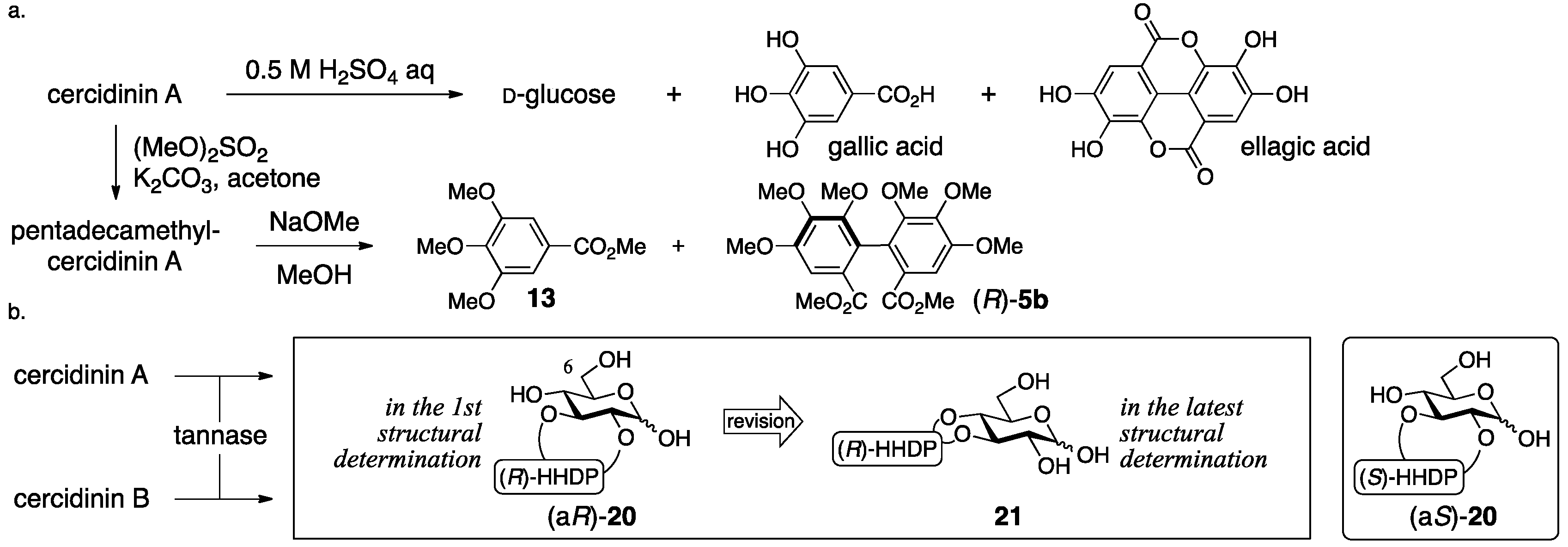
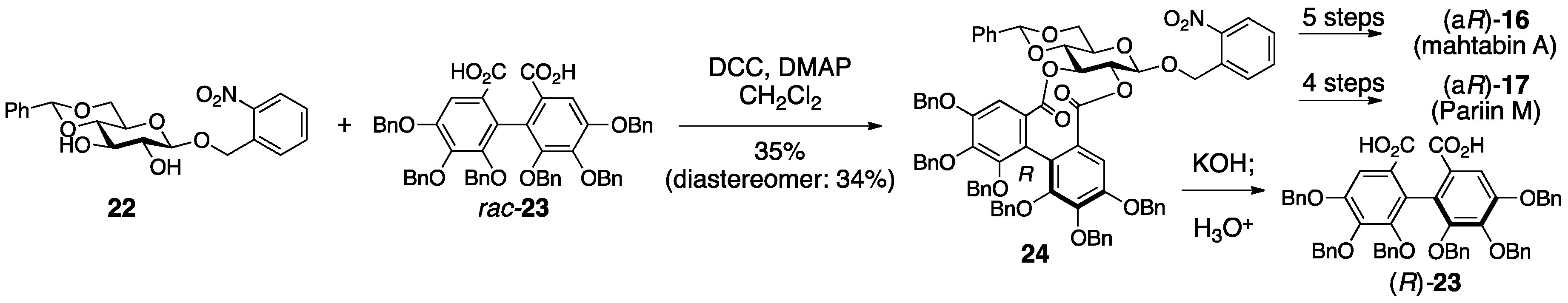
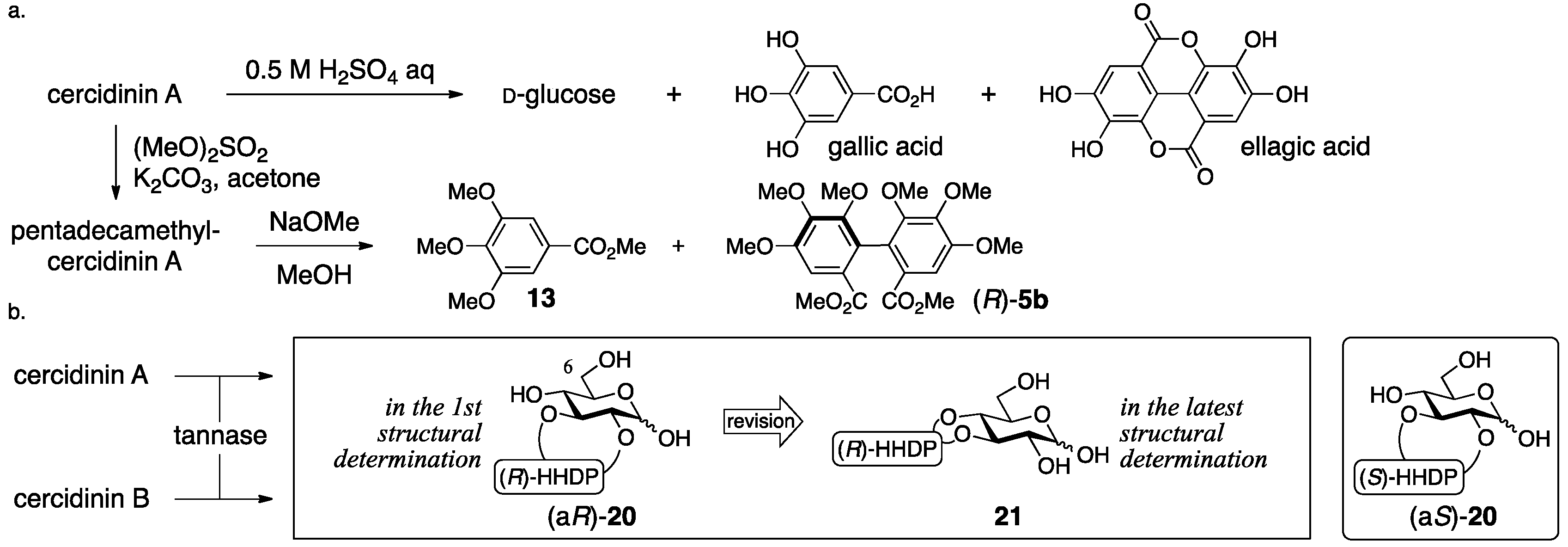
3.1.3. Cercidinin A and B
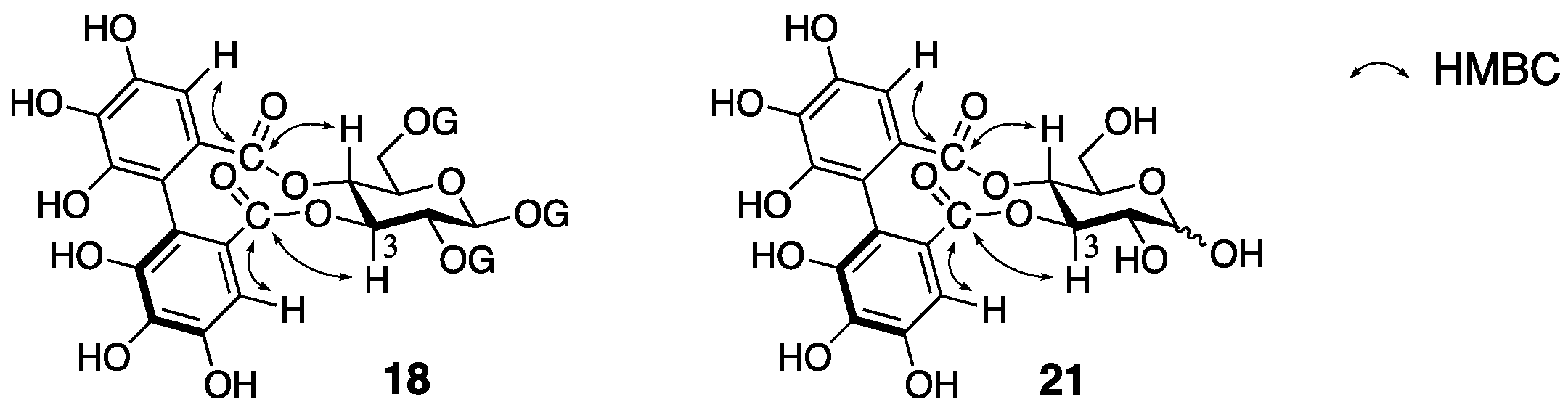
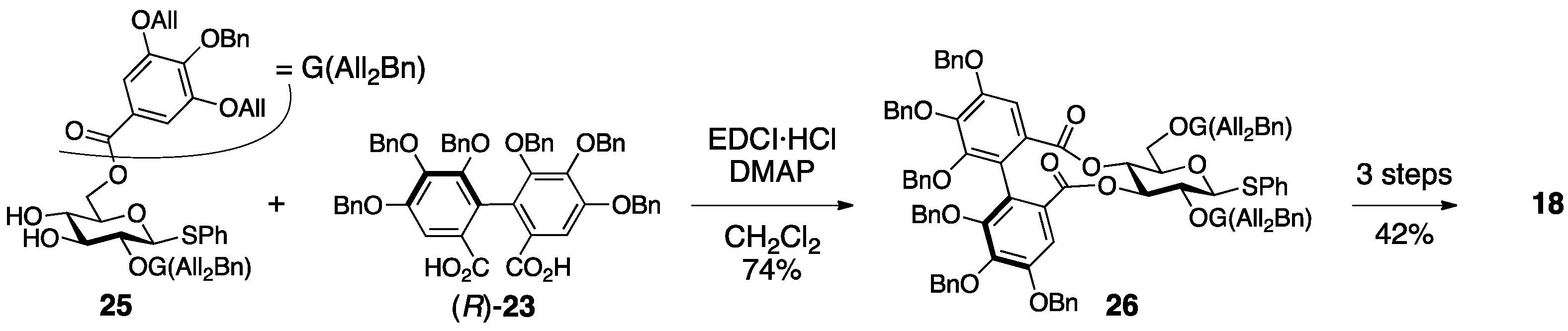
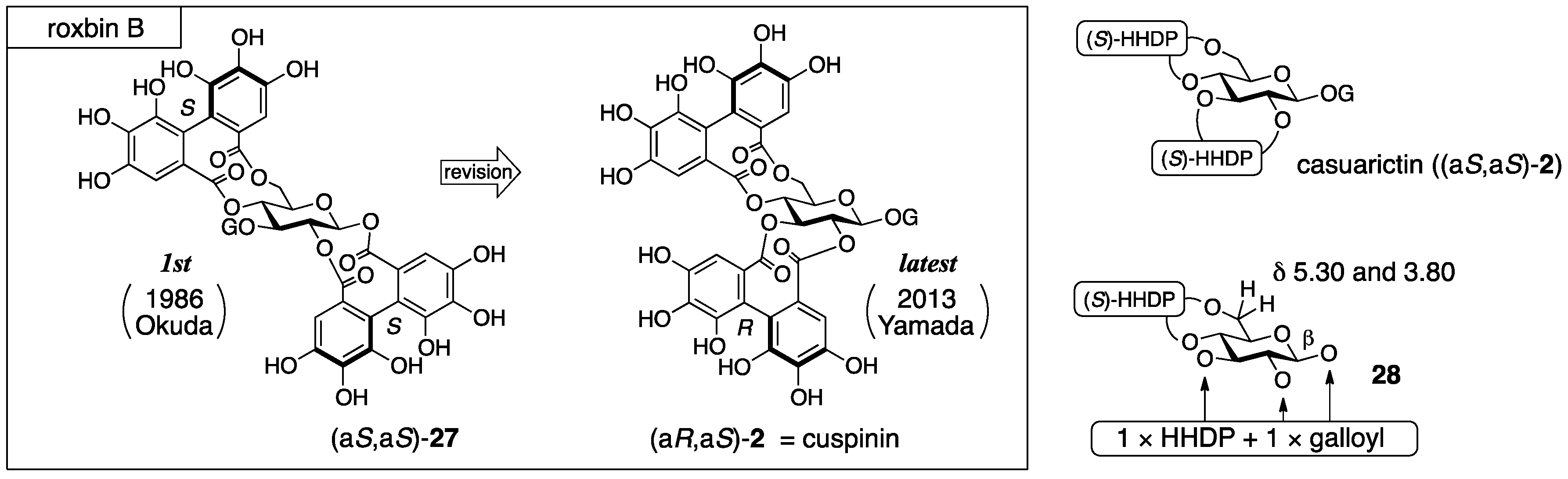
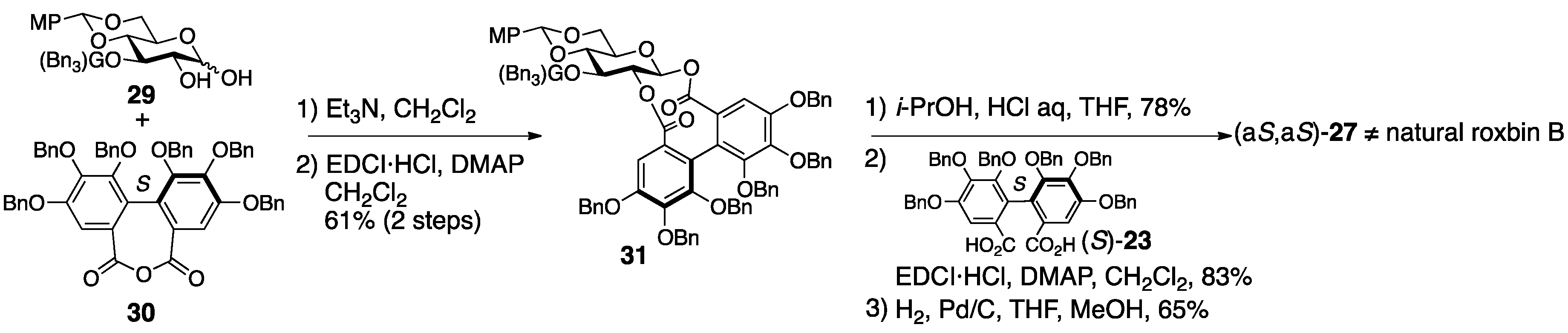
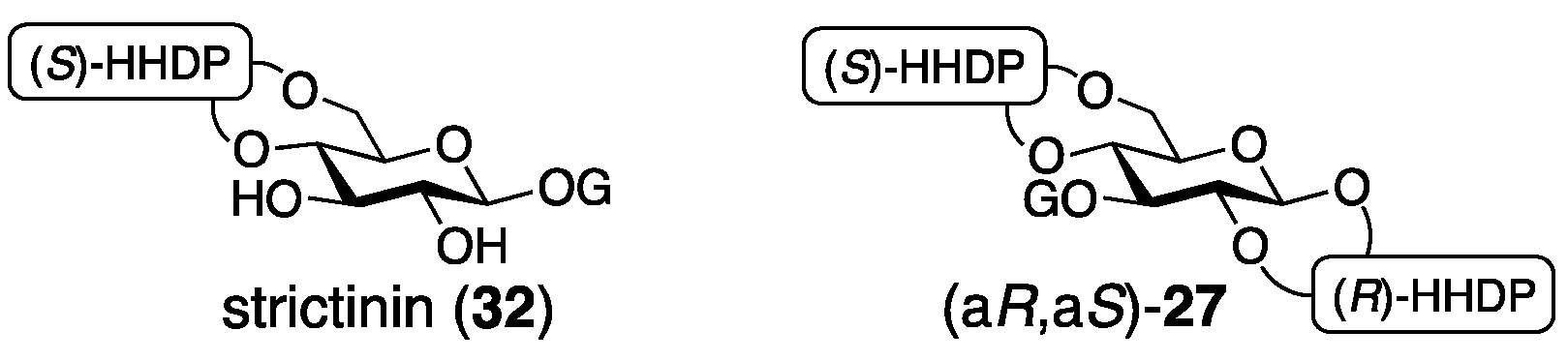
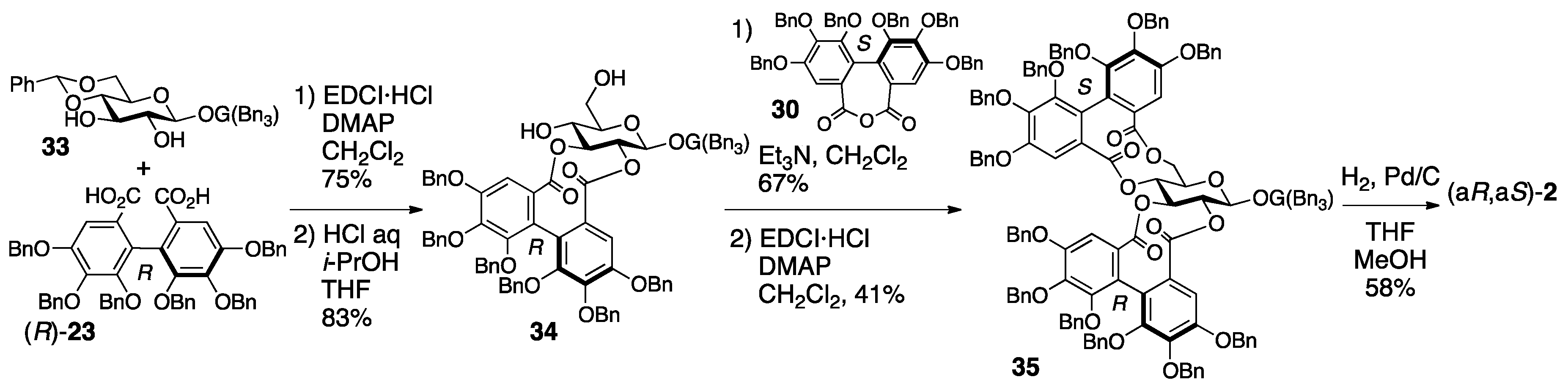
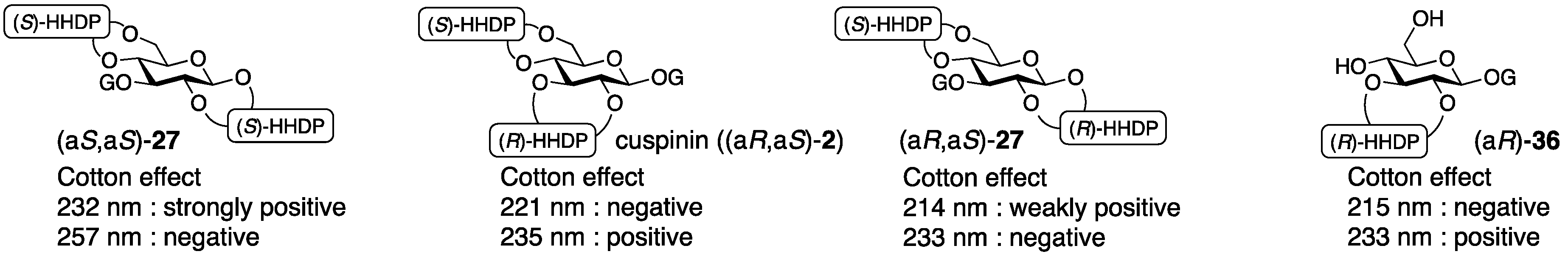
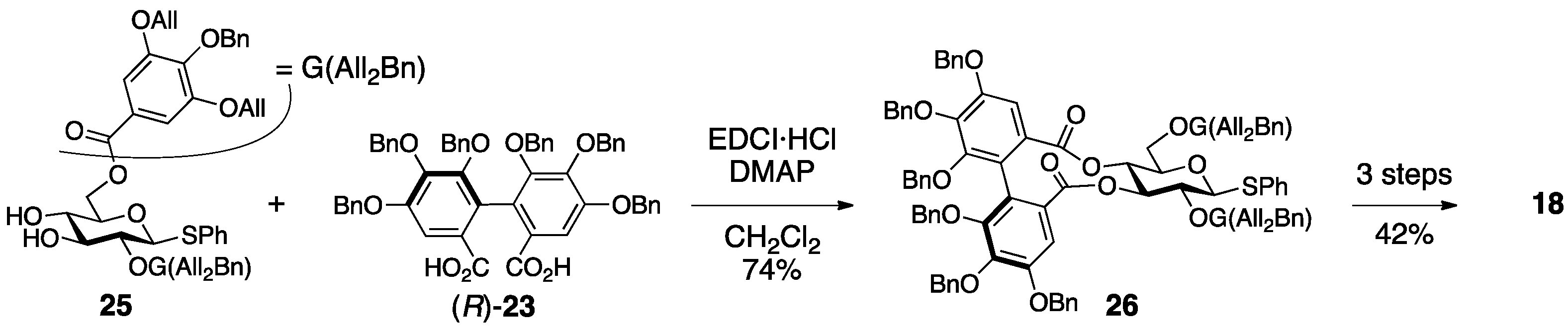
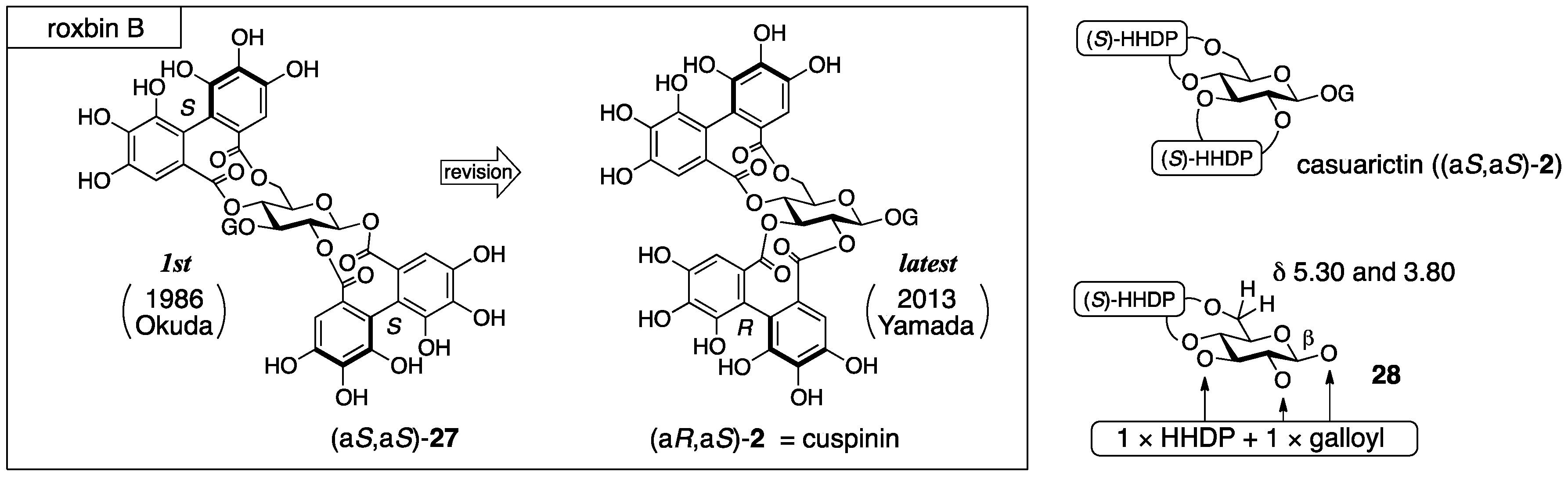
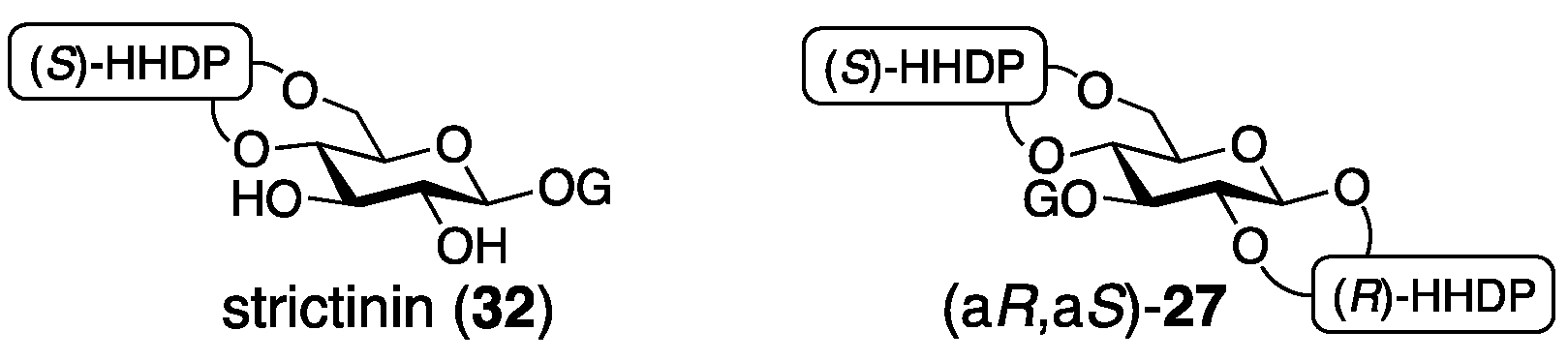
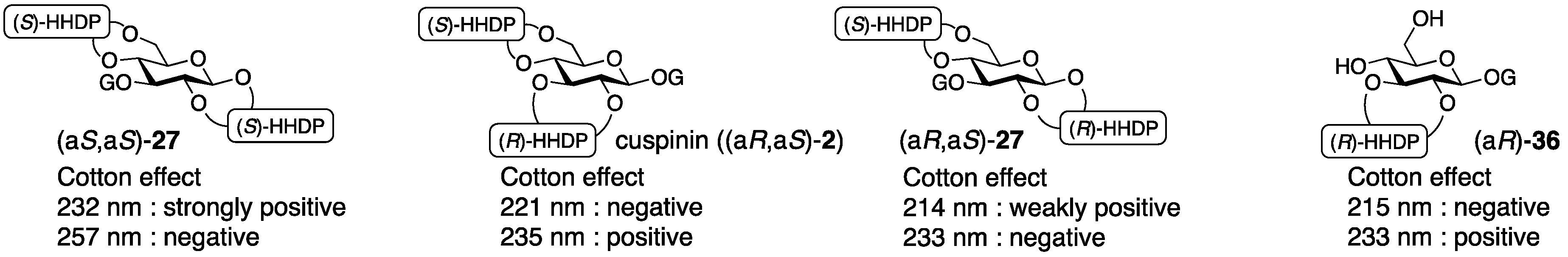
3.1.4. Roxbin B
3.2. Correction Based on the Structure of the DHHDP Group
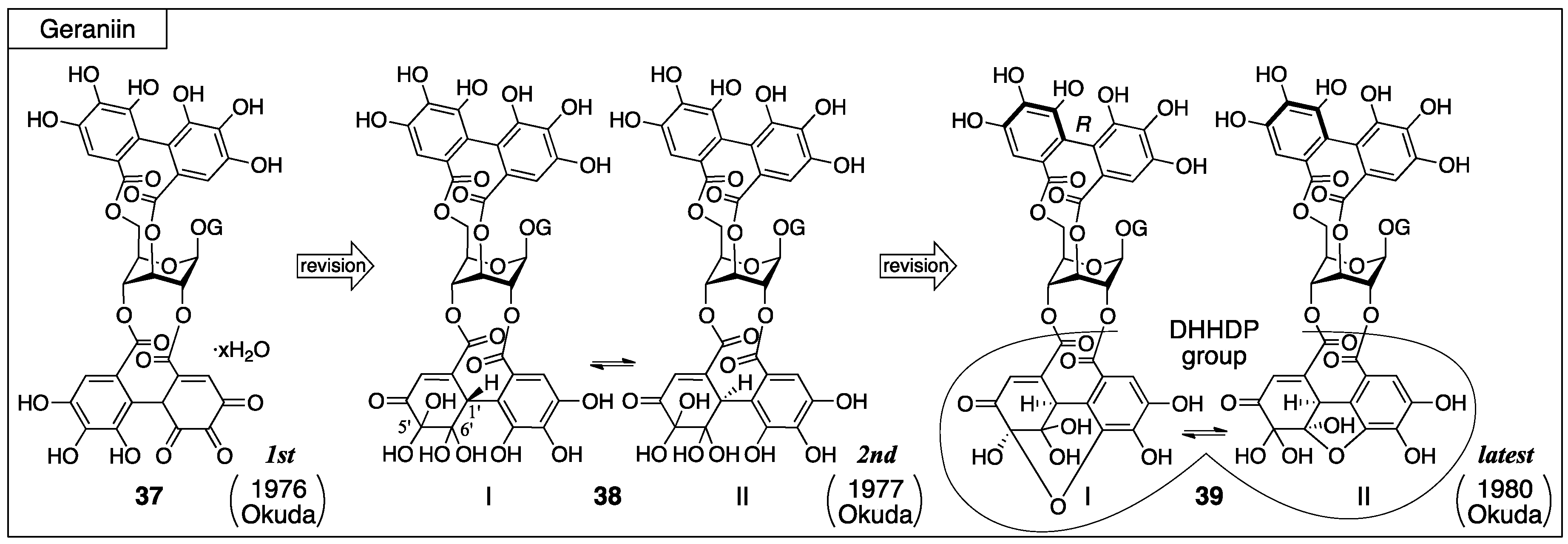
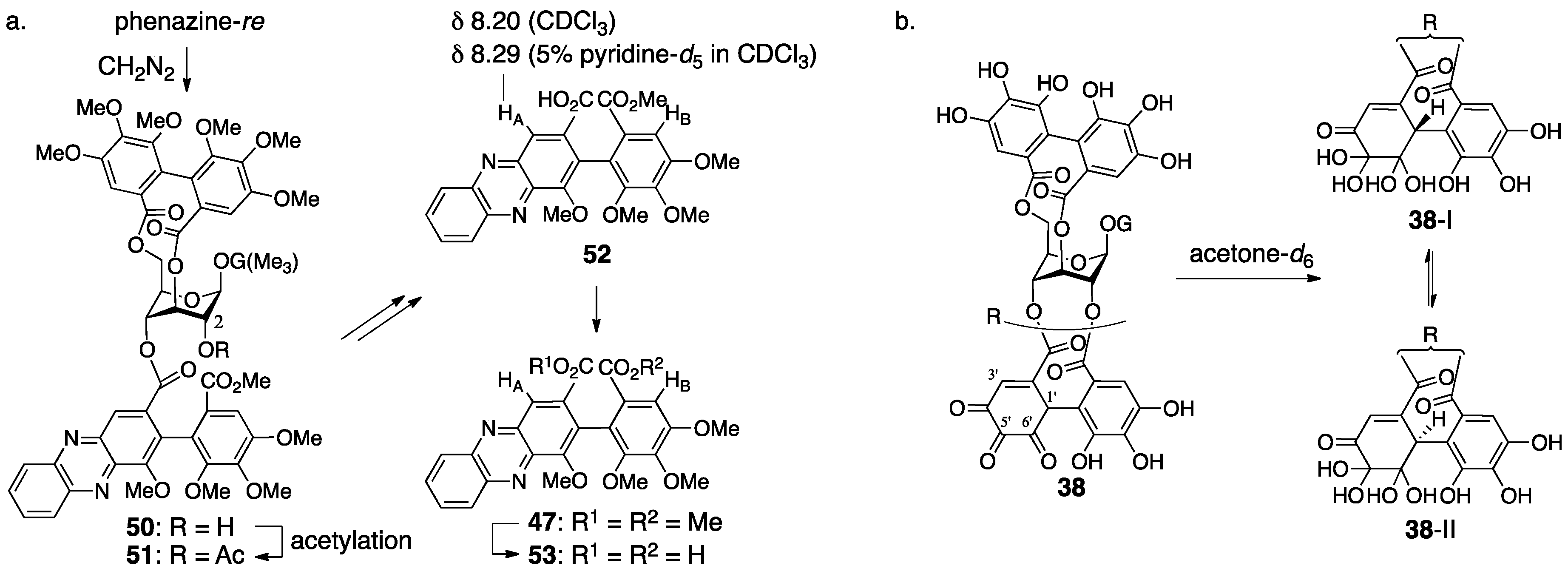
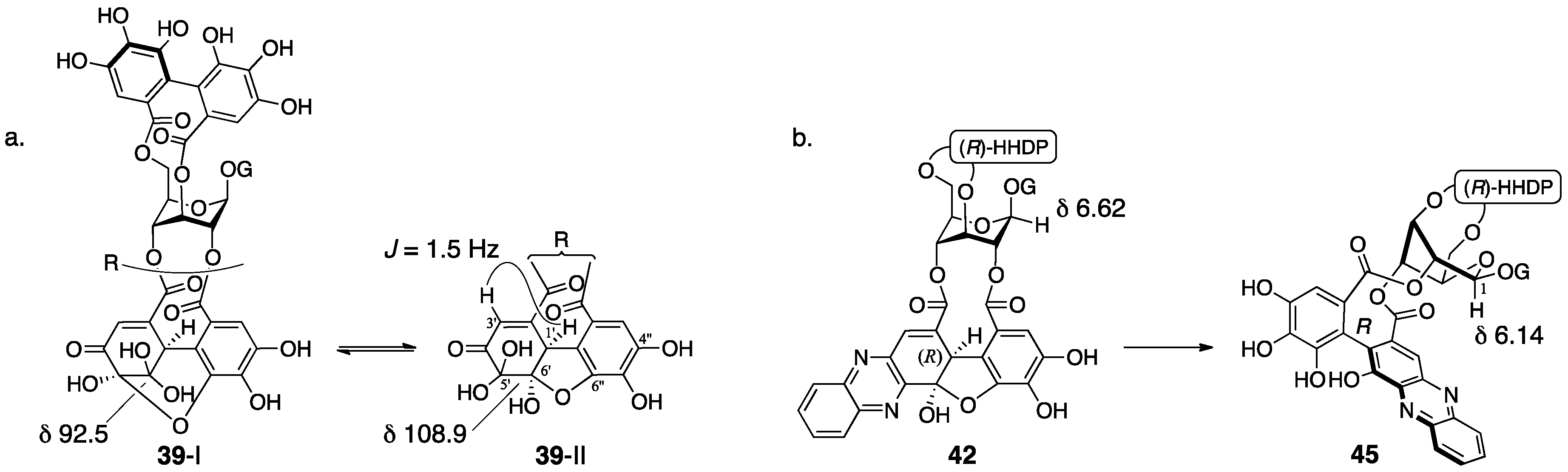
3.2.1. Geraniin
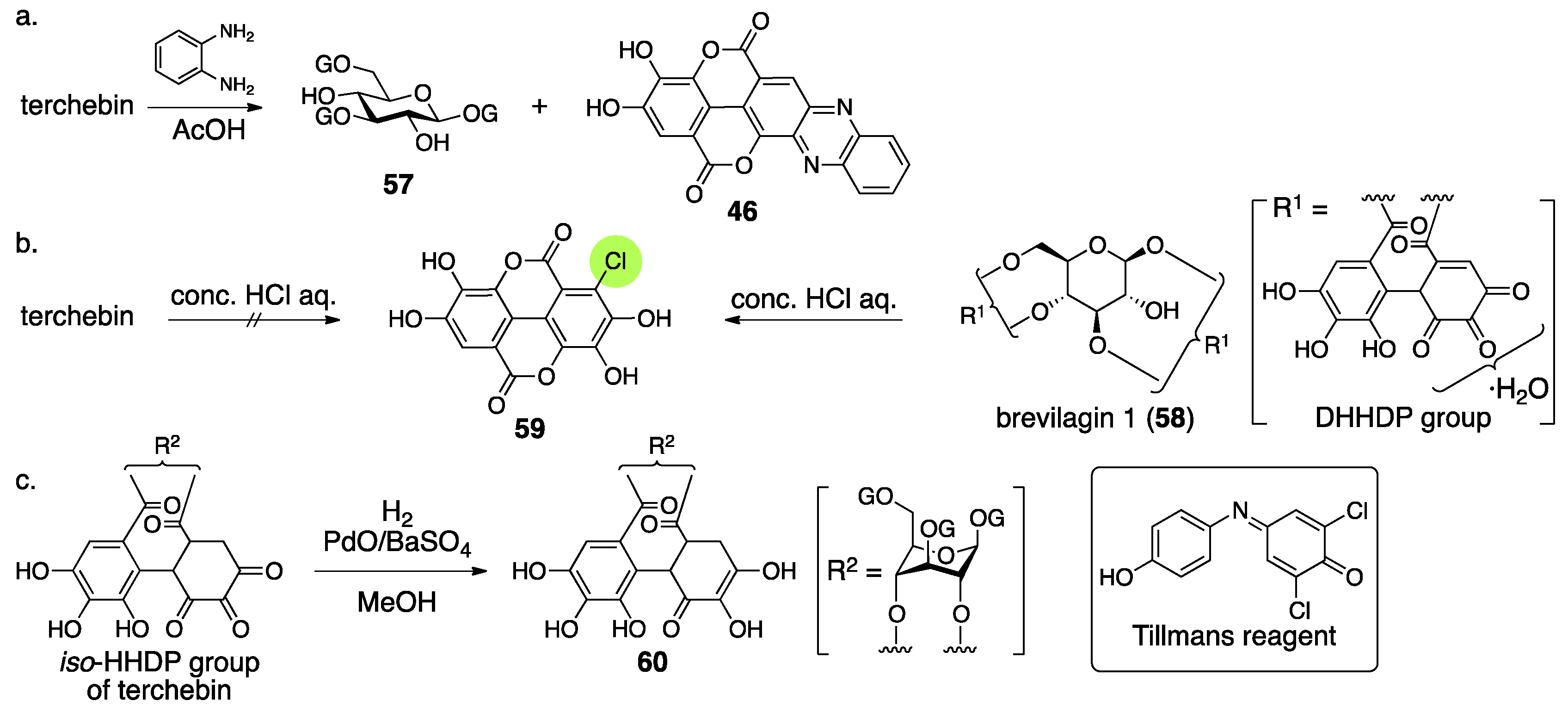
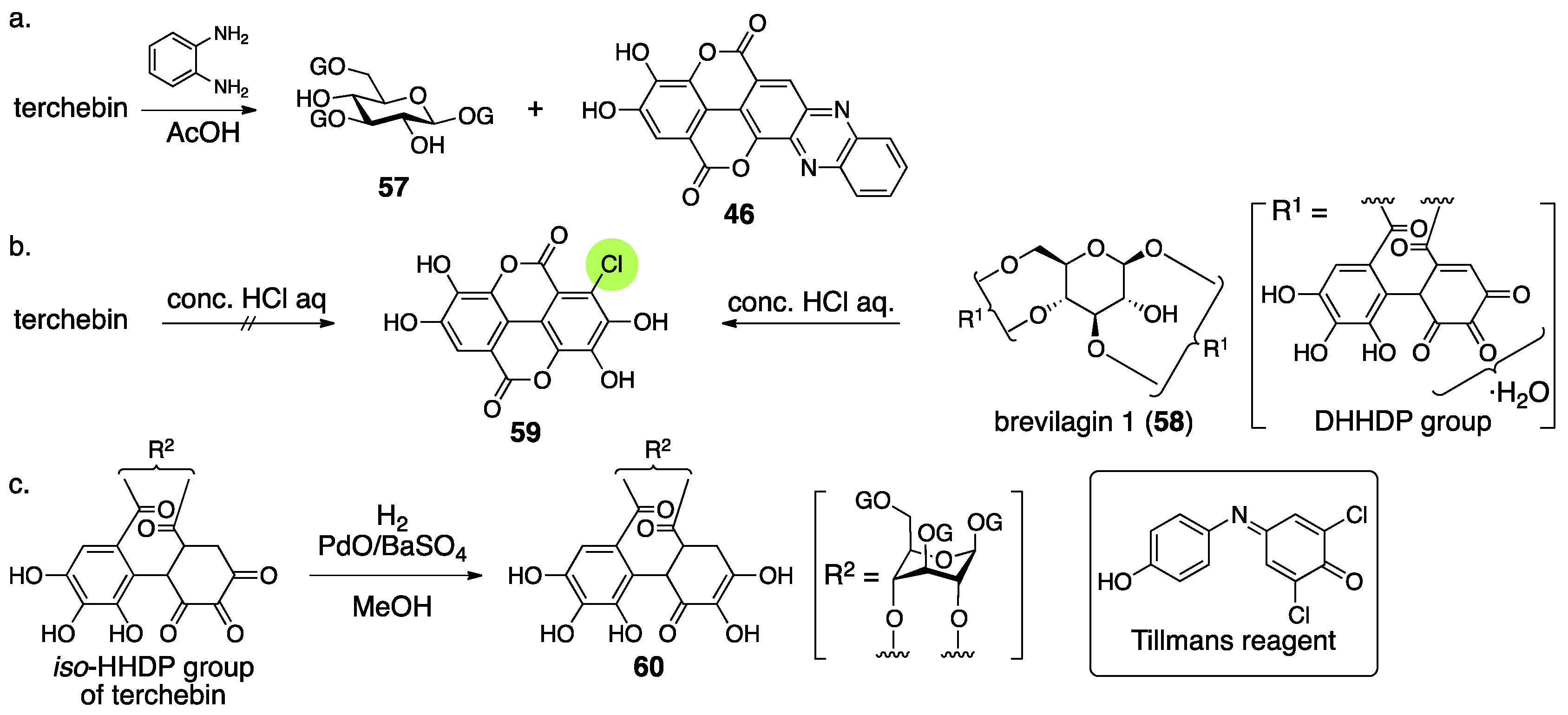
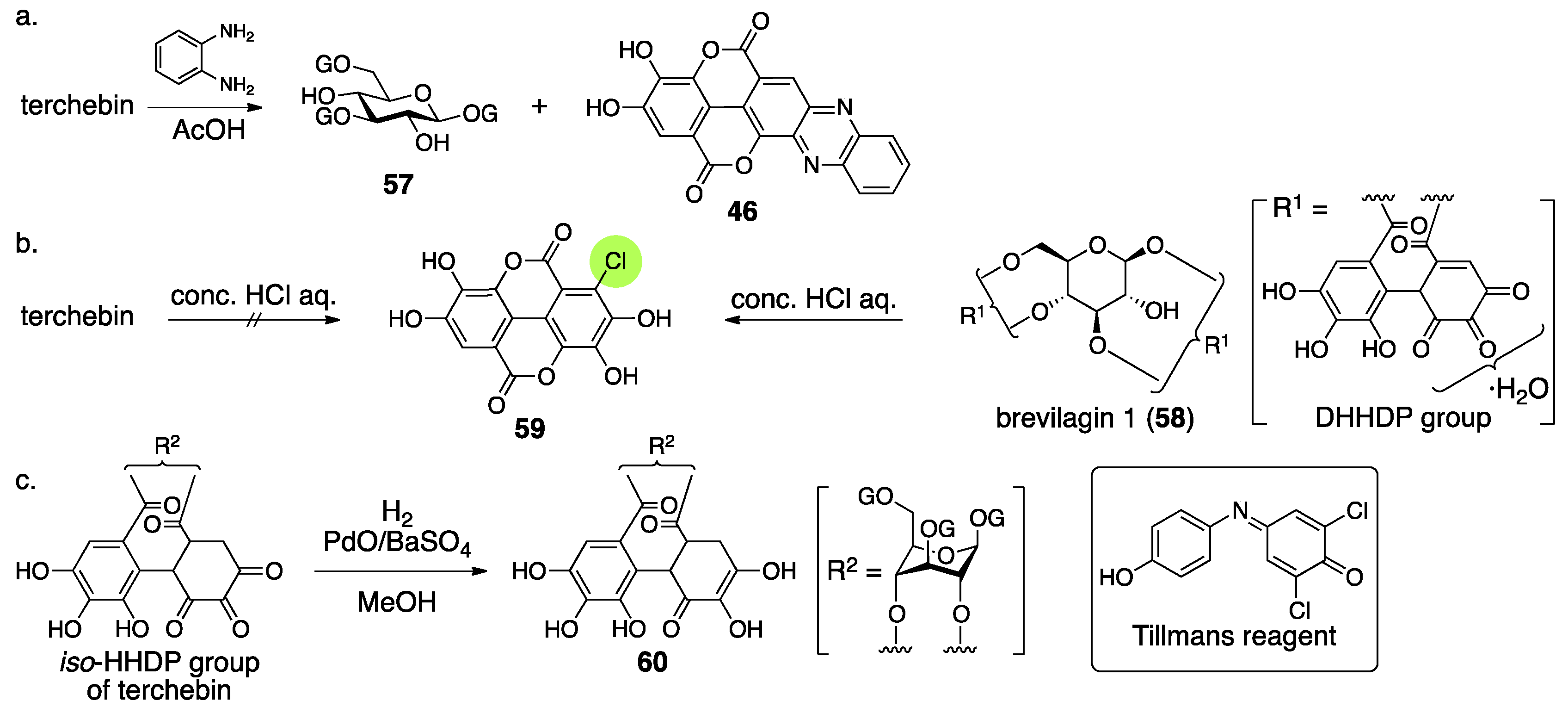
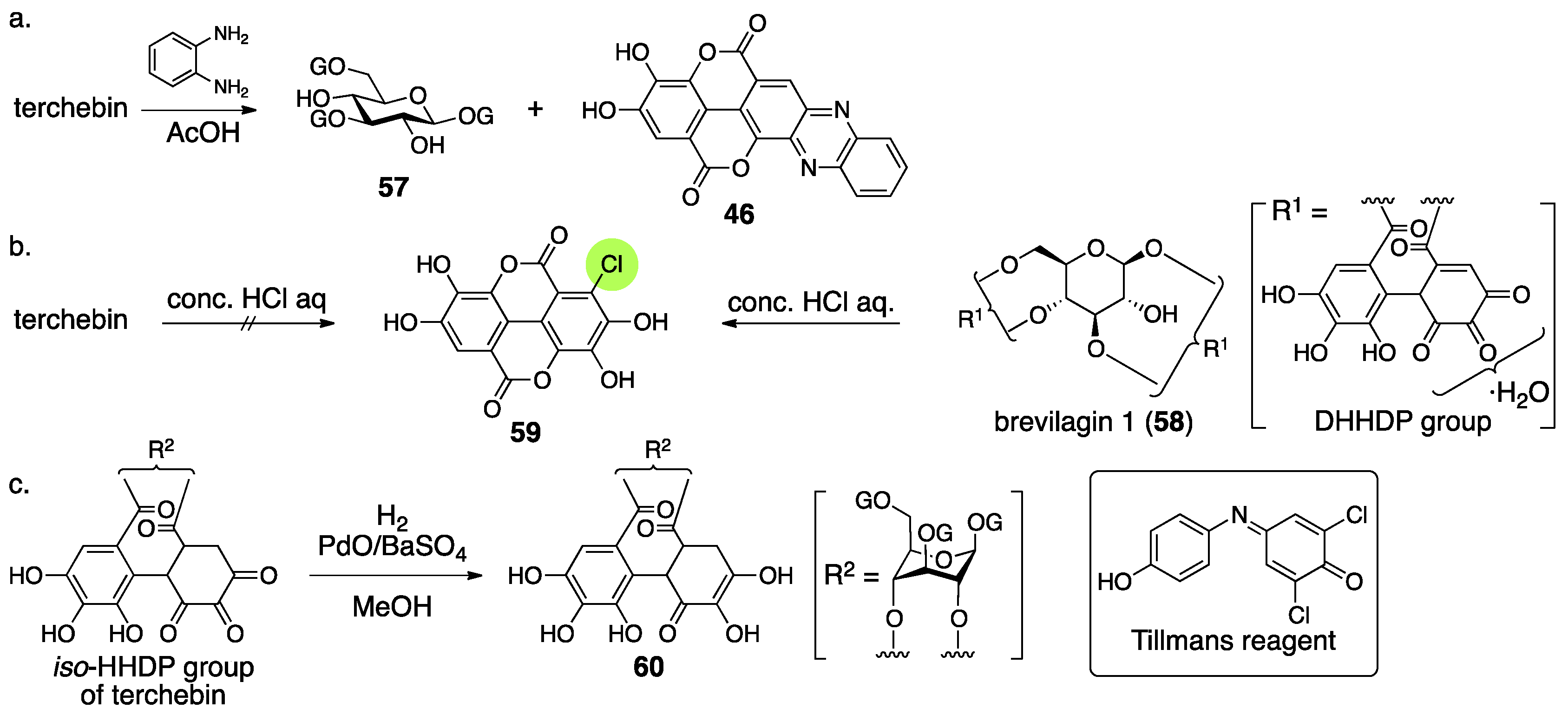
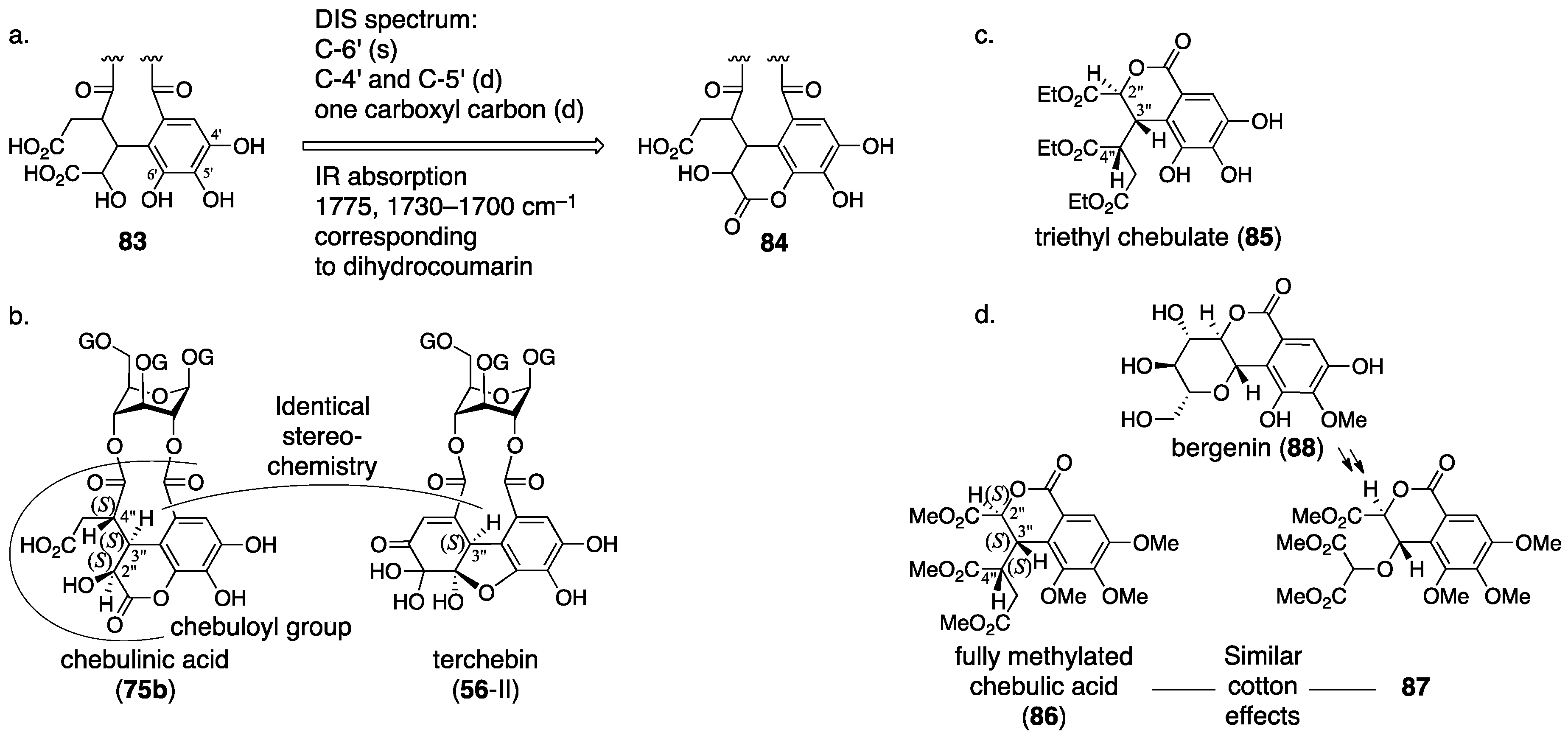
3.2.2. Terchebin
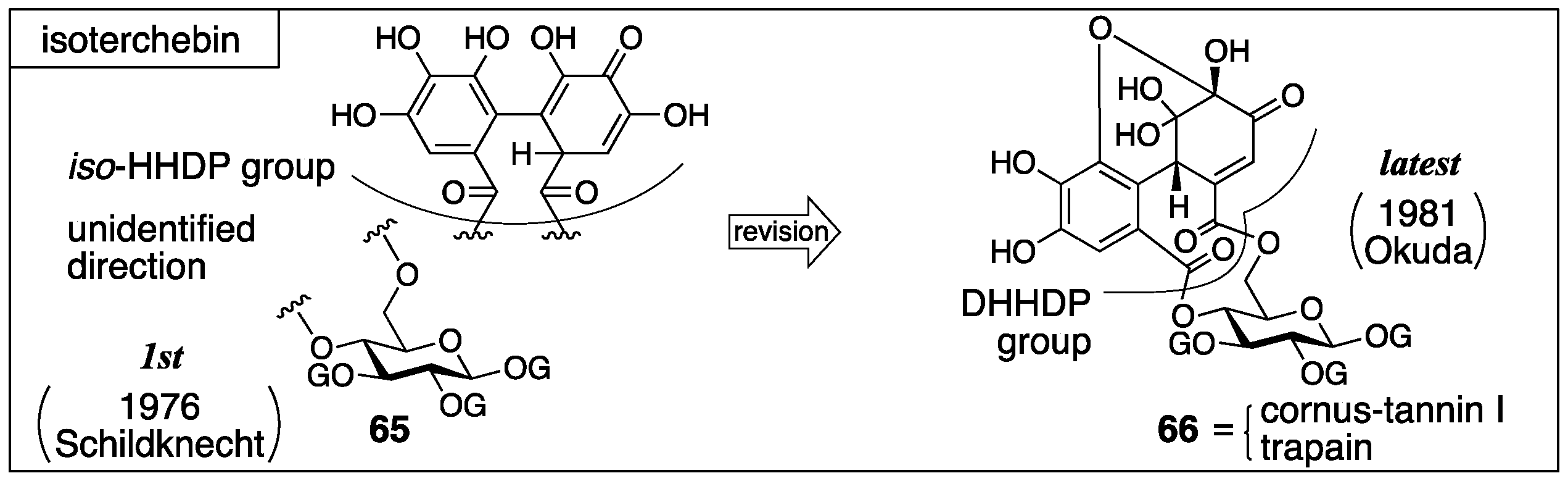
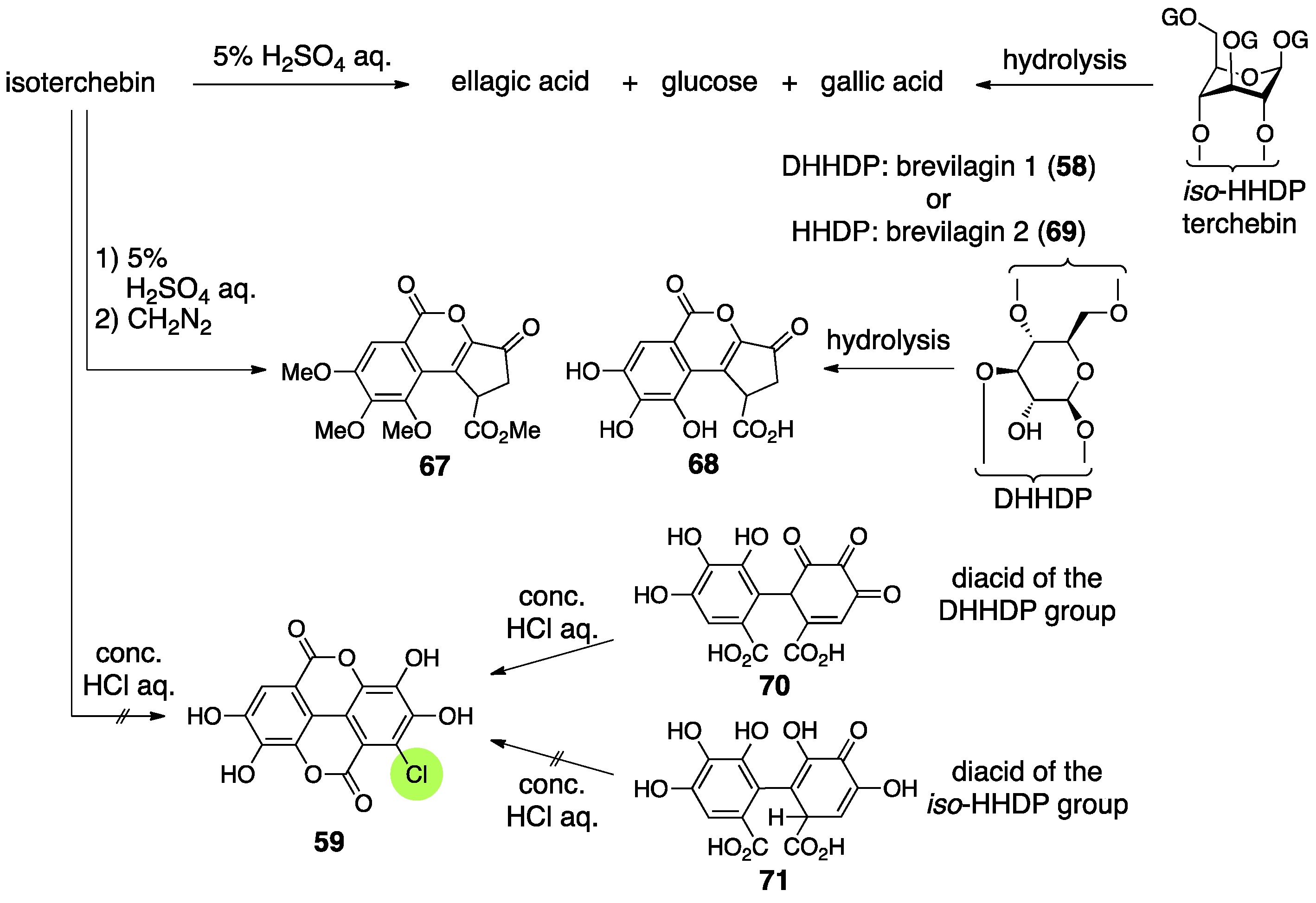
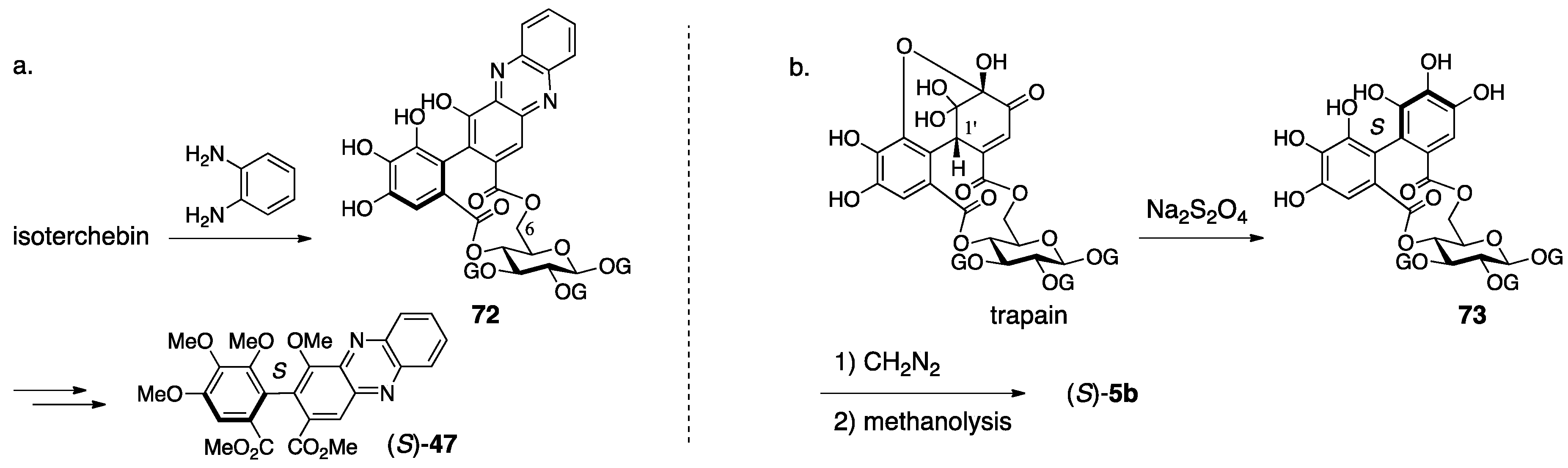
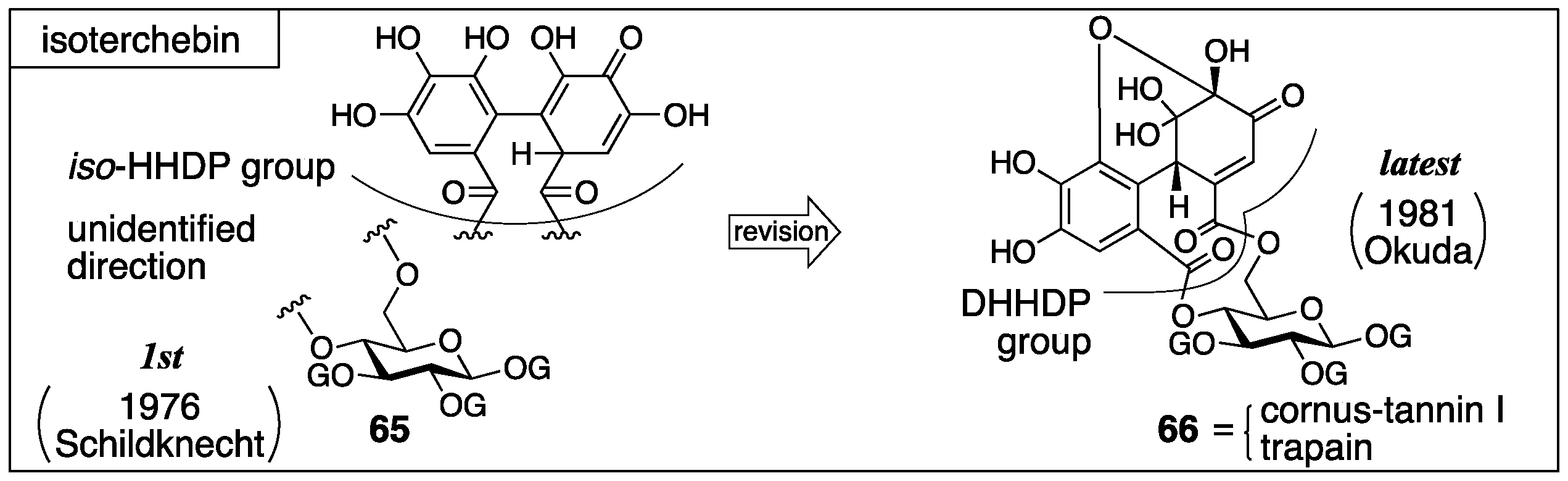
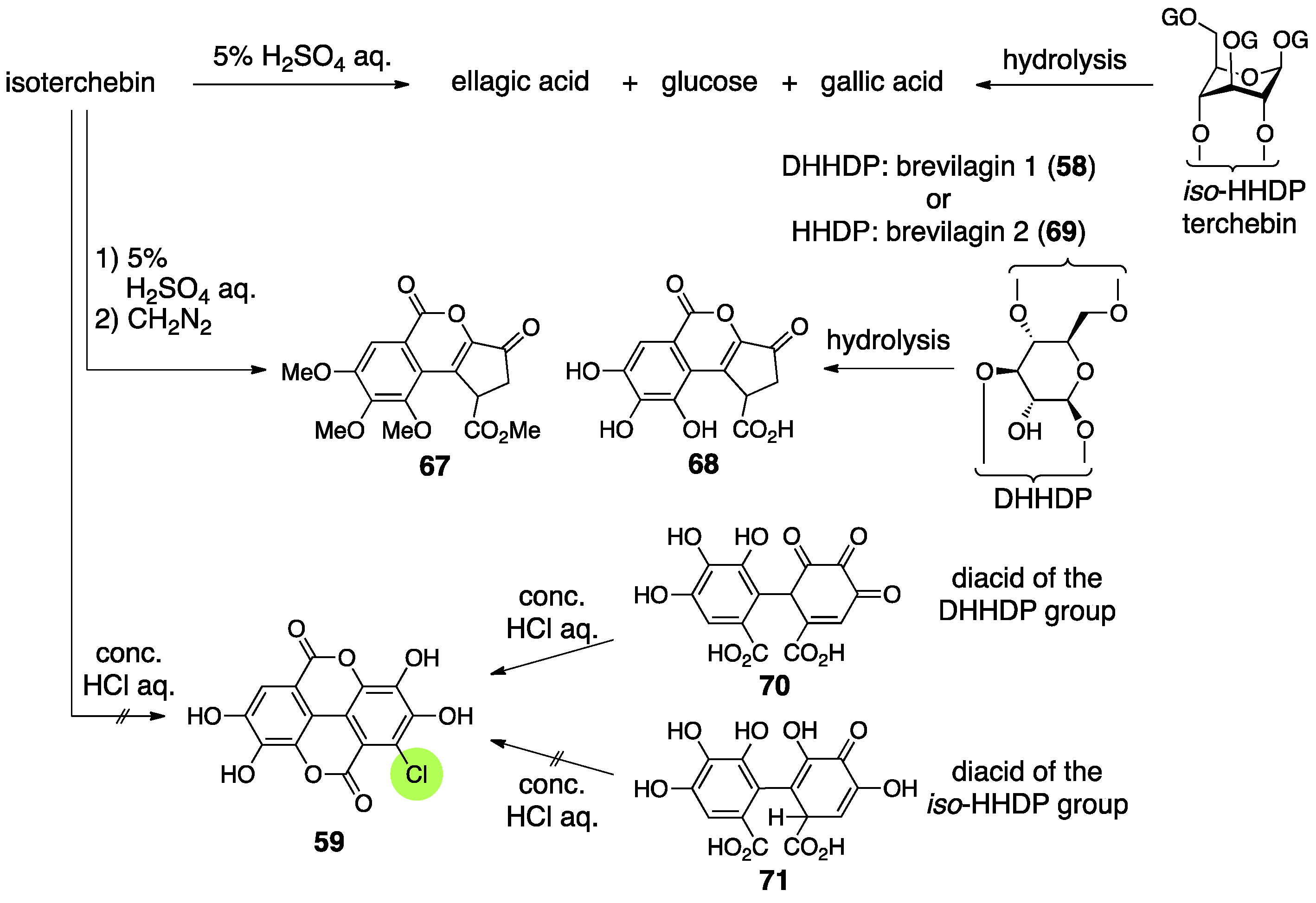
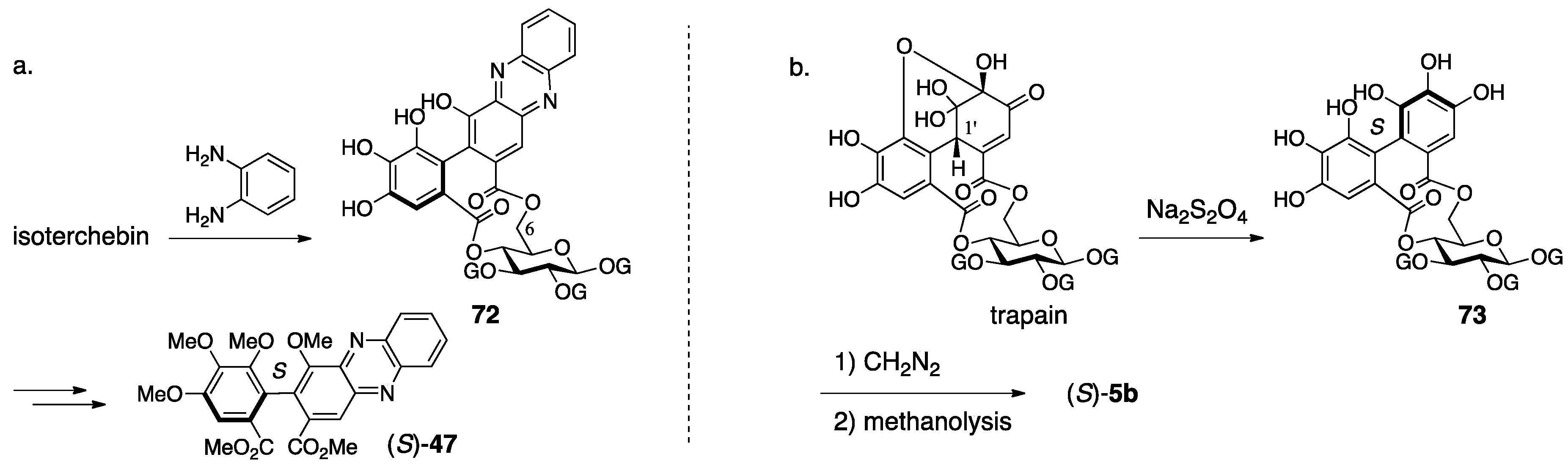
3.2.3. Isoterchebin
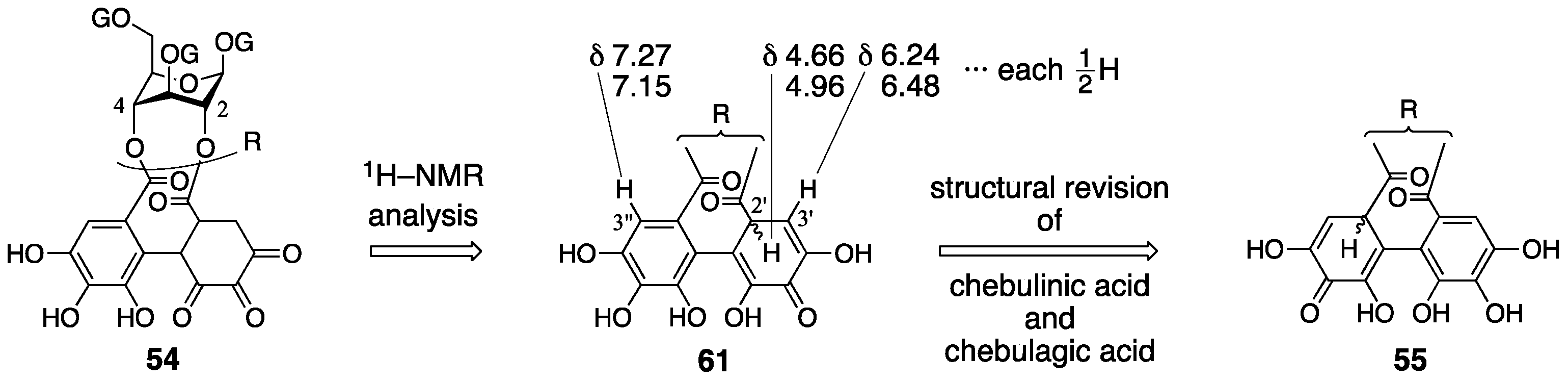
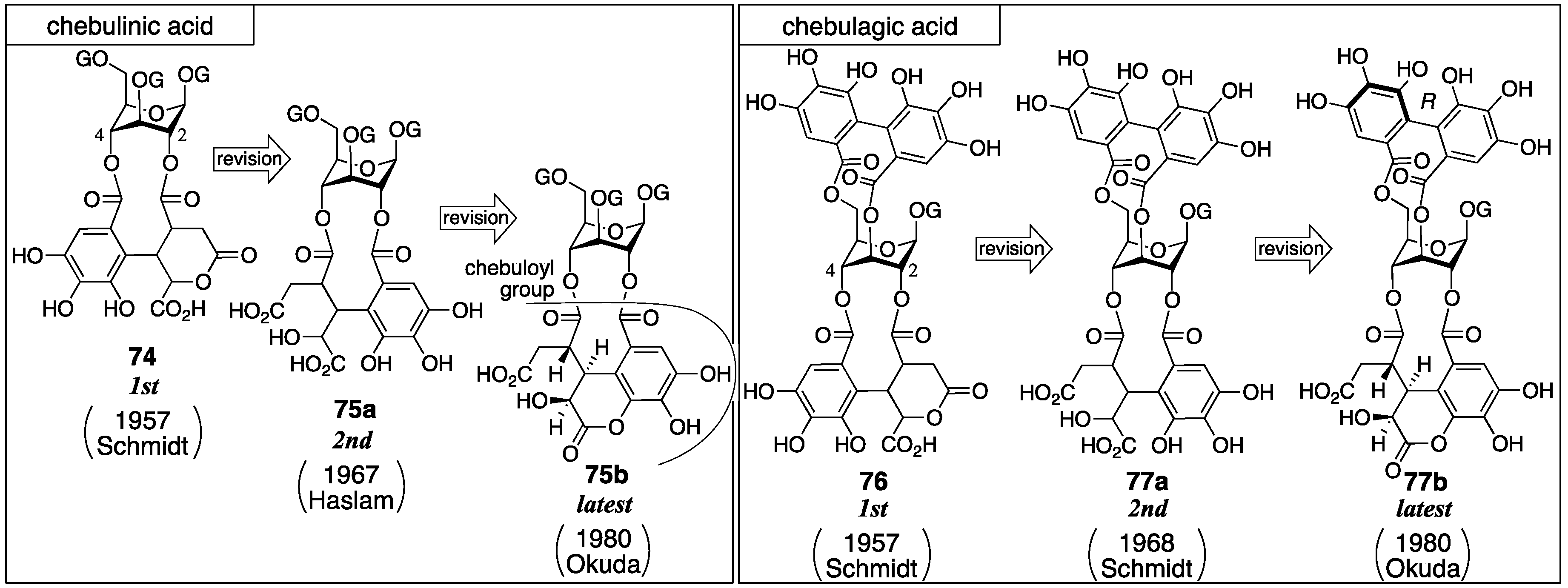
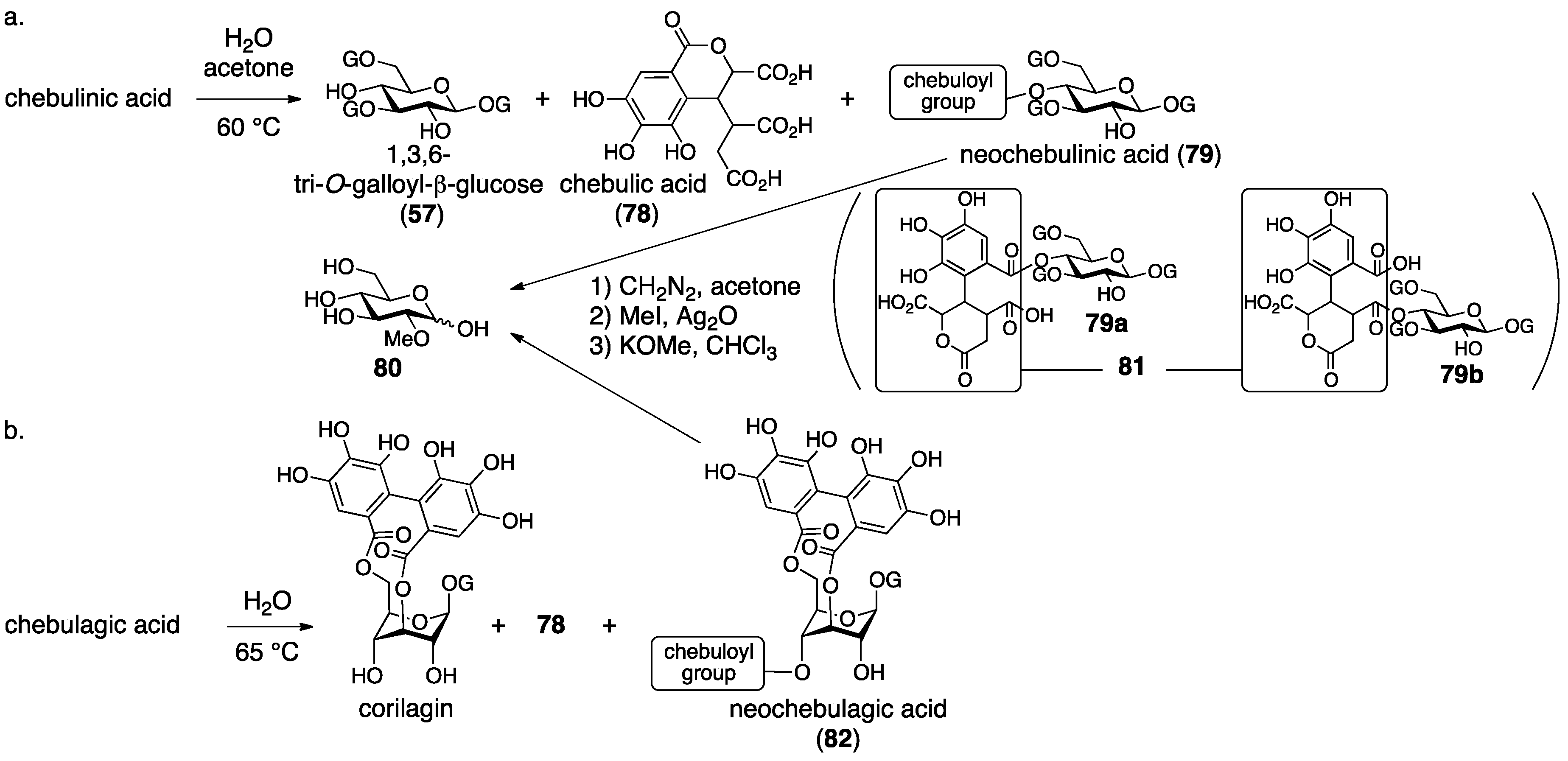
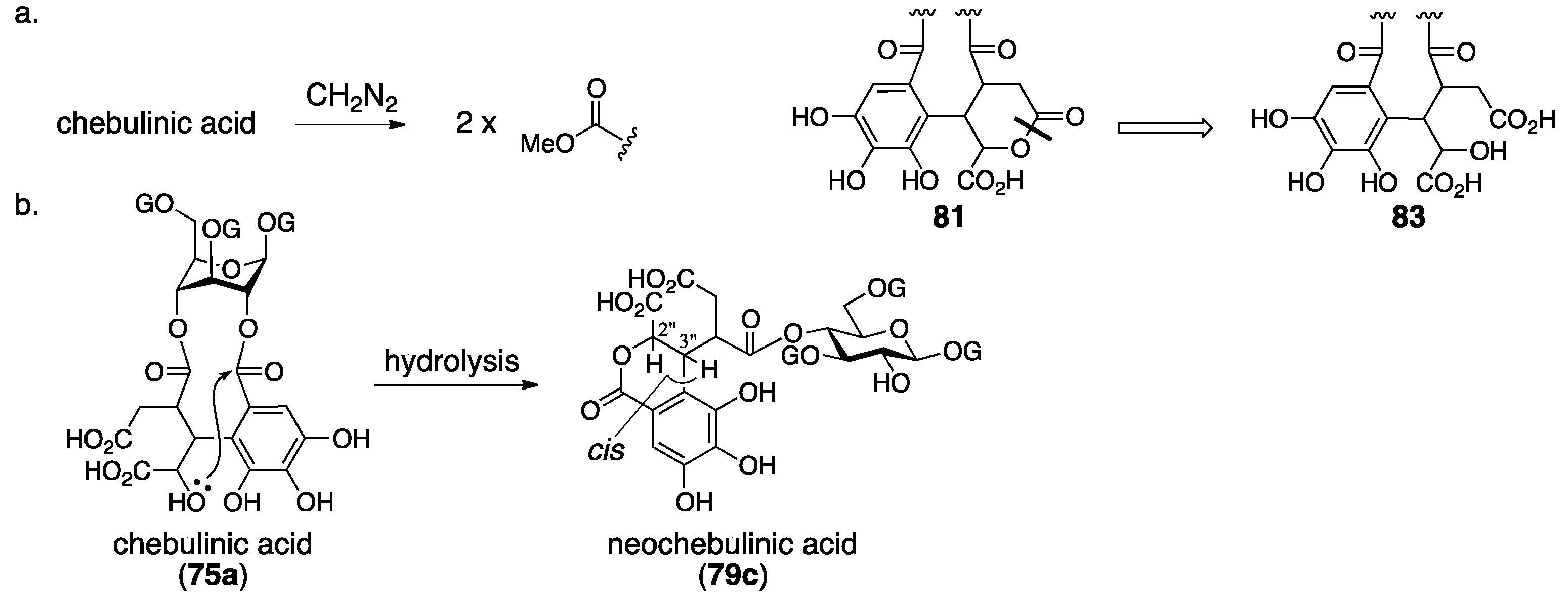
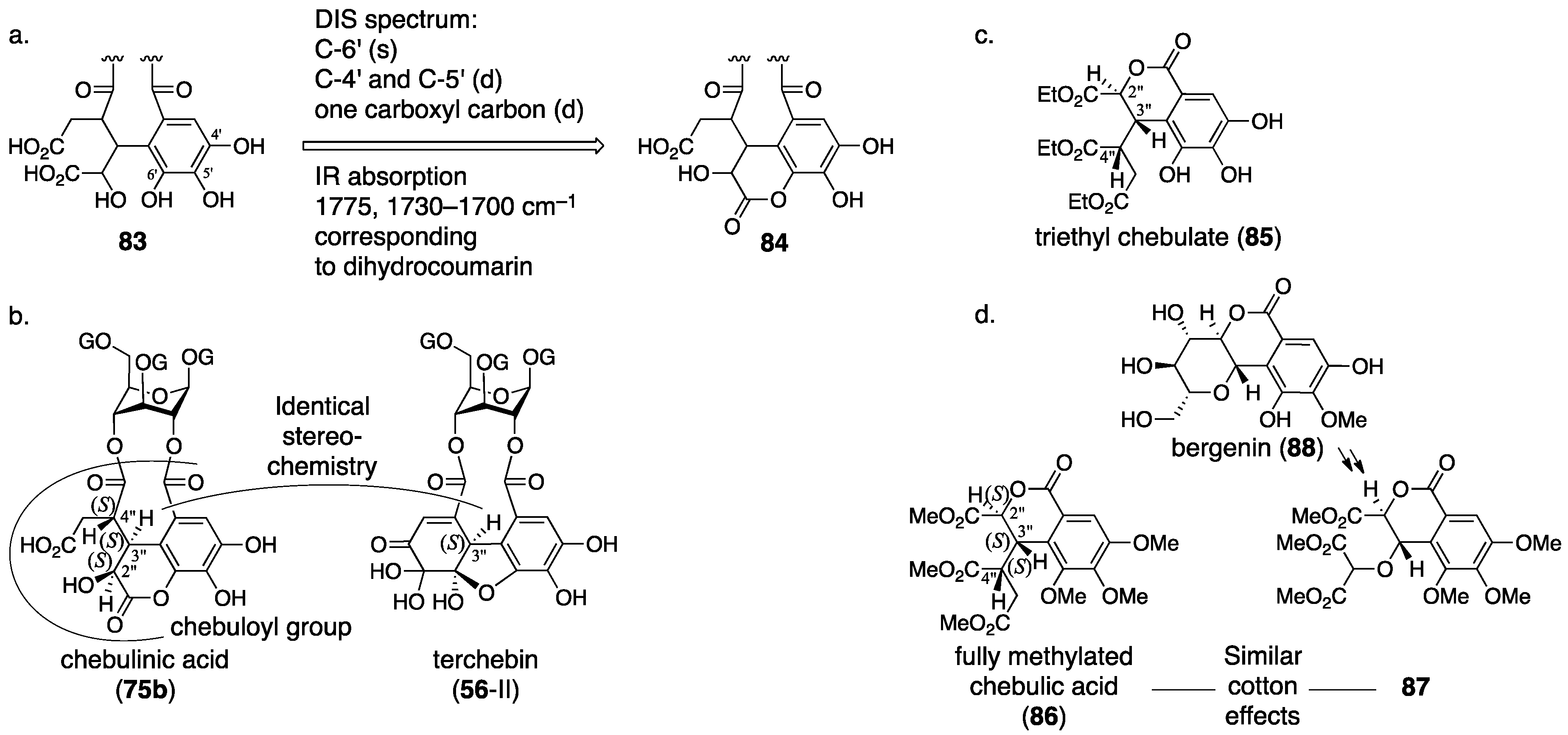
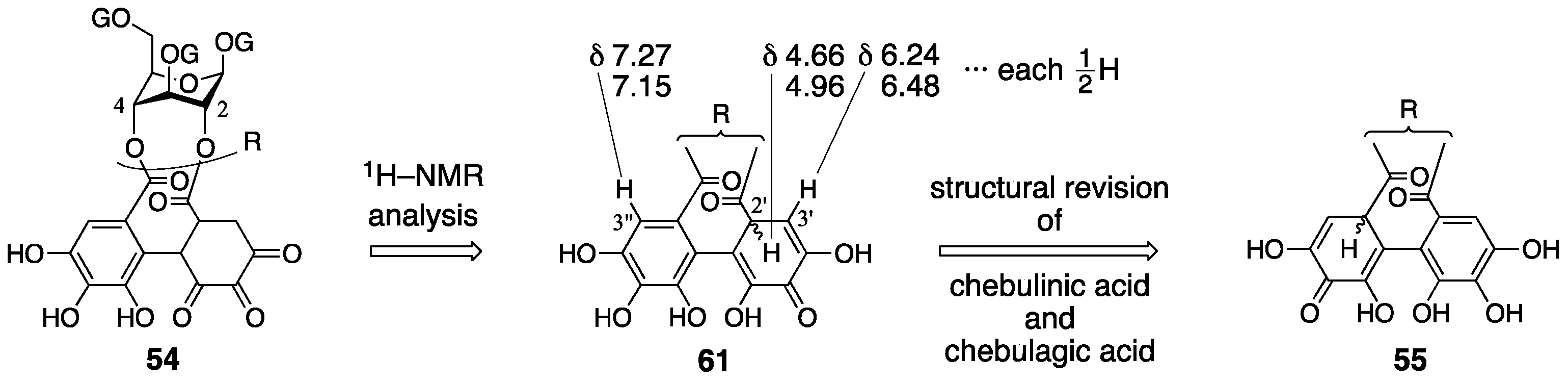
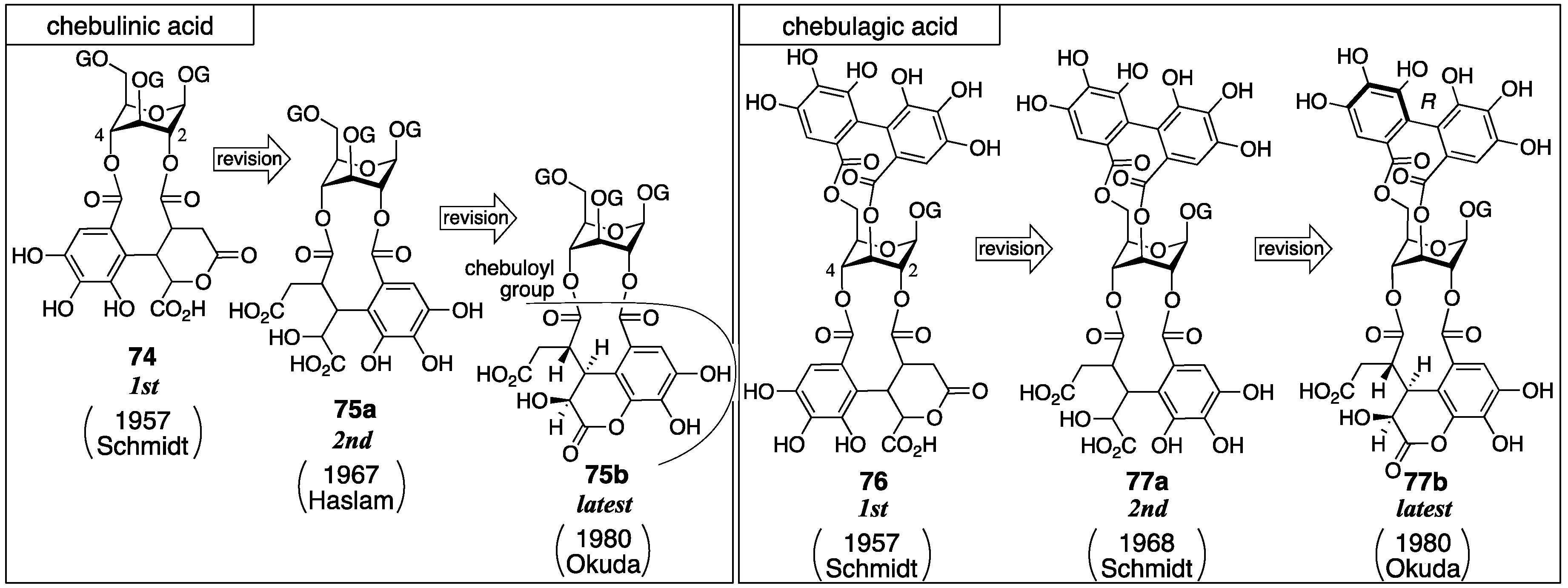
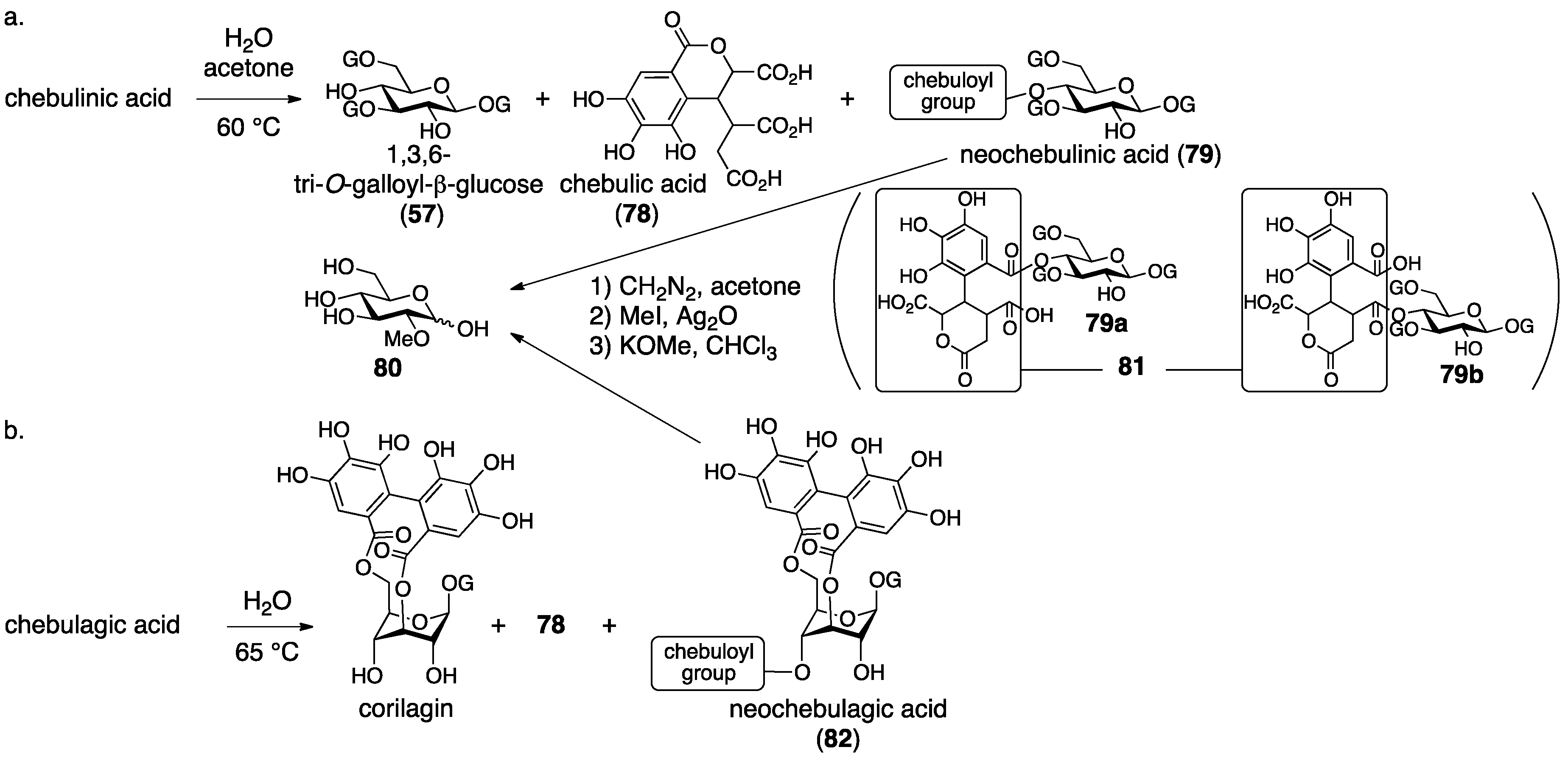
3.3. Correction Based on the Structure of the Chebuloyl Group: Chebulinic Acid and Chebulagic Acid
3.4. Compounds Containing a C–C-Connected Trimer and Tetramer of the Galloyl Group
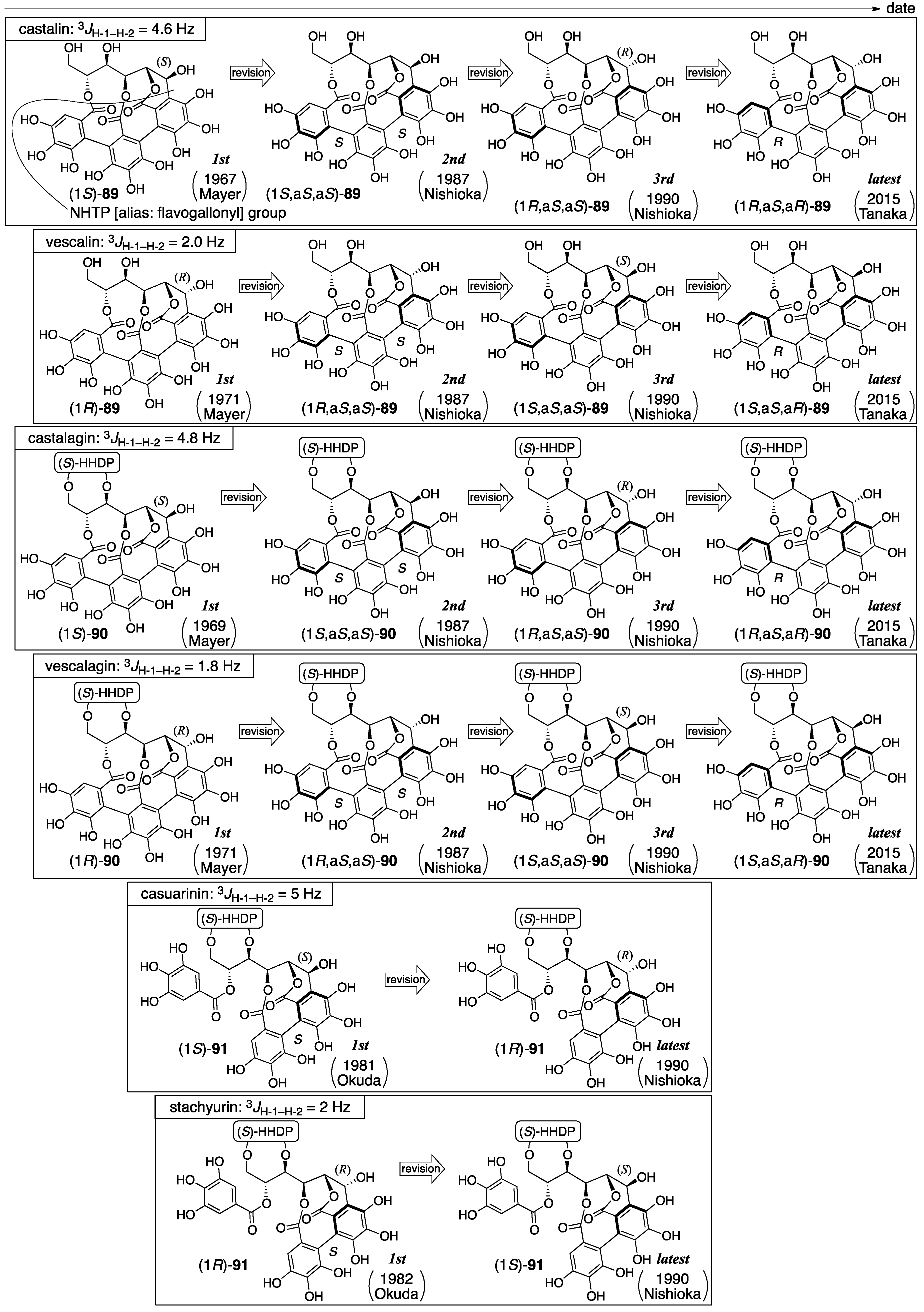
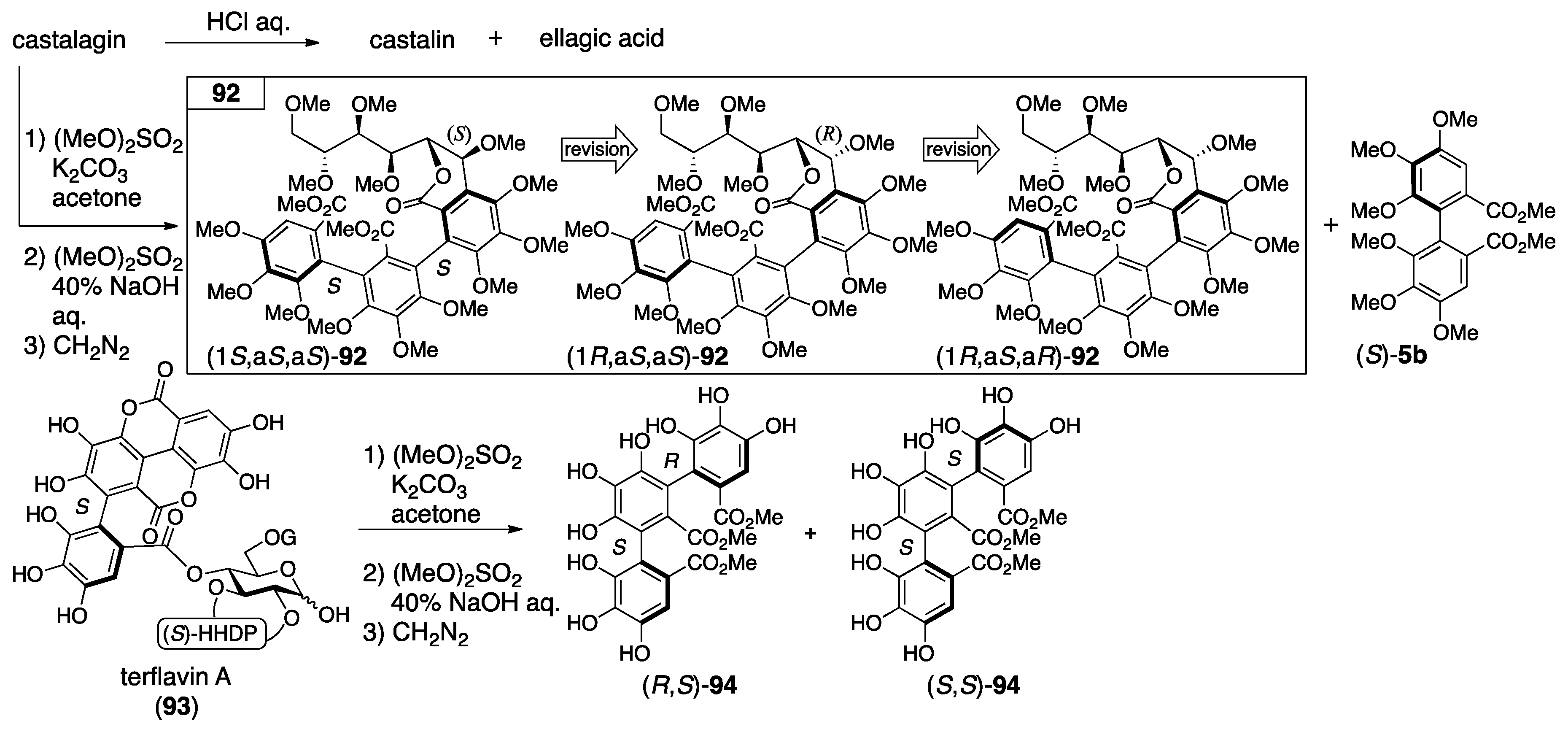
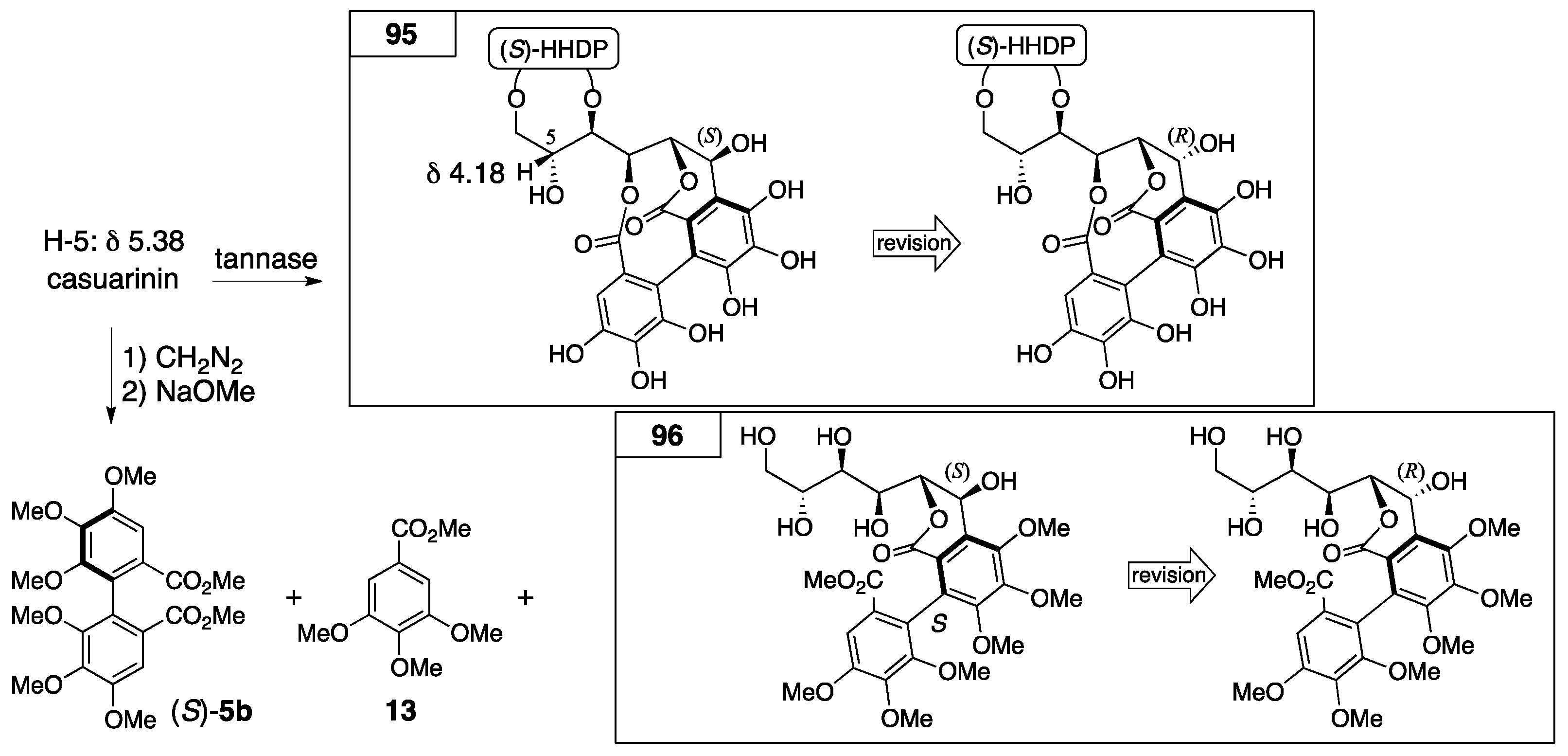
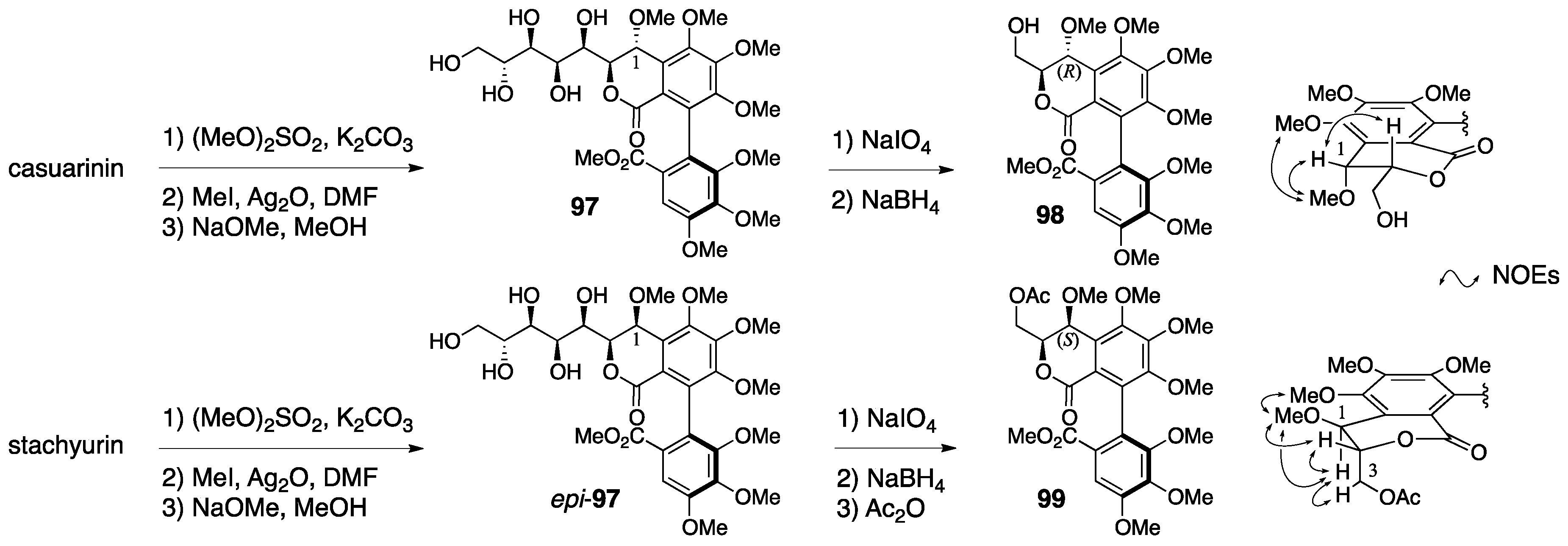
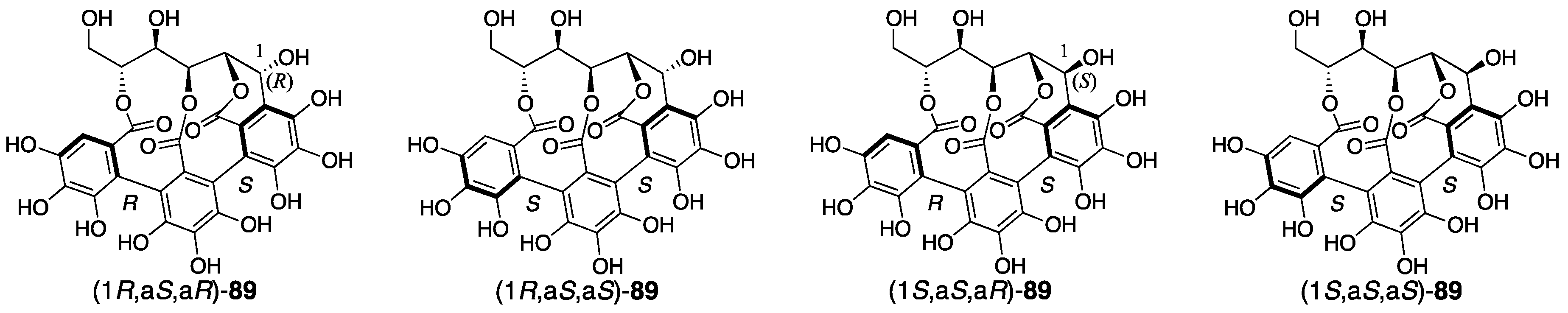
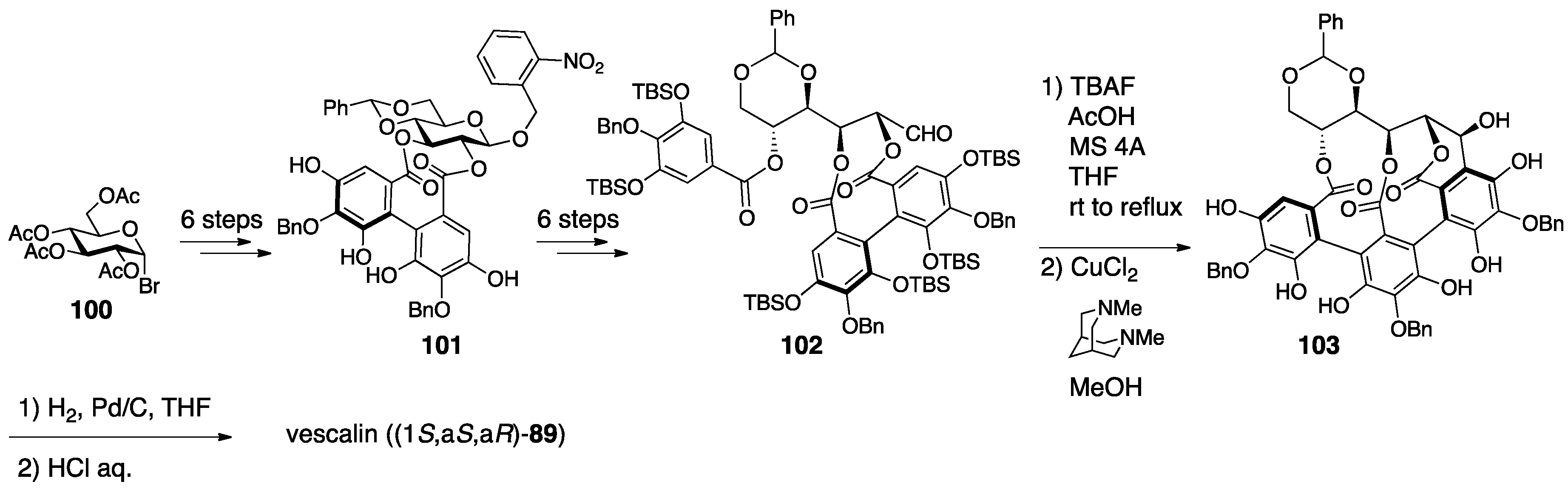
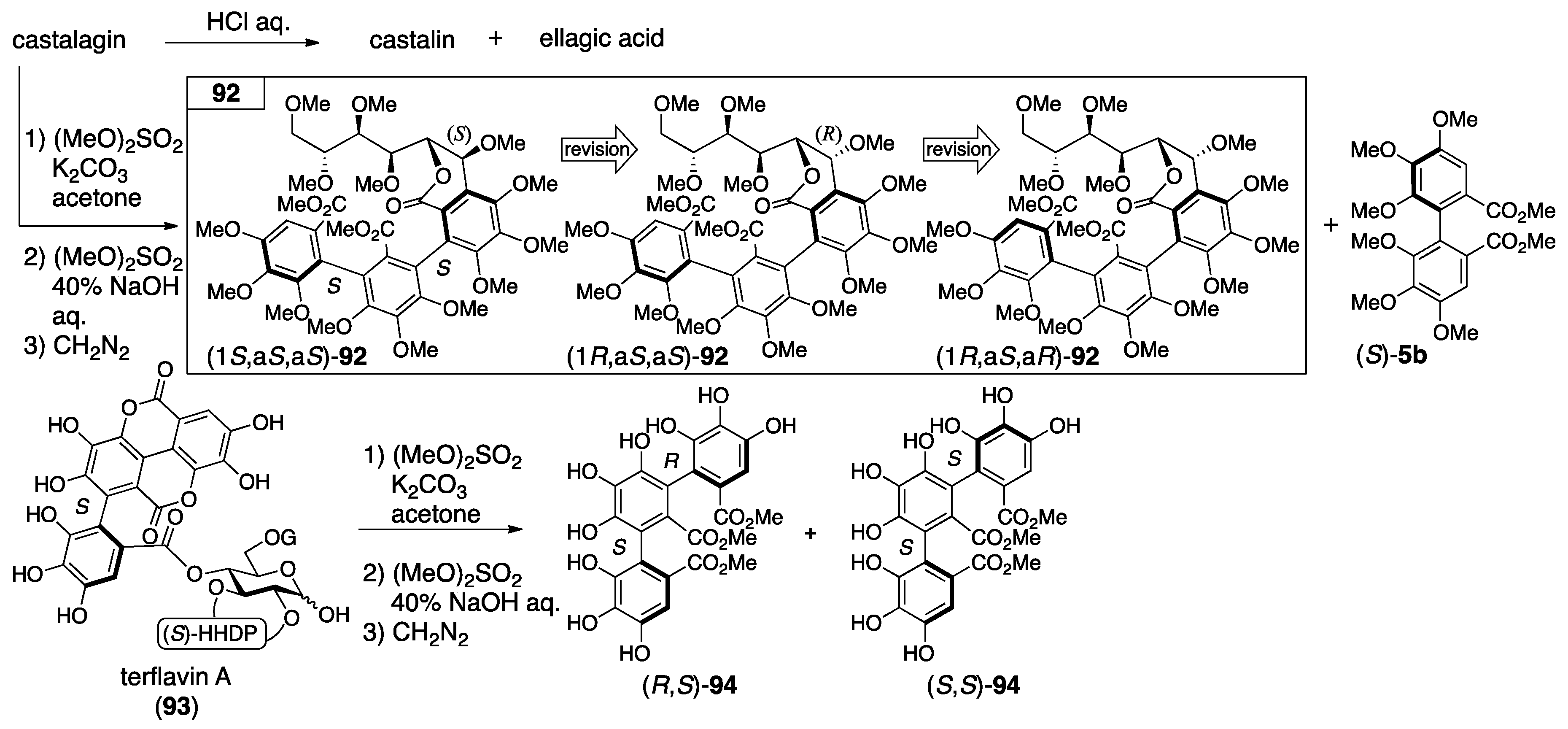
3.4.1. Castalin, Vescalin, Castalagin, Vescalagin, Casuarinin, and Stachyurin
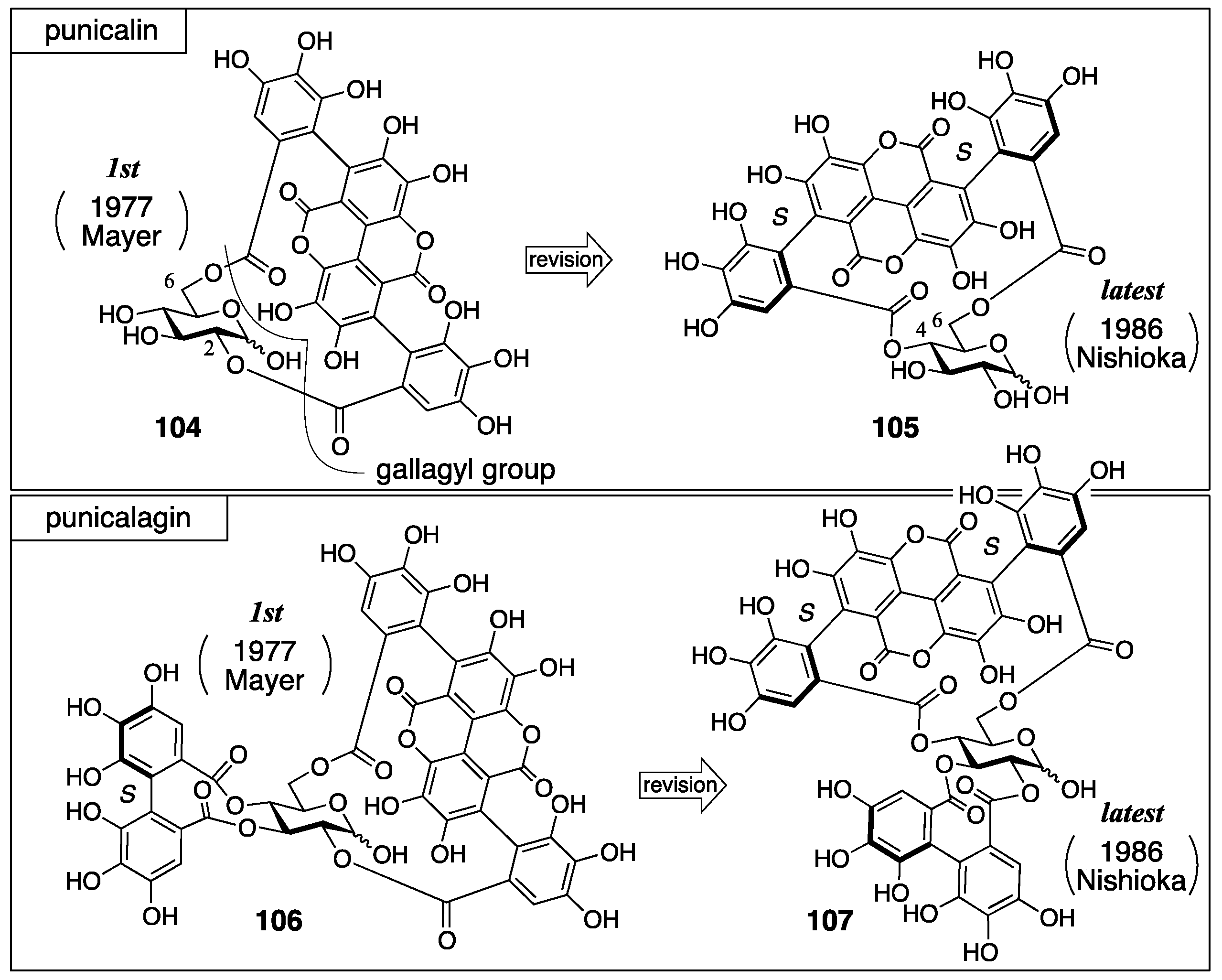
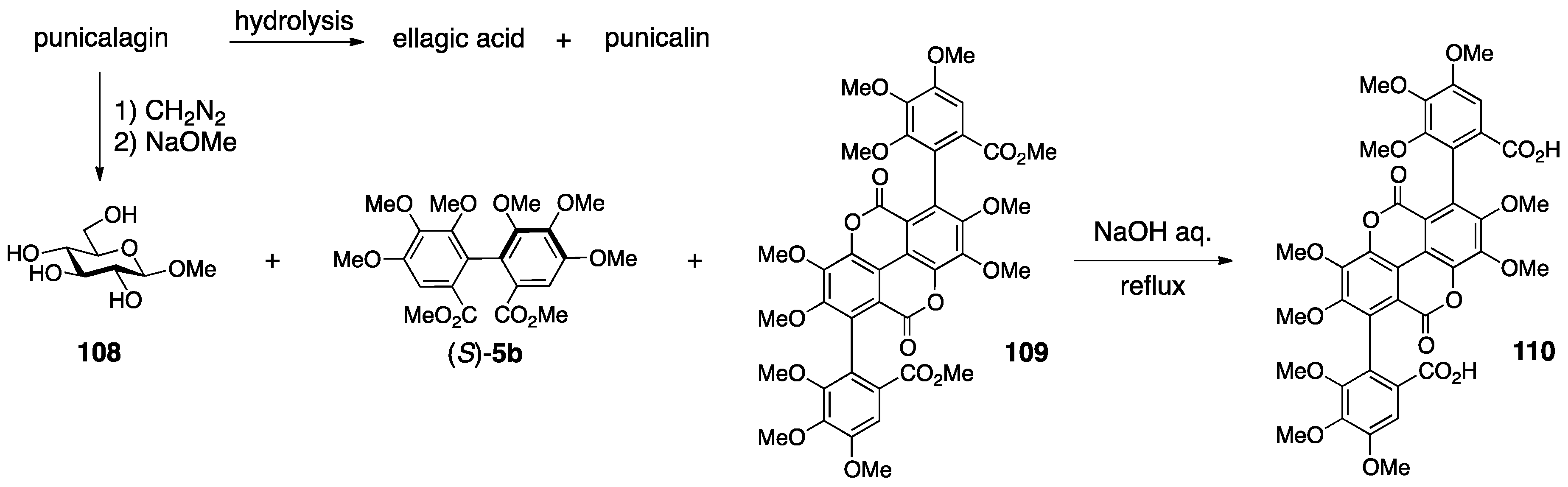
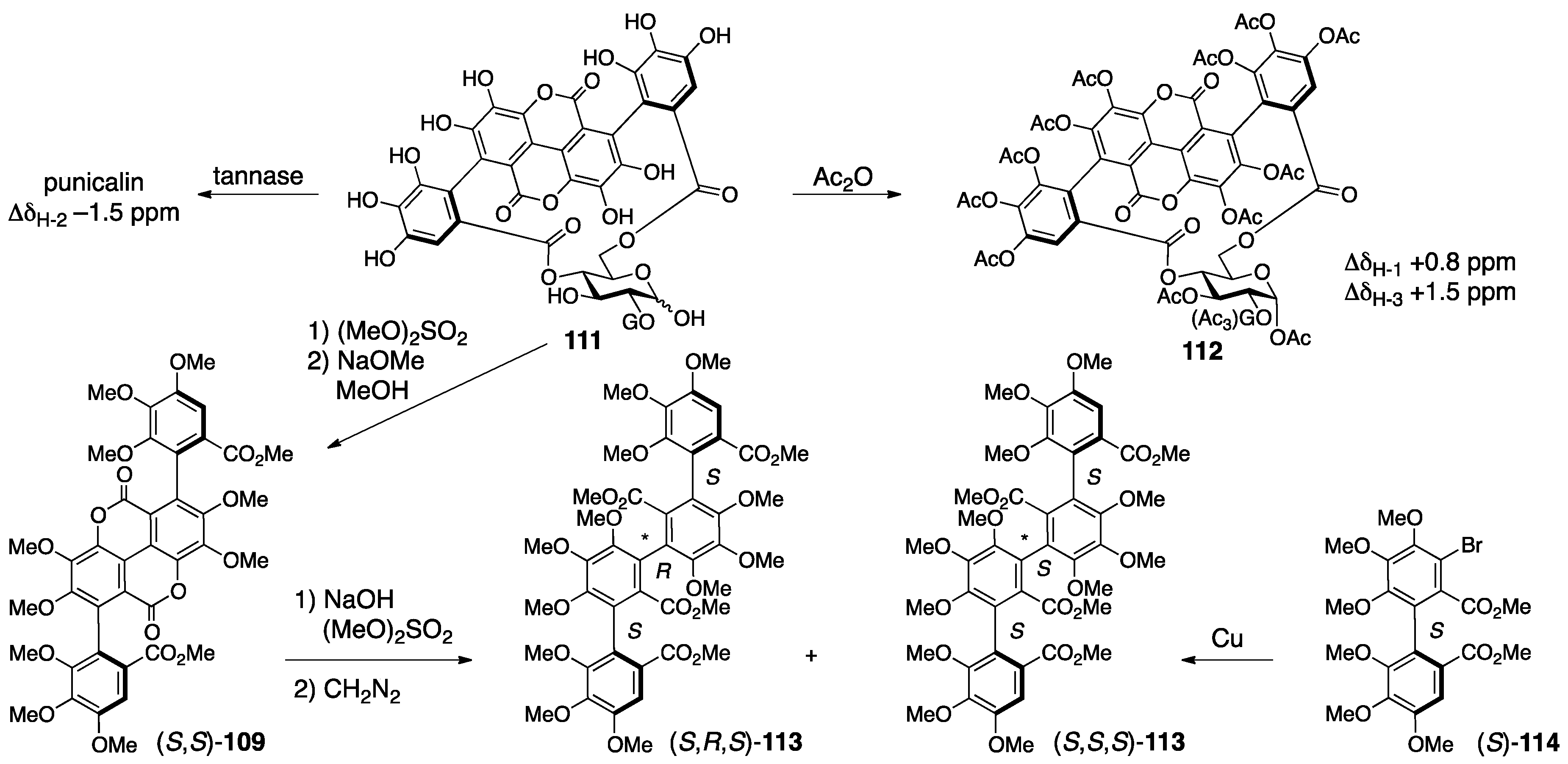
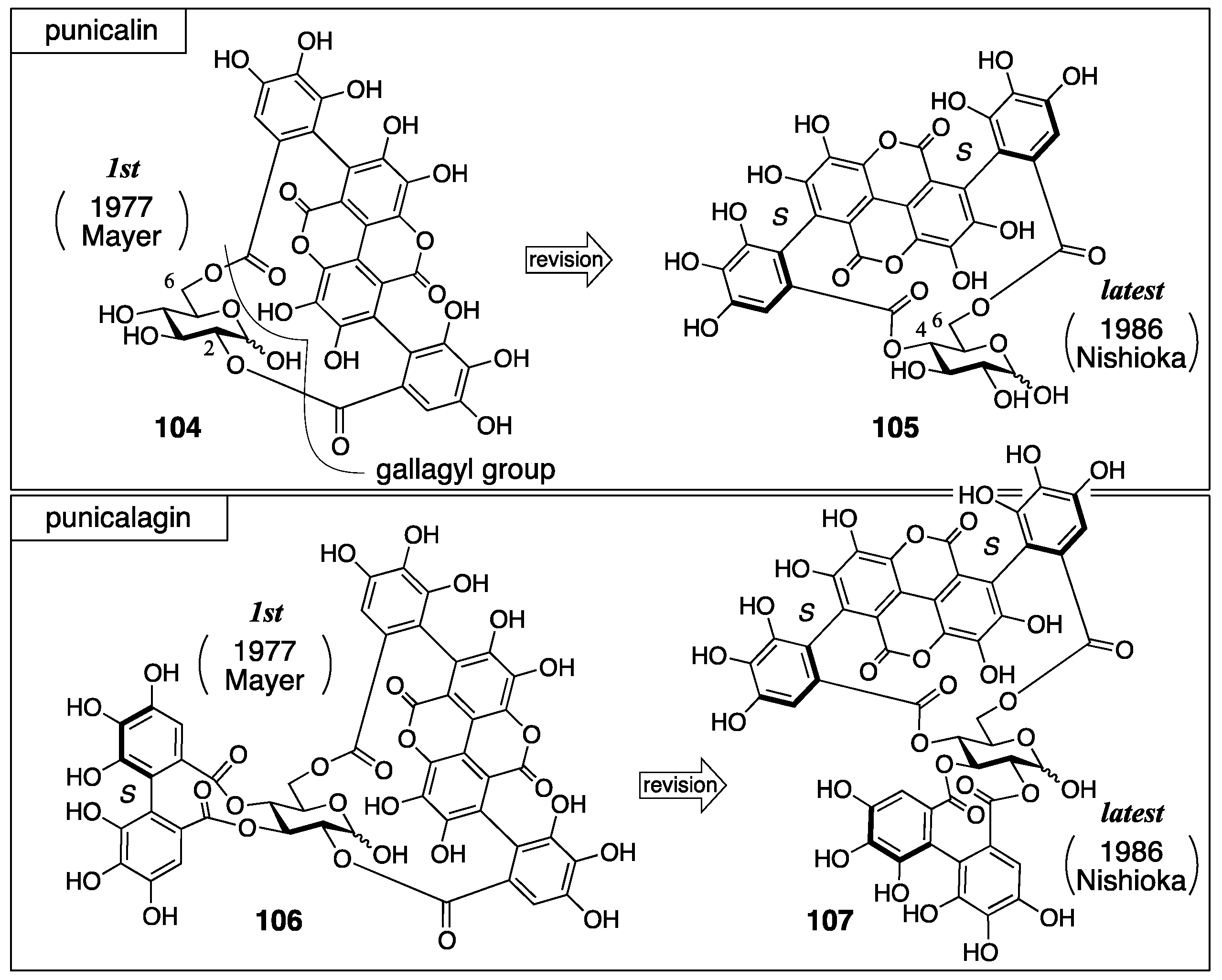
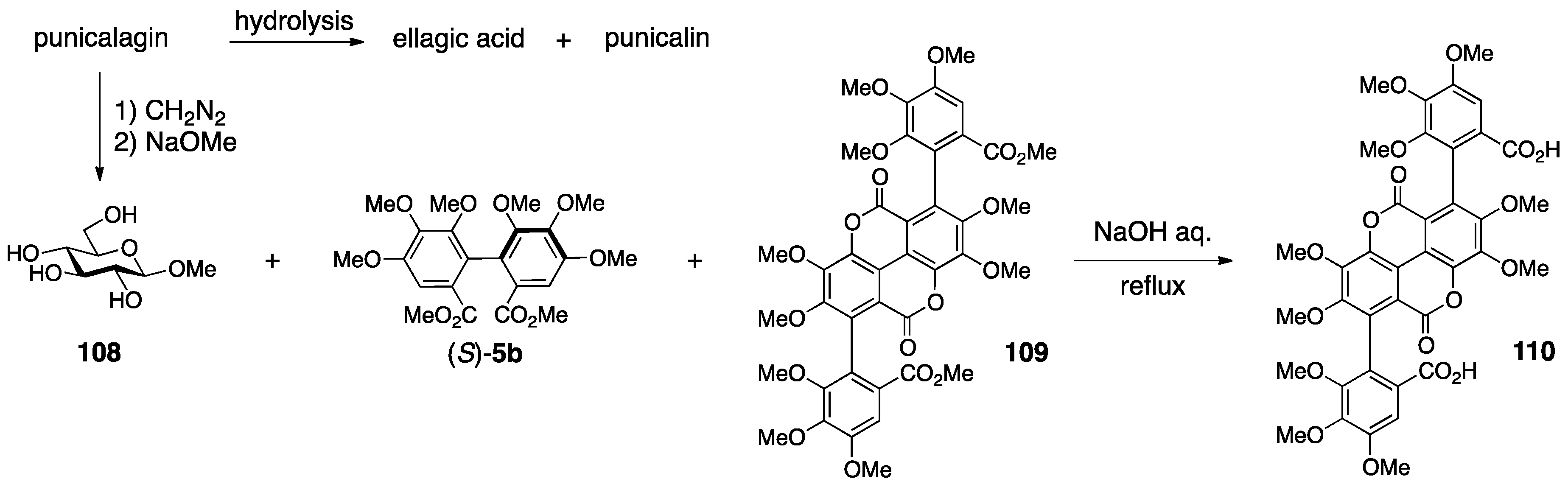
3.4.2. Punicalin and Punicalagin
3.5. Correction Based on the Bonding Position of C–O-Connected Components
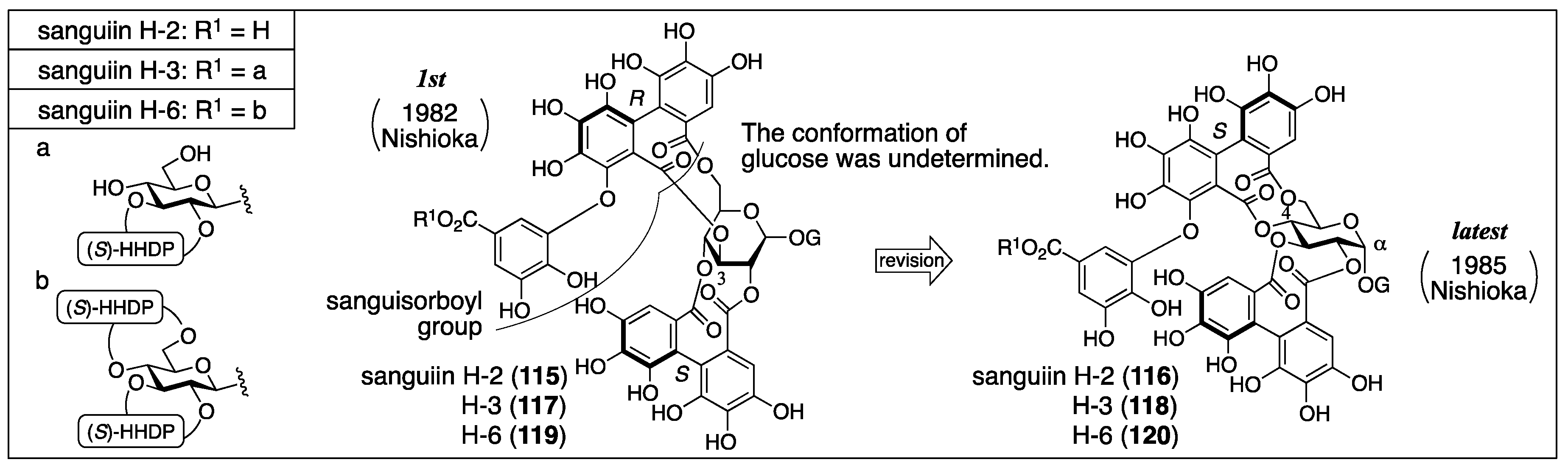
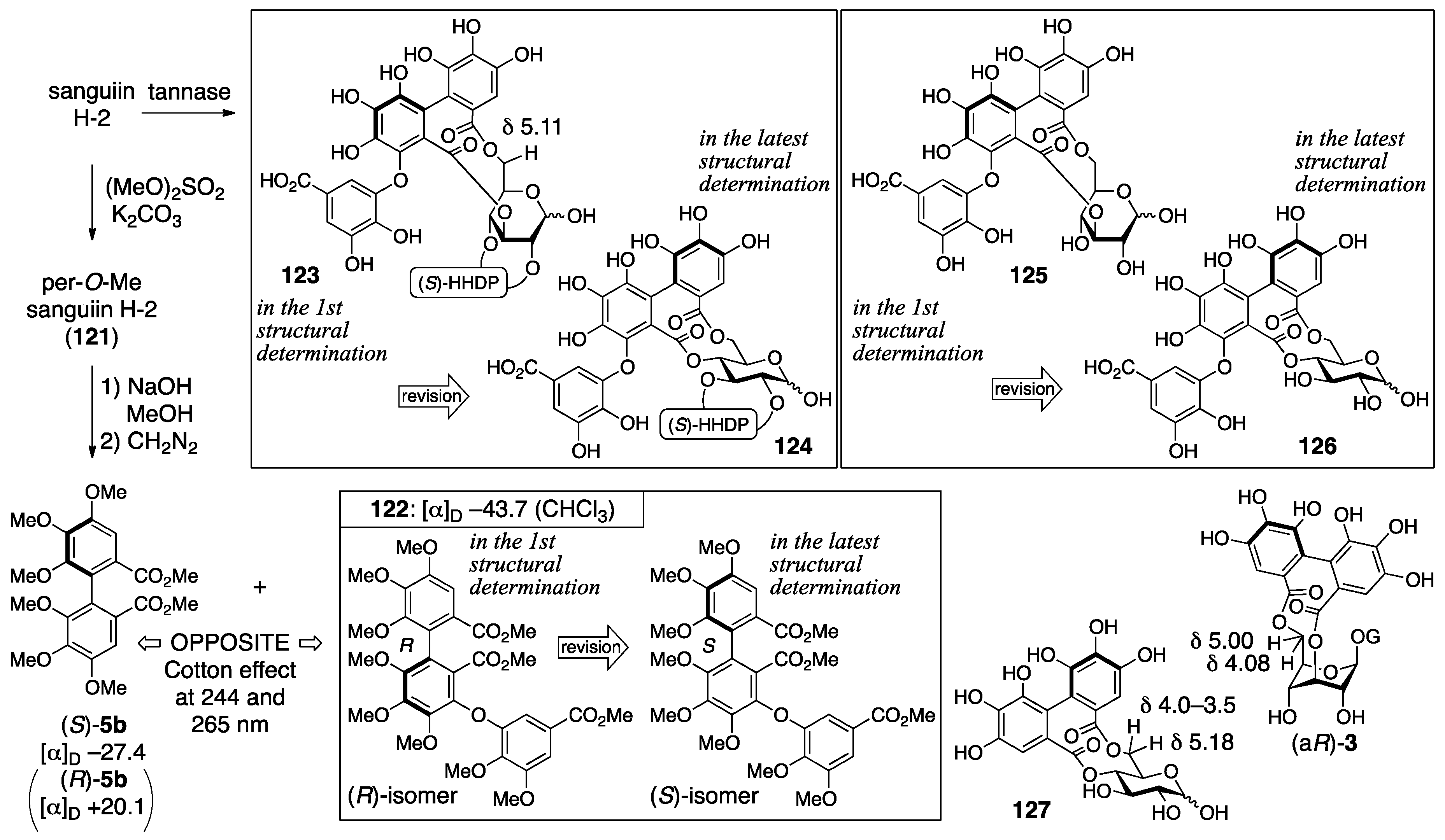
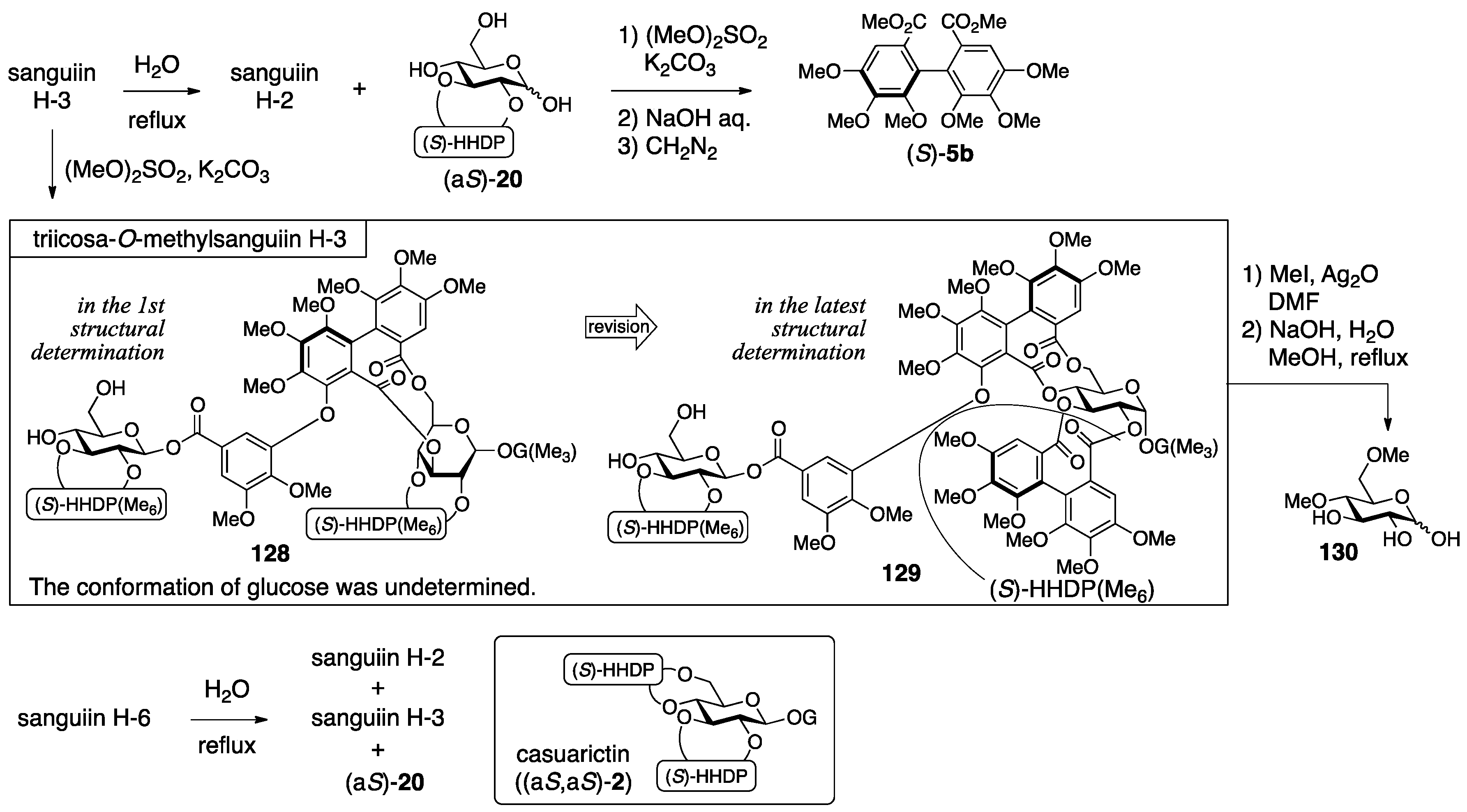
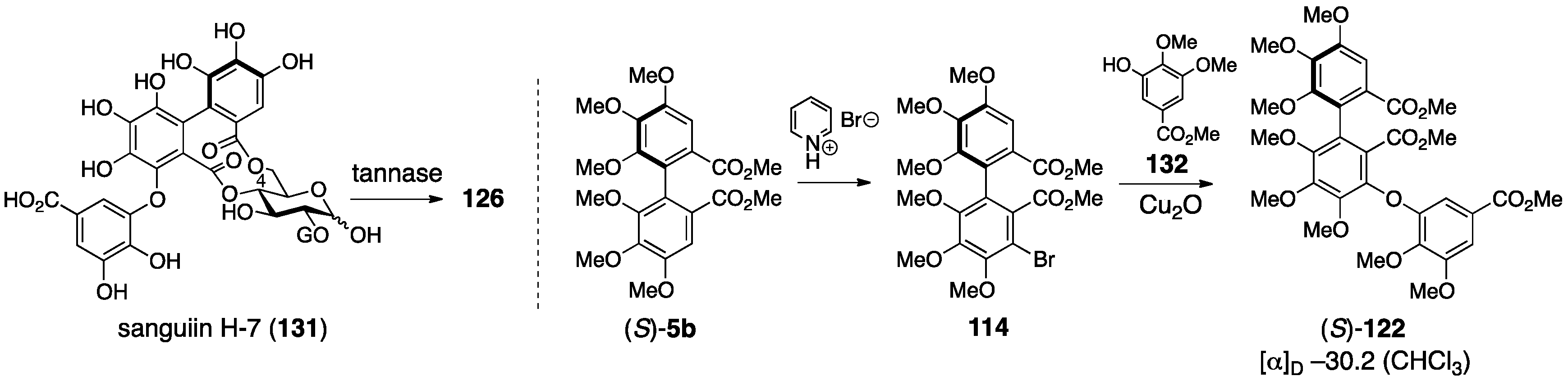
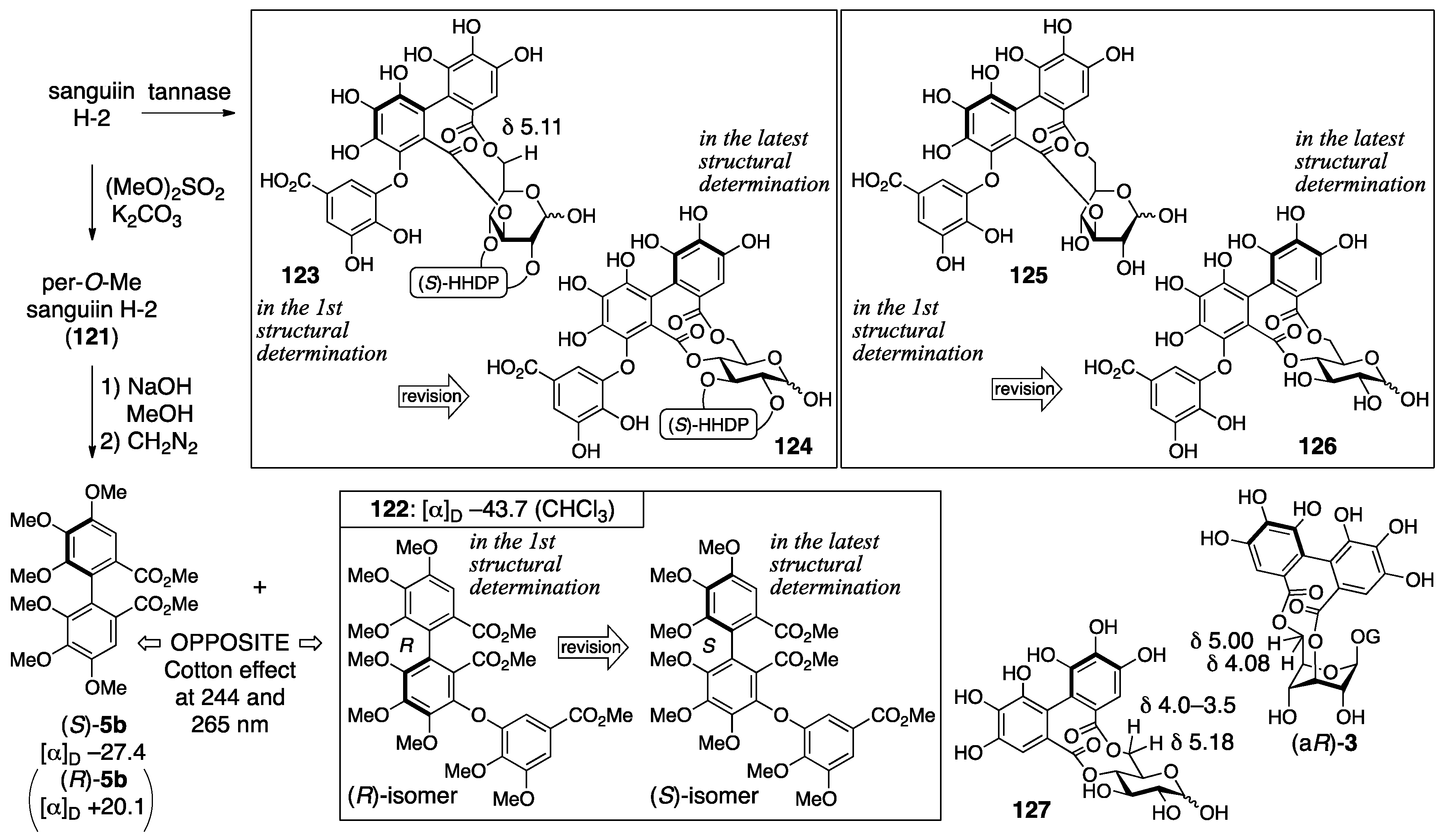
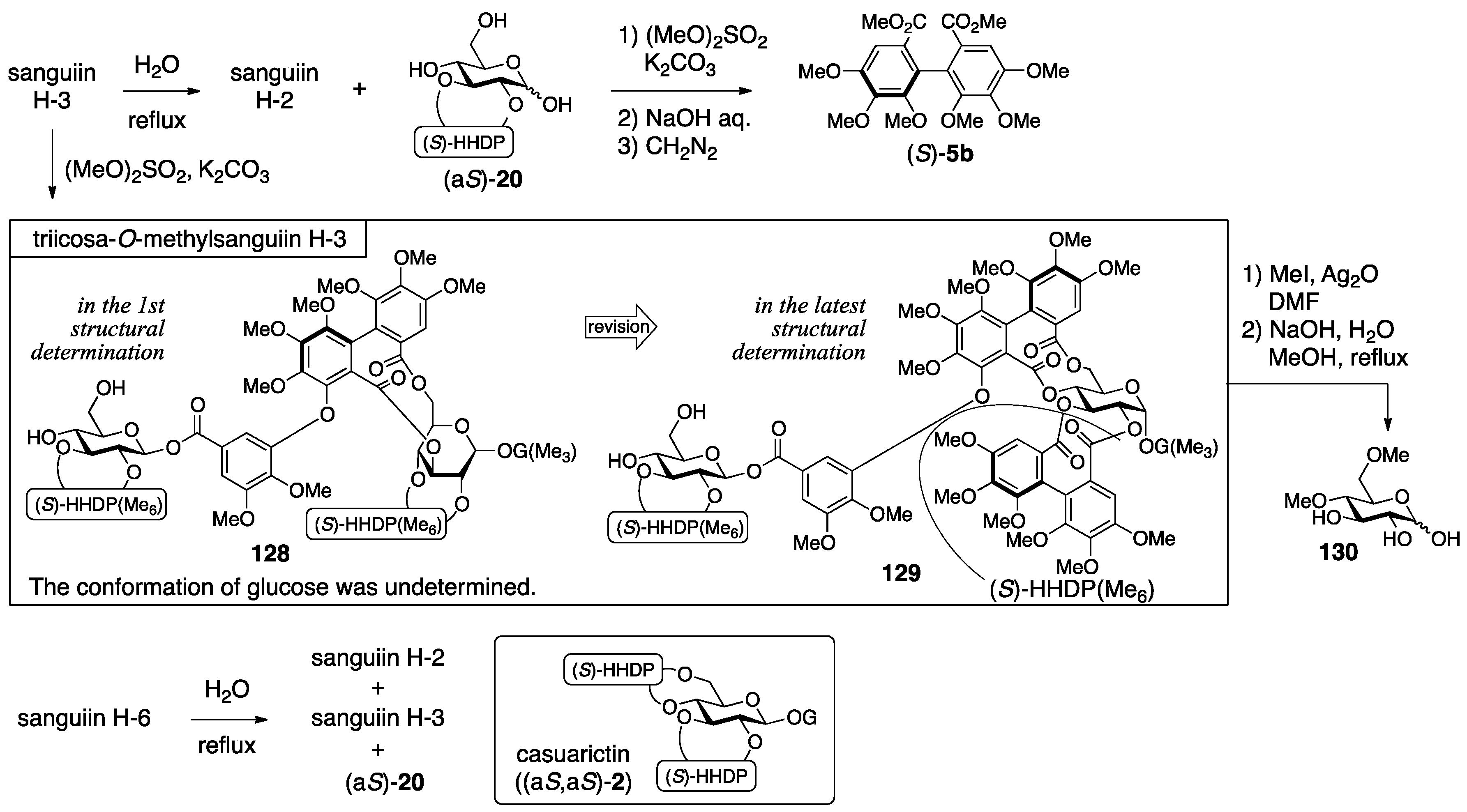
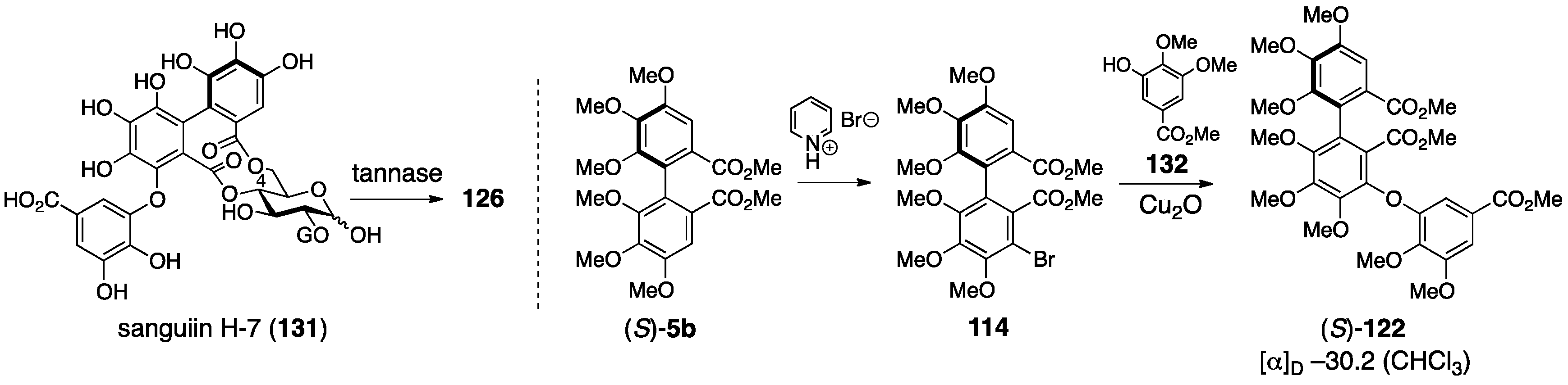
3.5.1. Sanguiin H-2, H-3, and H-6
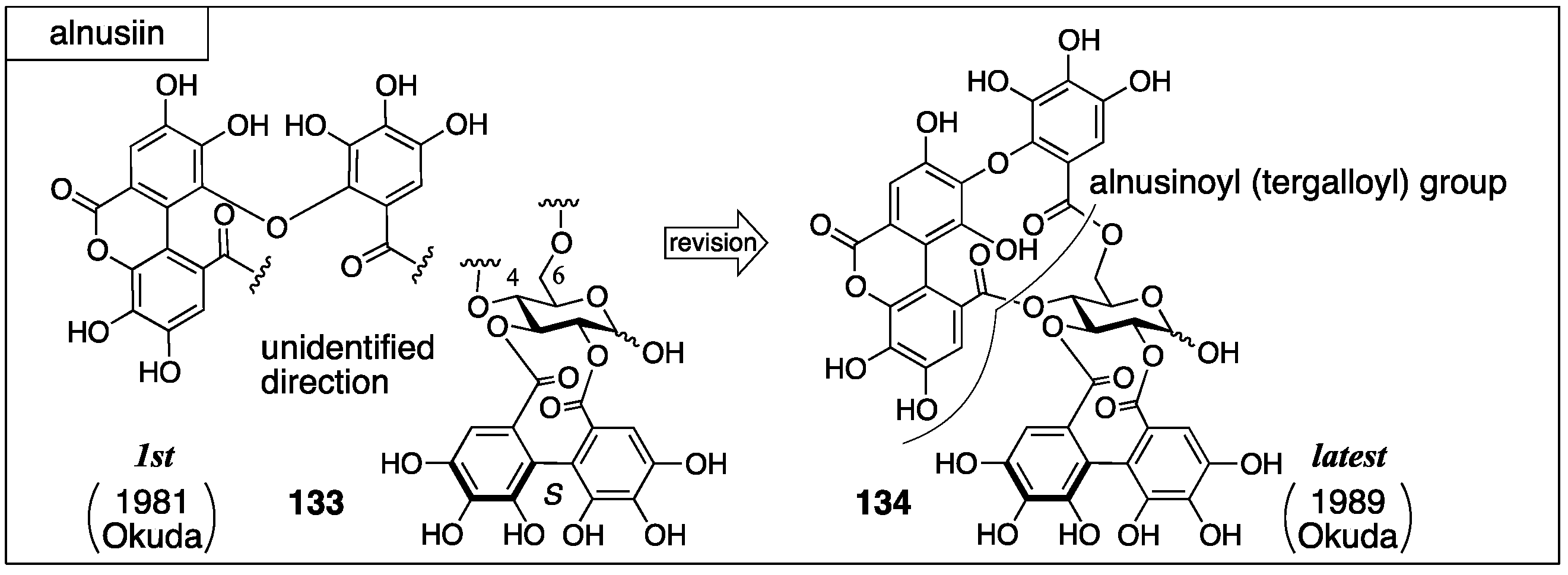
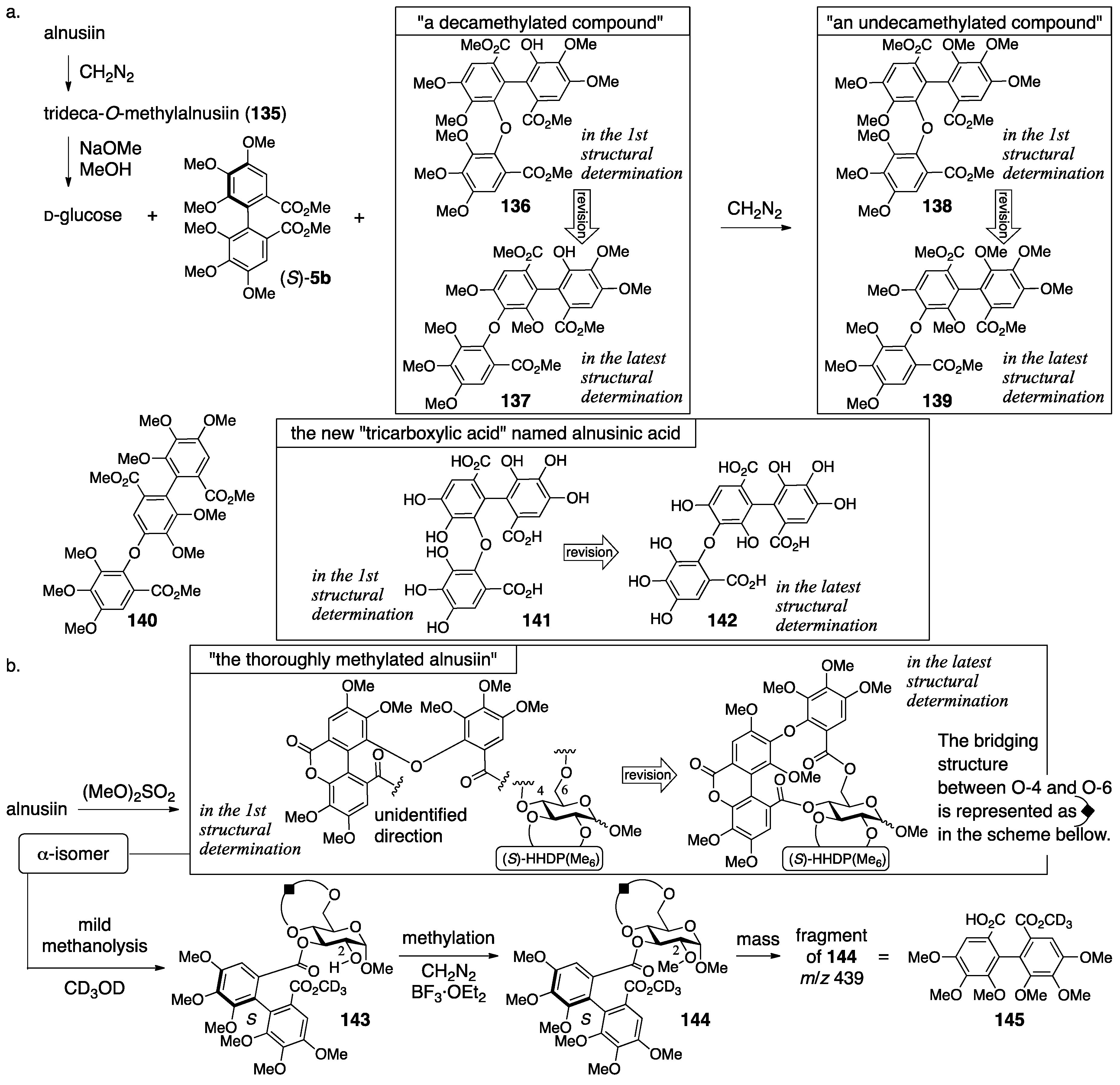
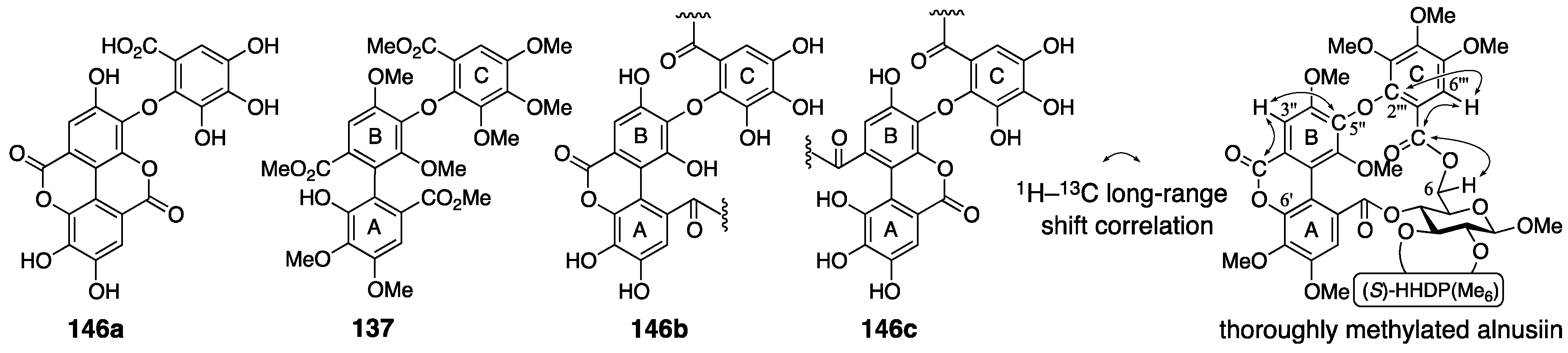
3.5.2. Alnusiin
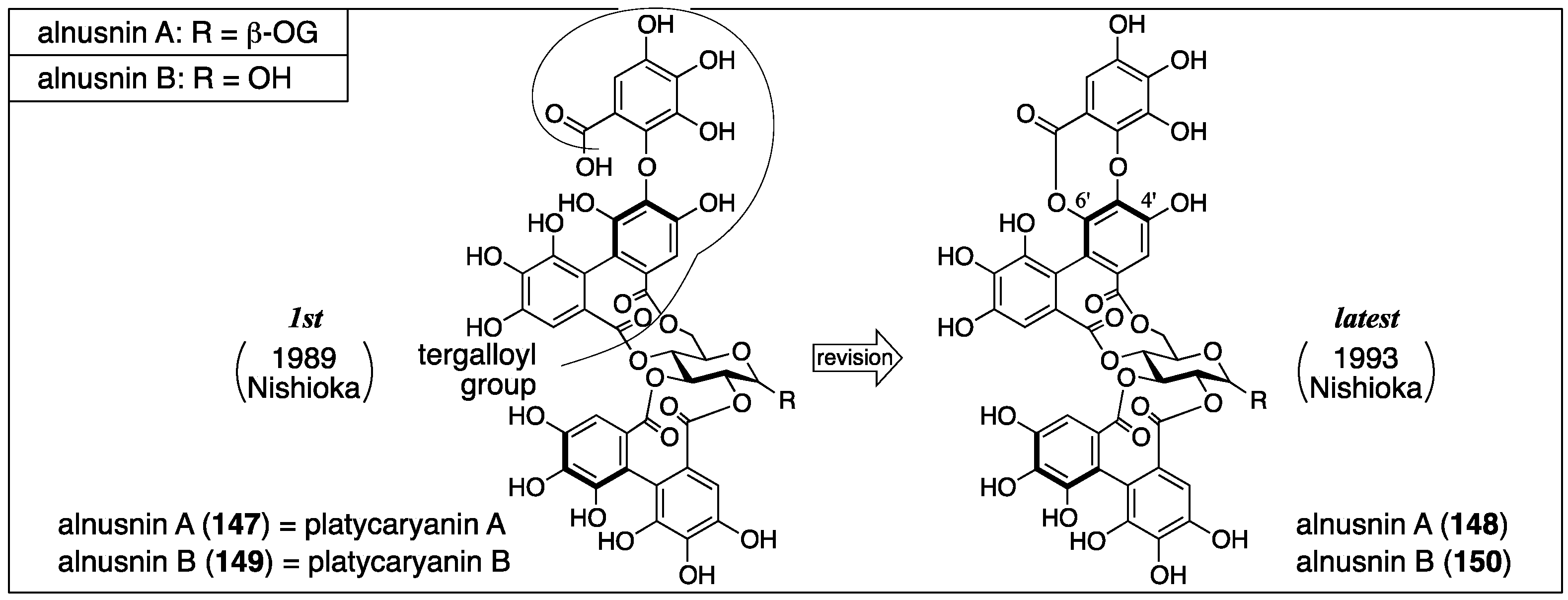
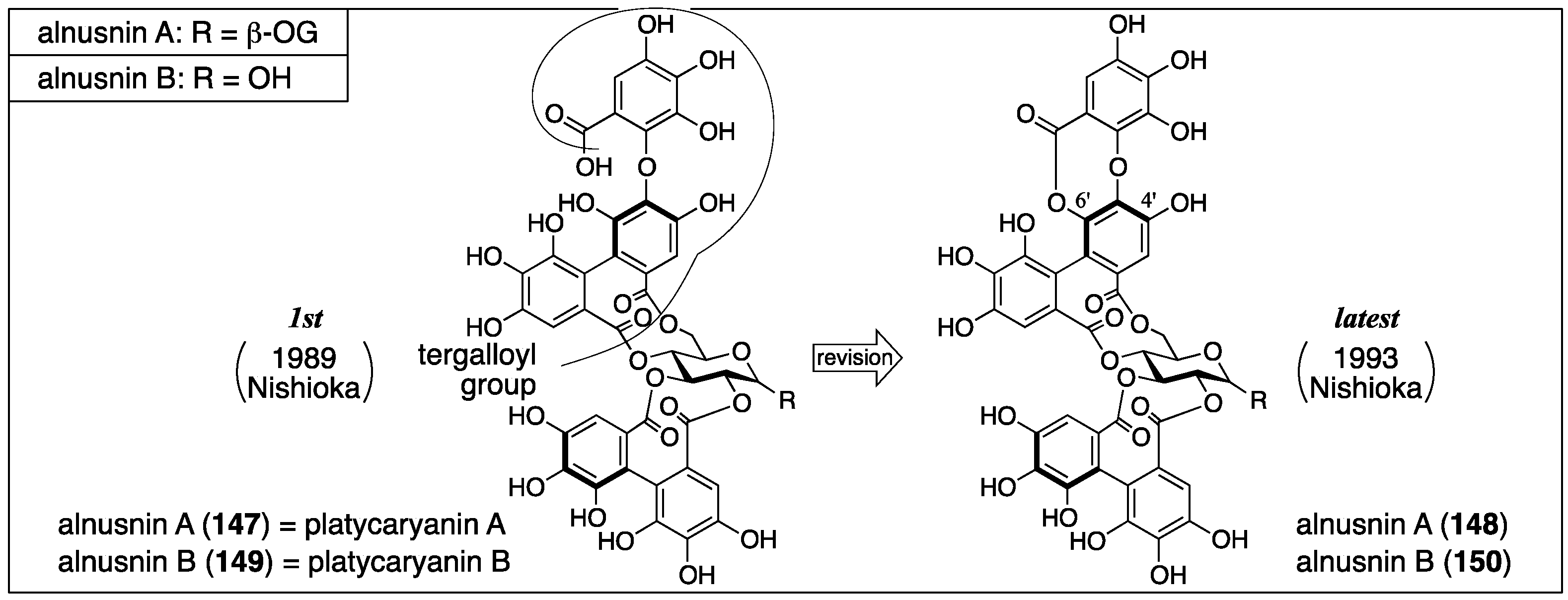
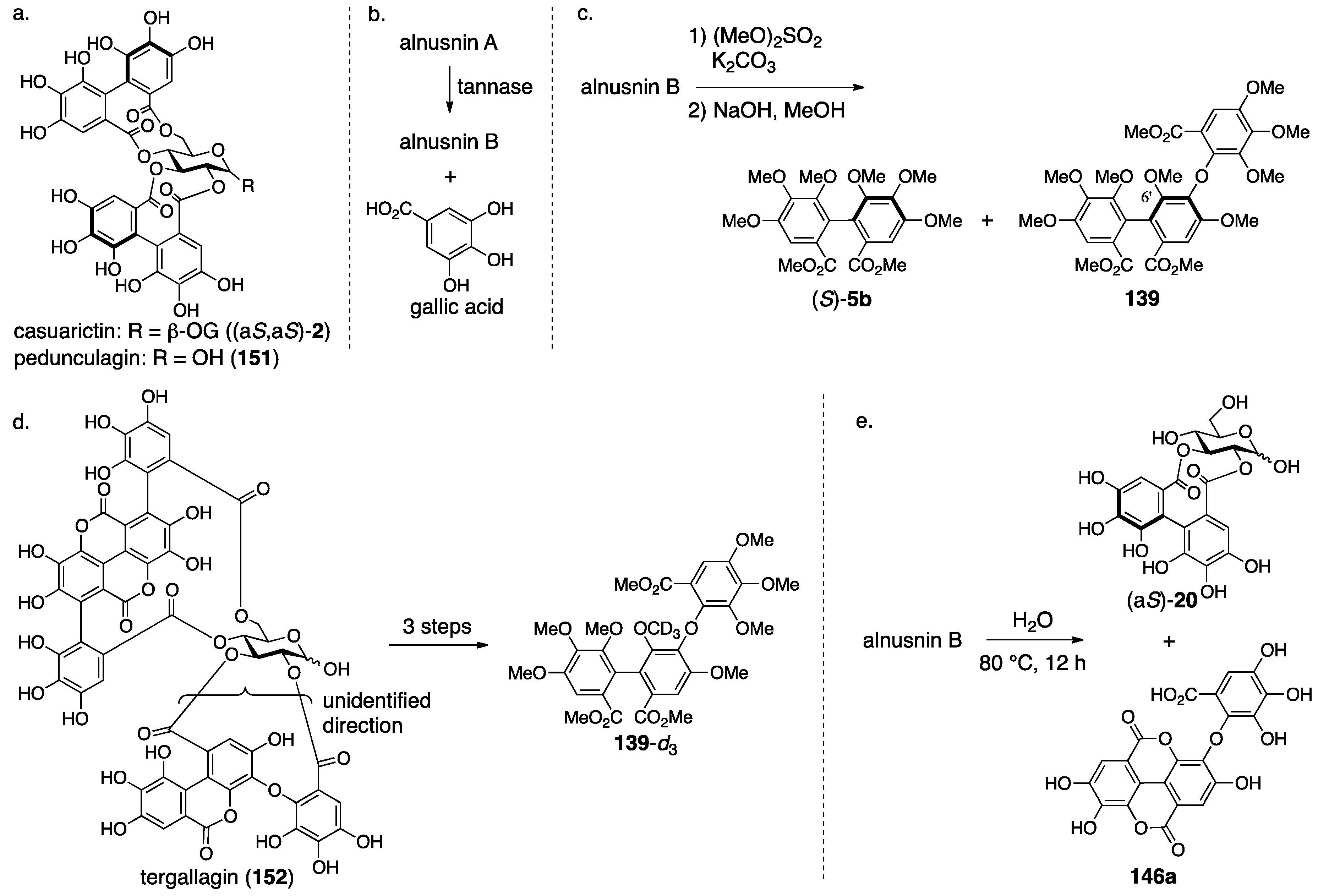
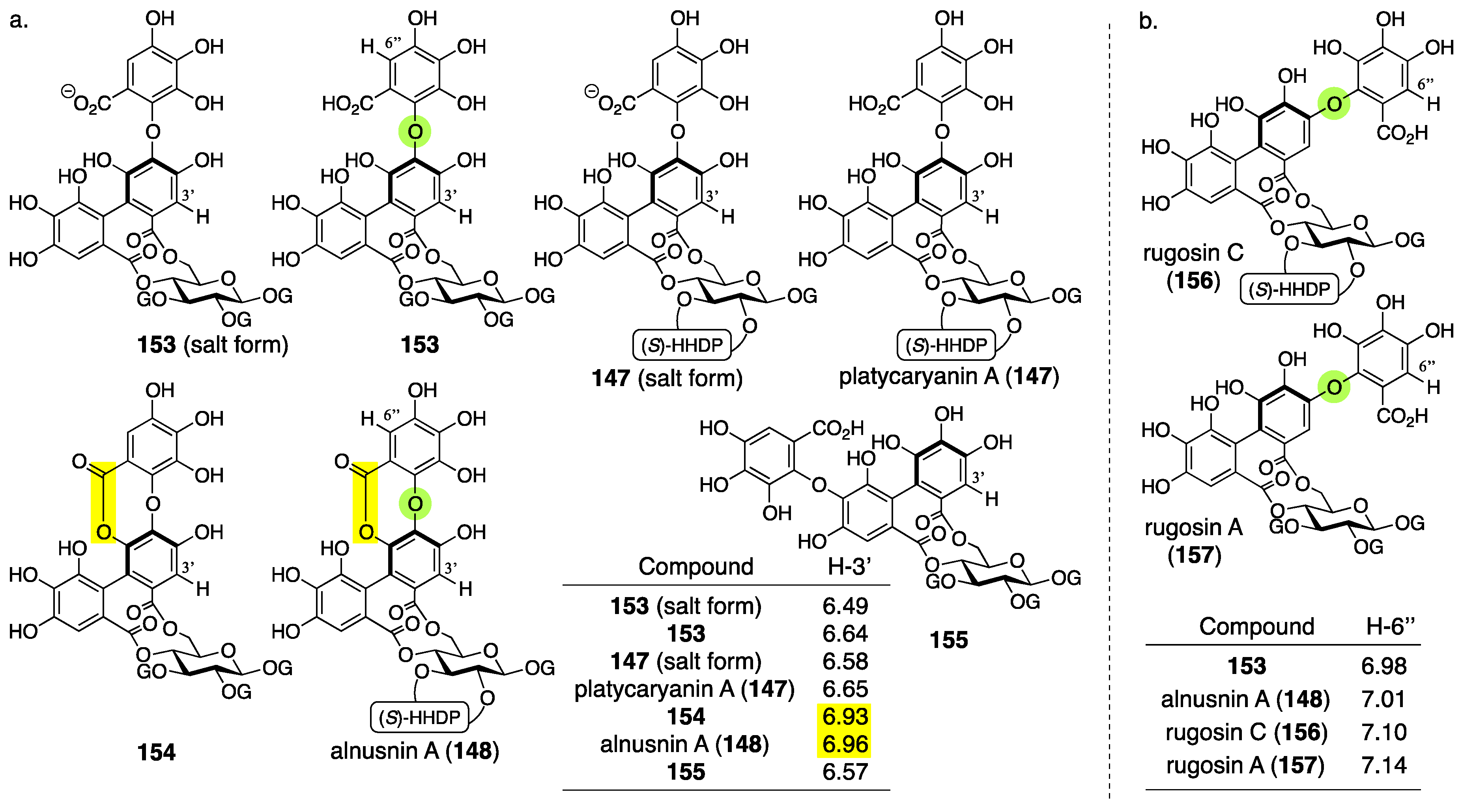
3.5.3. Alnusnin A and B
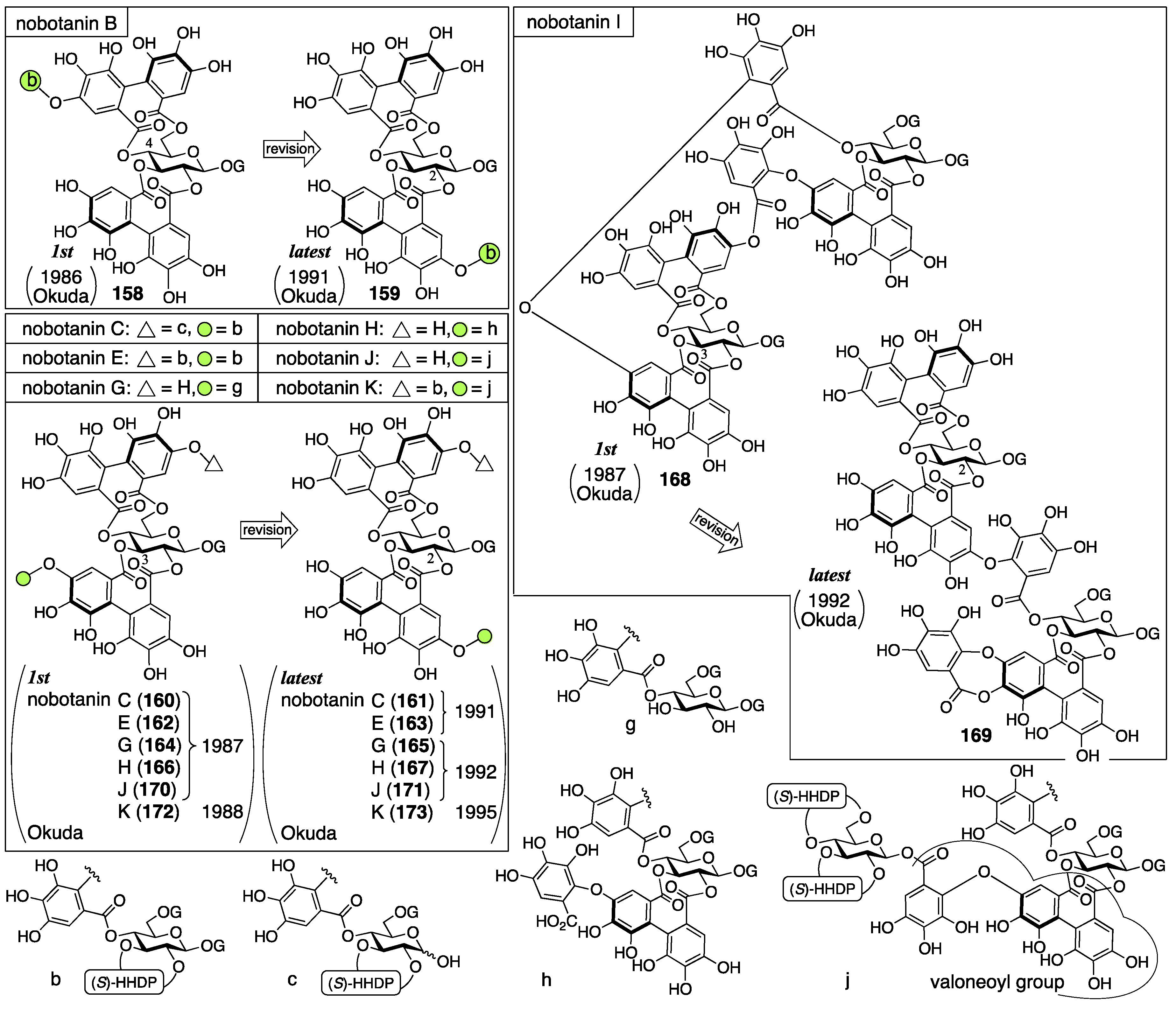
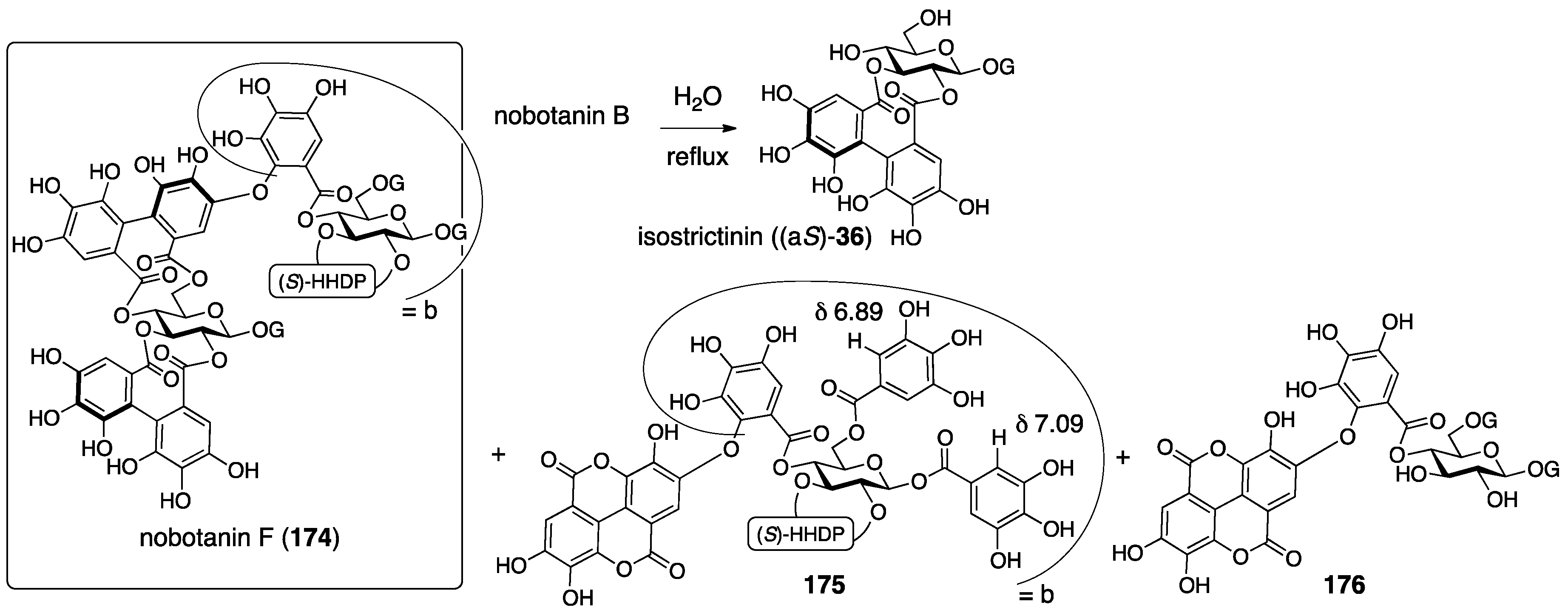
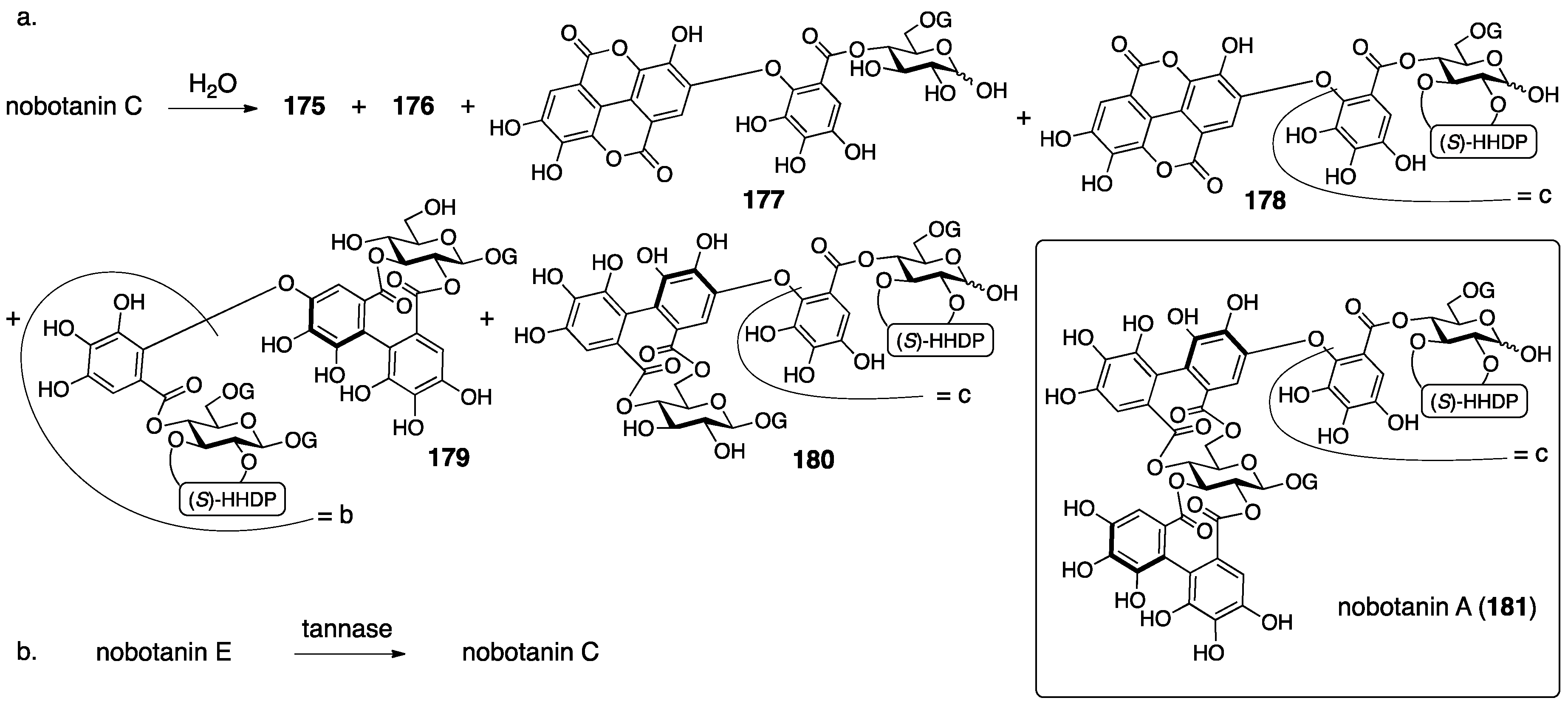
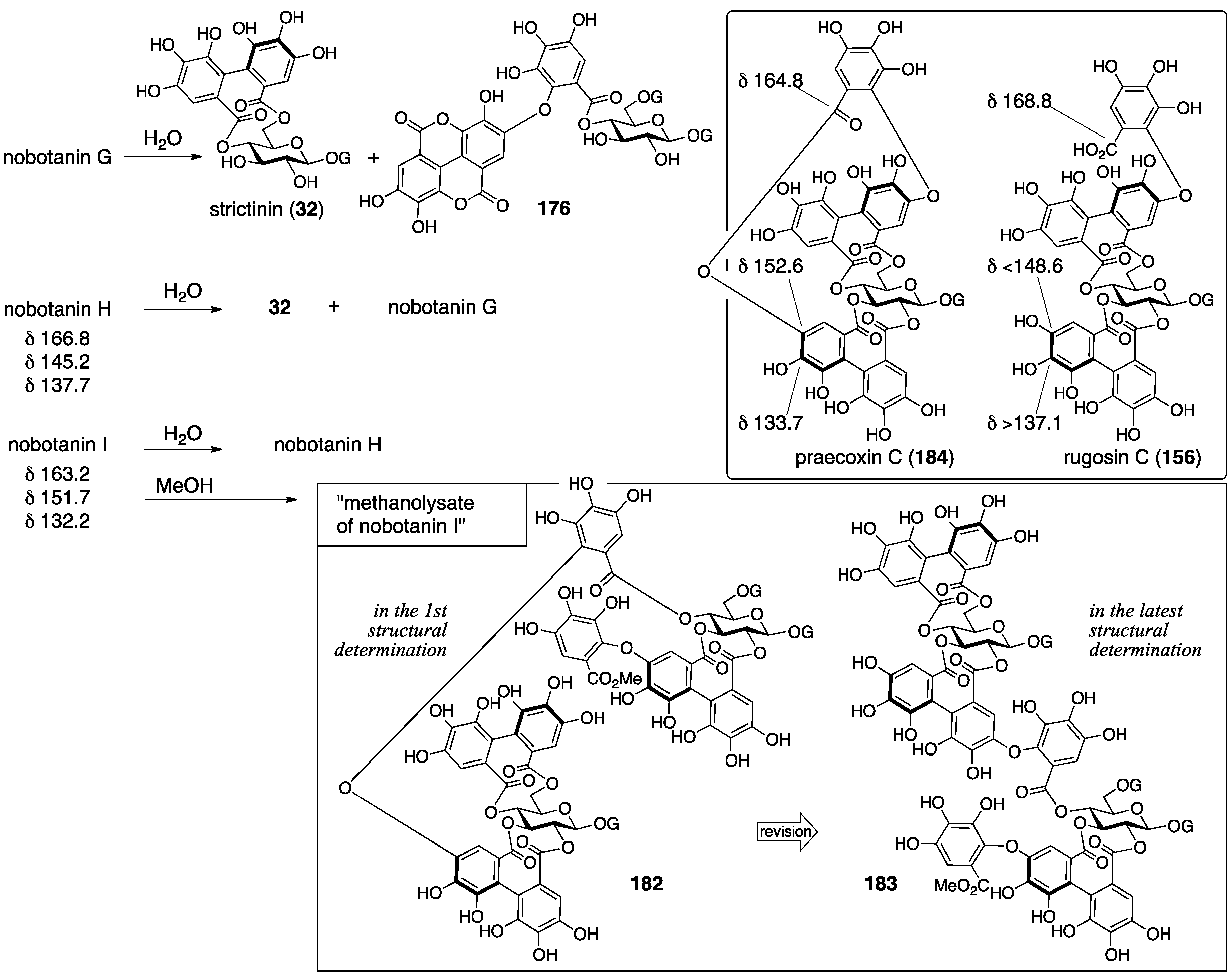
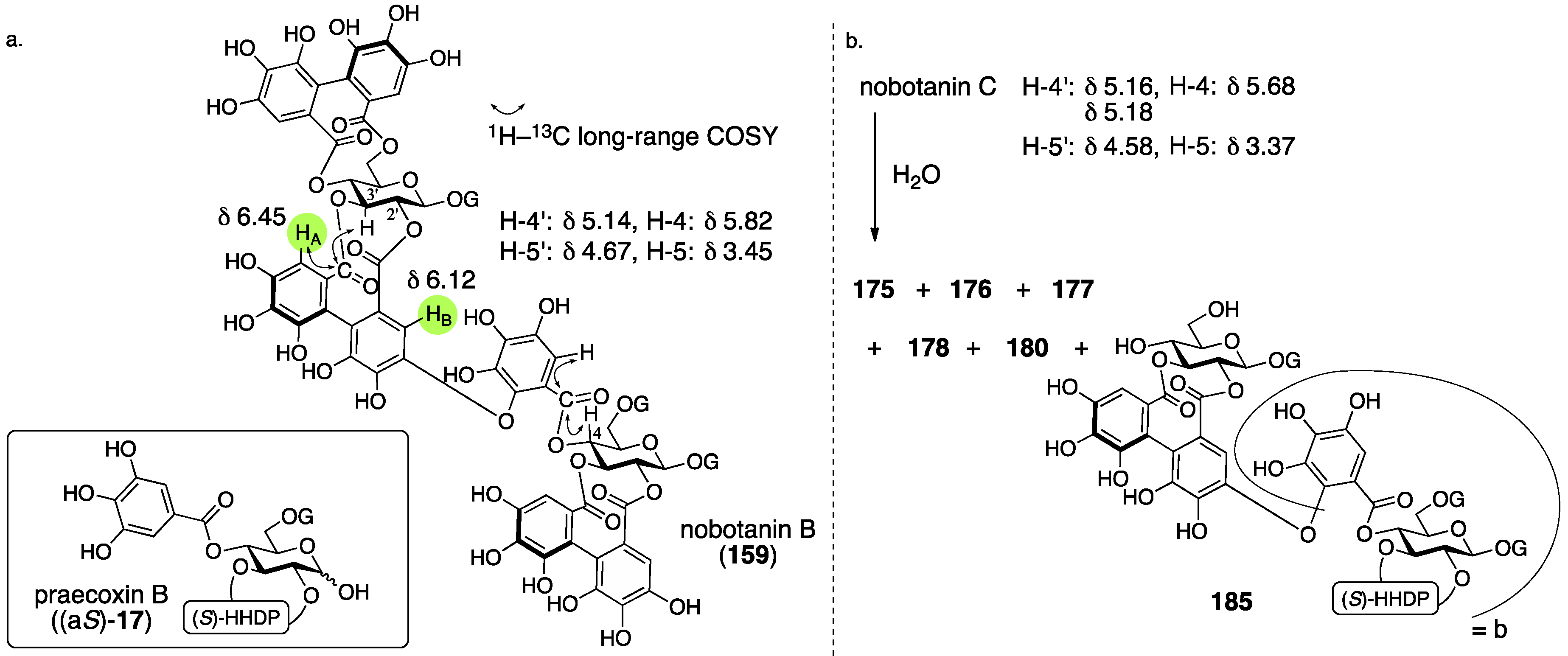
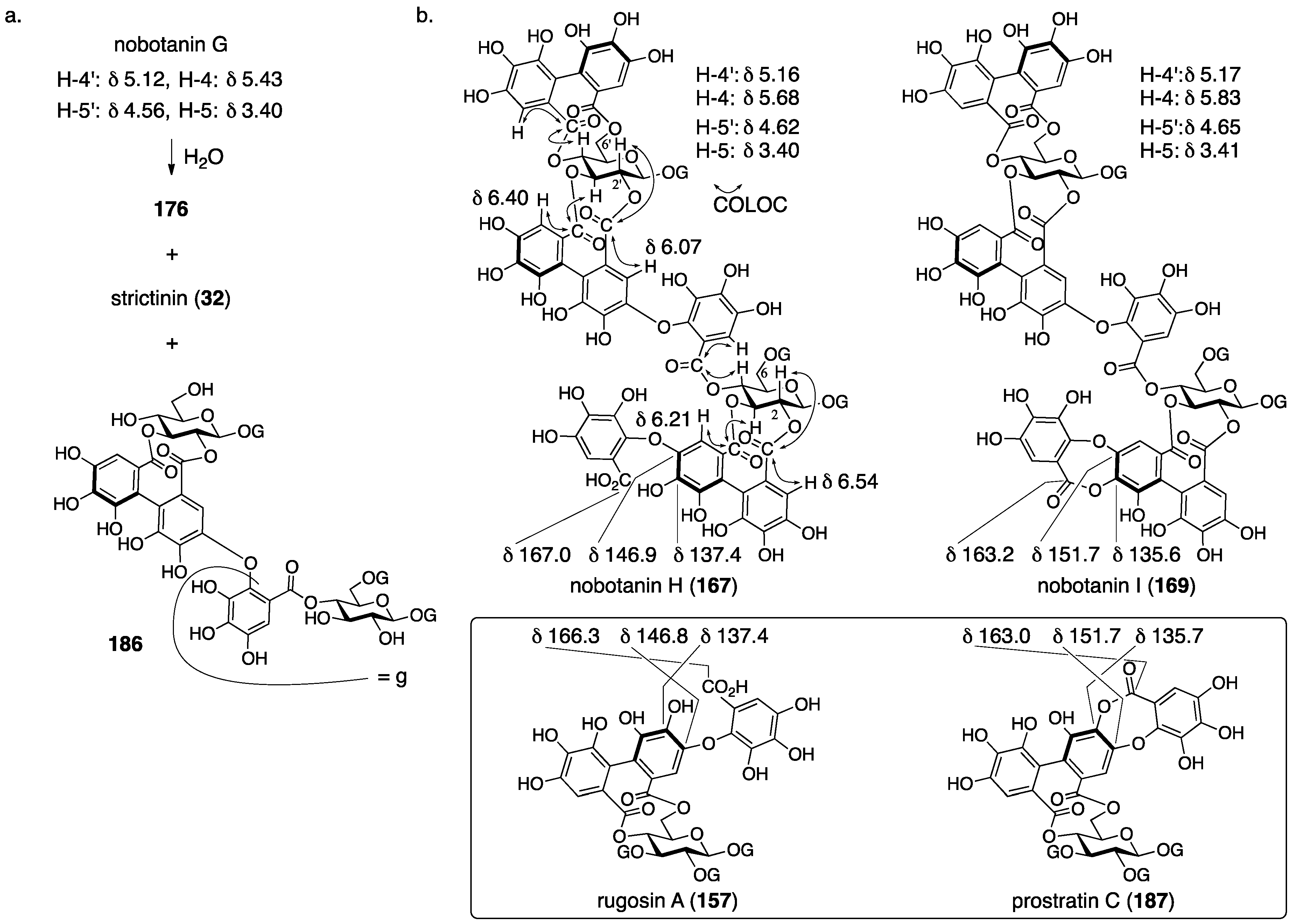
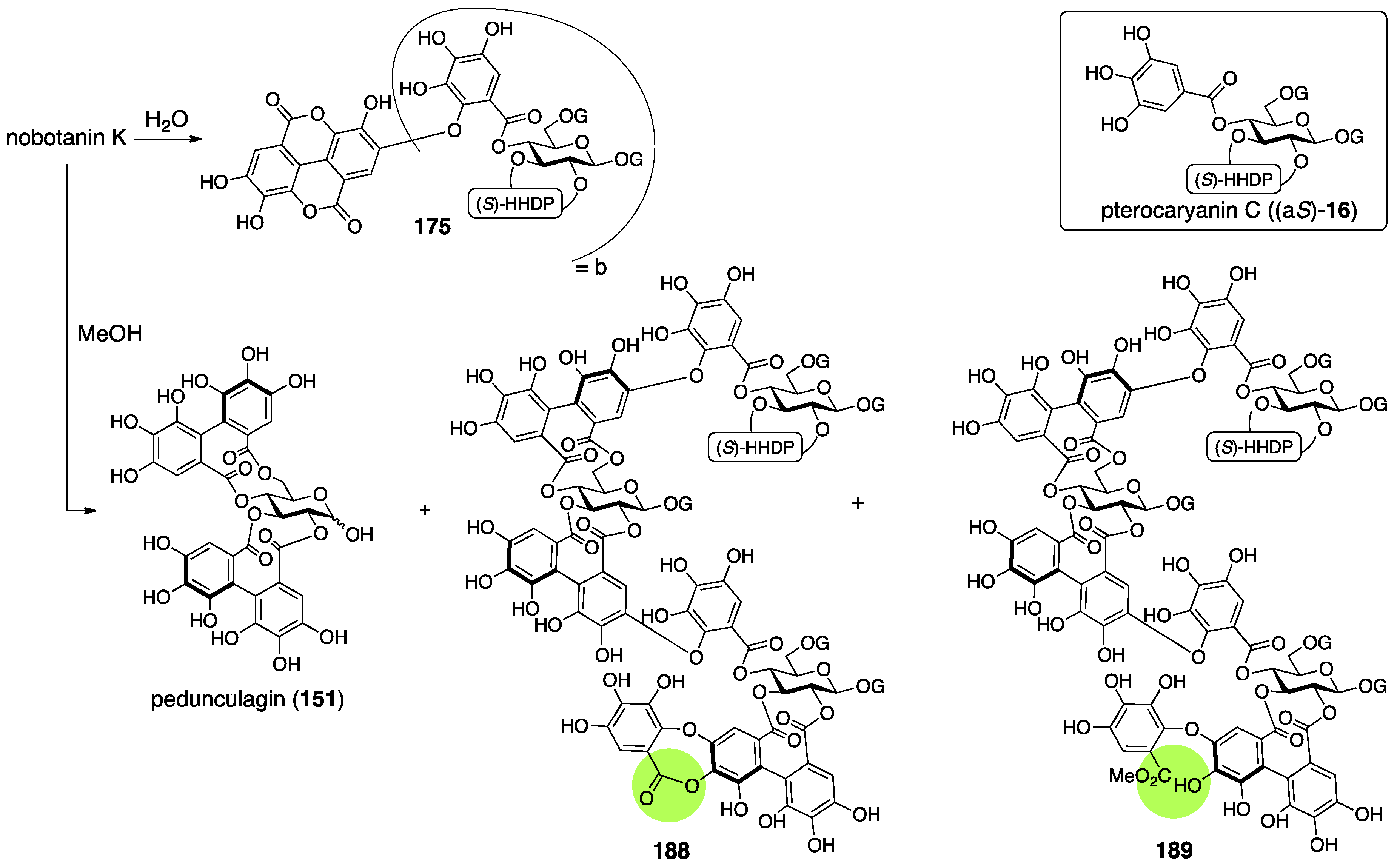
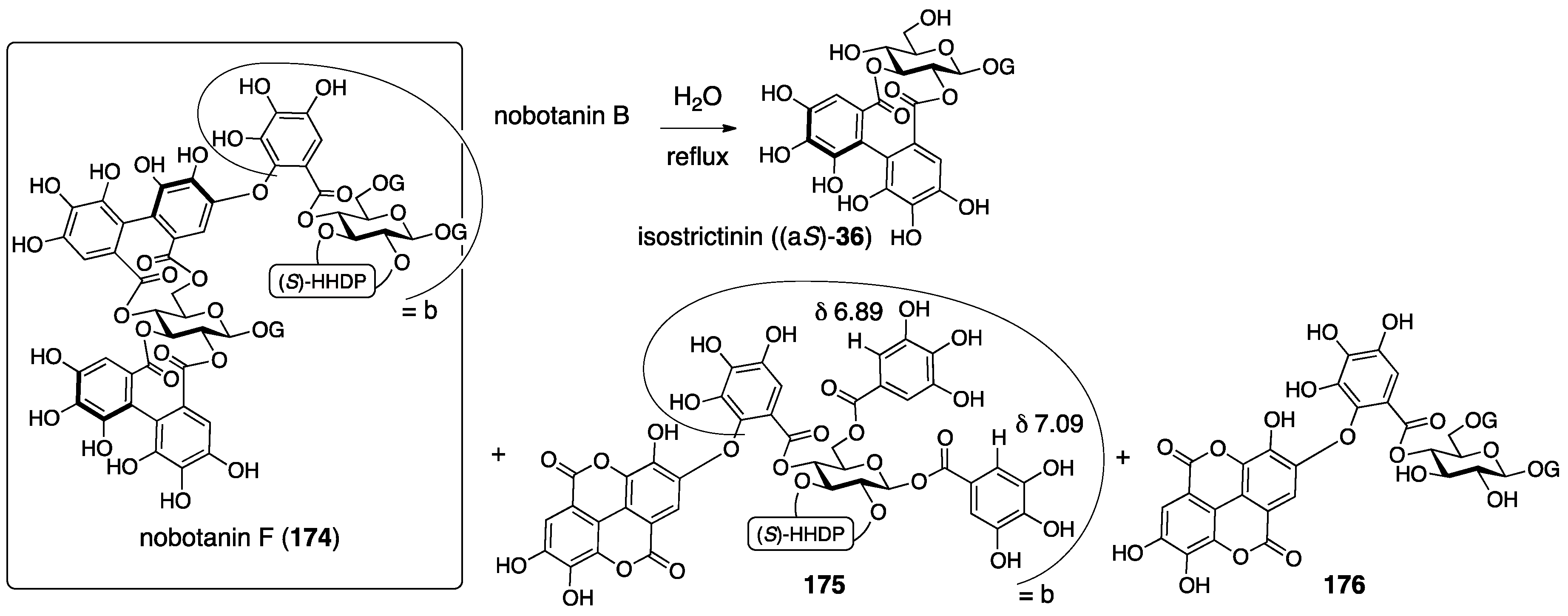
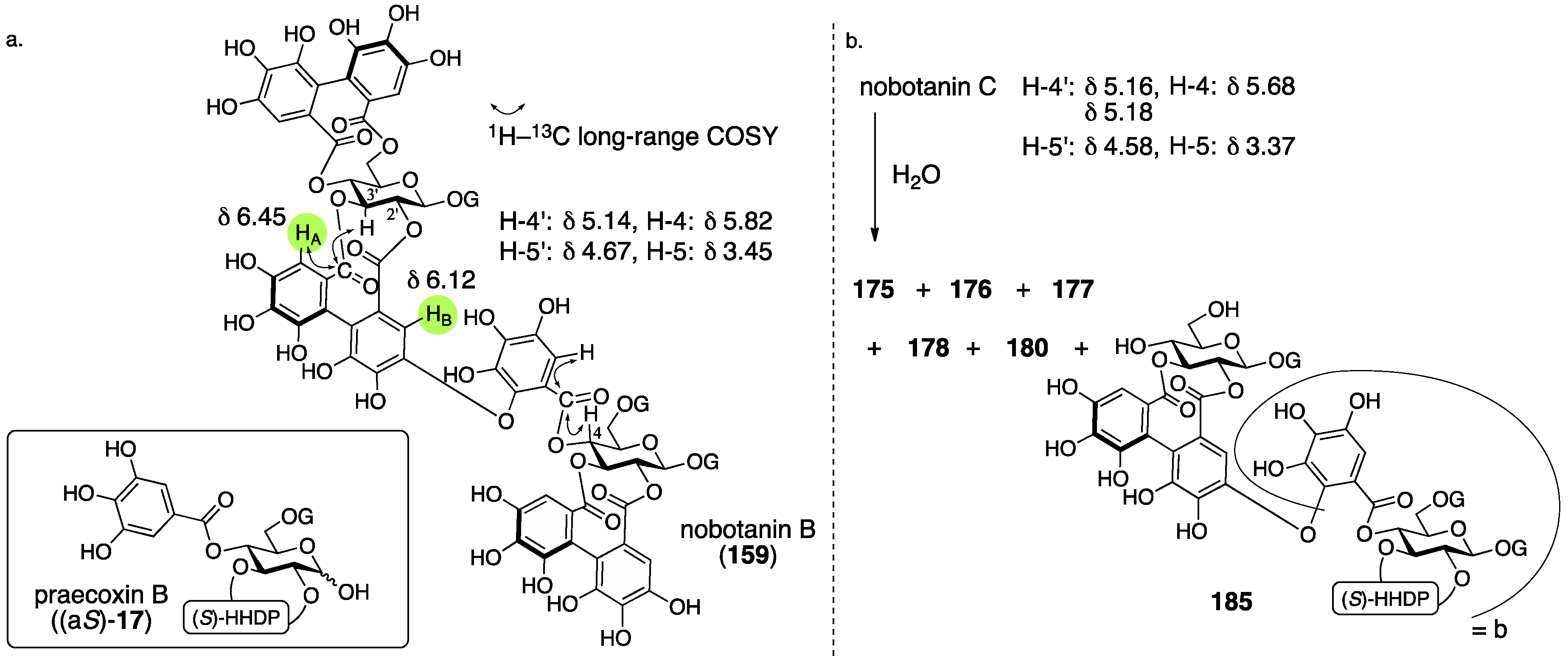
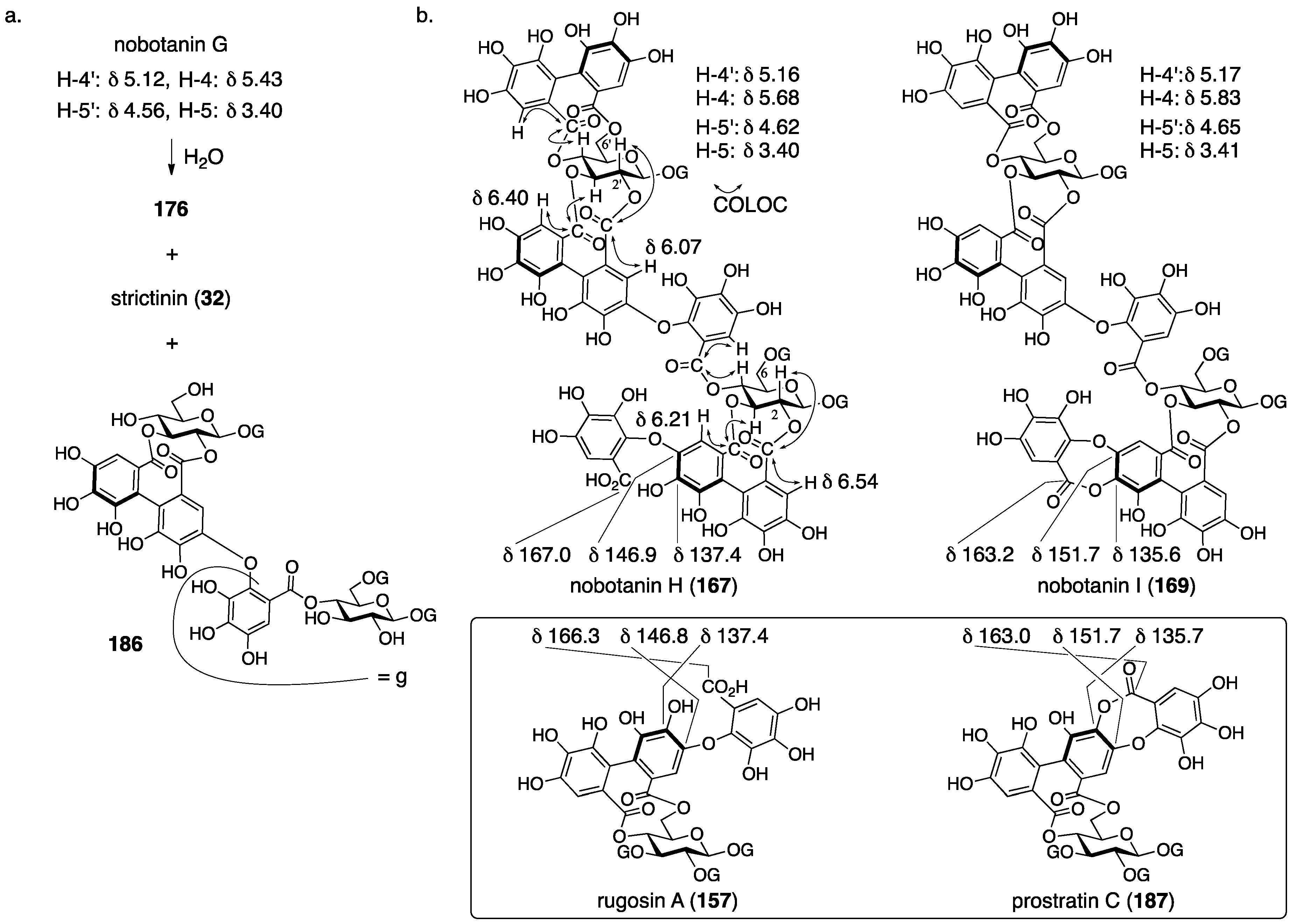
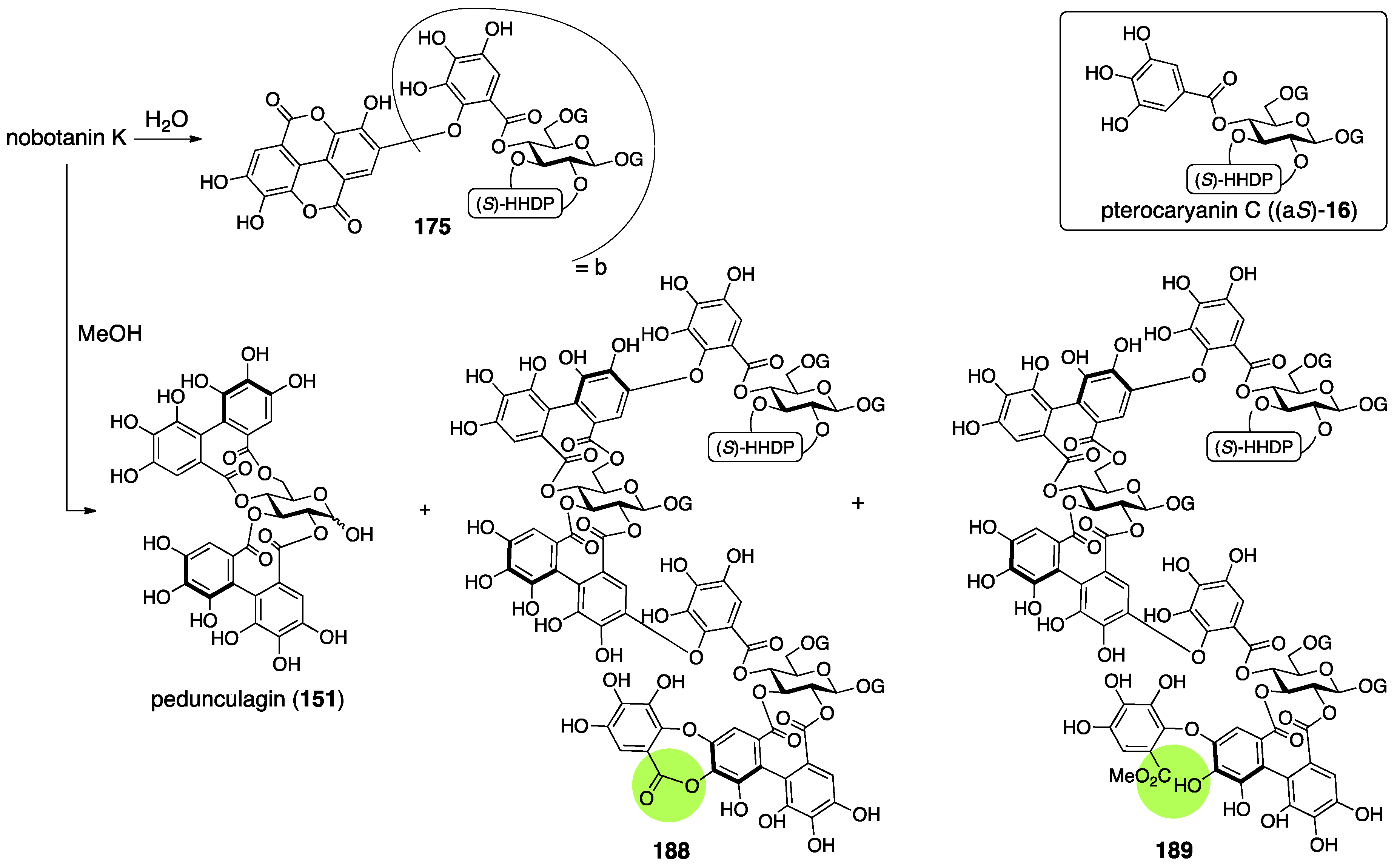
3.5.4. Nobotanin B, C, E, G, H, I, J, and K
4. Summary
Funding
Conflicts of Interest
Abbreviations
| Ac | acetyl |
| All | allyl |
| aq | aqueous |
| Bn | benzyl |
| CD | circular dichroism |
| COLOC | correlation spectroscopy via long-range coupling spectrum |
| conc | concentrated |
| COSY | correlation spectroscopy |
| D | deuterium, dextrorotatory |
| DCC | N,N′-dicyclohexylcarbodiimide |
| DFT | density functional theory |
| DHHDP | dehydrohexahydroxydiphenoyl |
| DIS | differential isotope shift |
| DMAP | 4-(dimethylamino)pyridine |
| DMF | N,N-dimethylformamide |
| DMSO | dimethyl sulfoxide |
| ECD | electronic circular dichroism |
| EDCI·HCl | 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride |
| Et | ethyl |
| epi | epimer |
| FAB-MS | fast atom bombardment-mass spectrometry |
| G | galloyl |
| G(Ac3) | tri-O-acetylgalloyl |
| G(Bn3) | tri-O-benzylgalloyl |
| G(All2Bn) | 3,5-di-O-allyl-4-O-benzylgalloyl |
| G(Me3) | tri-O-methylgalloyl |
| GPC | gel permeation chromatography |
| h | hour(s) |
| HHDP | hexahydroxydiphenoyl |
| HHDP(Me6) | hexa-O-methyl-hexahydroxydiphenoyl |
| HMBC | hetero-nuclear multiple-bond coherence |
| i | iso |
| IR | infrared |
| Me | methyl |
| MP | 4-methoxyphenyl |
| MS | mass spectrum (or spectra) |
| MS 4A | molecular sieves 4A |
| n | normal |
| NHTP | nonahydroxytriphenoyl |
| NMR | nuclear magnetic resonance |
| NOE | nuclear Overhauser effect |
| ORD | optical rotatory dispersion |
| Ph | phenyl |
| Pr | propyl |
| rac | racemic |
| rt | room temperature |
| t | tertiary |
| TBAF | tetra-n-butylammonium fluoride |
| TBS | t-butyldimethylsilyl |
| THF | tetrahydrofuran |
| UV | ultraviolet |
References
- Okuda, T. Novel aspects of tannins–Renewed concepts and structure-activity relationships. Curr. Org. Chem. 1999, 3, 609–622. [Google Scholar]
- Ascacio-Valdés, J.A.; Fuenrostro-Figueroa, J.J.; Aguilera-Carbo, A.; Prado-Barragán, A.; Rodríguez-Harrera, R.; Aguilar, C.N. Ellagitannins: Biosynthesis, biodegradation and biological properties. J. Med. Plants Res. 2011, 5, 4696–4703. [Google Scholar]
- Quideau, S. Chemistry and Biology of Ellagitannins—An Underestimated Class of Bioactive Plant Polyphenols; World Scientific: Singapore, 2009. [Google Scholar]
- Okuda, T.; Ito, H. Tannins of constant structure in medicinal and food plants—Hydrolyzable tannins and polyphenols related to tannins. Molecules 2011, 16, 2191–2217. [Google Scholar] [CrossRef]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Plant polyphenols: Chemical properties, biological activities, and synthesis. Angew. Chem. Int. Ed. 2011, 50, 586–621. [Google Scholar]
- Schmidt, O.T.; Mayer, W. Natürliche Gerbstoffe. Angew. Chem. 1956, 68, 103–115. [Google Scholar]
- Haslam, E. Gallic acid and its metabolites. In Plant Polyphenols; Hemingway, R.W., Laks, P.E., Eds.; Springer: New York, NY, USA, 1992; pp. 169–194. [Google Scholar]
- Haslam, E. Che Faro Senza Polifenoli: Plant Polyphenols 2; Gross, G.G., Hemingway, R.W., Yoshida, T., Eds.; Springer: New York, NY, USA, 1999; pp. 15–40. [Google Scholar]
- Niemetz, R.; Schilling, G.; Gross, G.G. Ellagitannin biosynthesis: Oxidation of pentagalloylglucose to tellimagrandin II by an enzyme from Tellima grandiflora leaves. Chem. Commun. 2001, 1, 35–36. [Google Scholar] [CrossRef]
- Niemetz, R.; Gross, G.G. Oxidation of pentagalloylglucose to the ellagitannin, tellimagrandin II, by a phenol oxidase from Tellima grandiflora leaves. Phytochemistry 2003, 62, 301–306. [Google Scholar] [CrossRef]
- Buckingham, J. Tannins. In Dictionary of Natural Products; Chapman & Hall: London, UK, 1994. [Google Scholar]
- Pouységu, L.; Deffieux, D.; Malik, G.; Natangelo, A.; Quideau, S. Synthesis of ellagitannin natural products. Nat. Prod. Rep. 2011, 28, 853–874. [Google Scholar] [PubMed]
- Li, H.; Tanaka, T.; Zhang, Y.J.; Yang, C.R.; Kouno, I.; Rubusuaviins, A.F. Monomeric and Oligomeric Ellagitannins from Chinese Sweet Tea and Their α-Amylase Inhibitory Activity. Chem. Pharm. Bull. 2007, 55, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, K.; Nonaka, G.I.; Nishioka, I. Tannins and related compounds. LV. Isolation and characterization of acutissimins A and B, novel tannins from Quercus and Castanea species. Chem. Pharm. Bull. 1987, 35, 602–610. [Google Scholar] [CrossRef]
- Lee, M.W.; Tanaka, T.; Nonaka, G.I.; Nishioka, I. Hirsunin, an ellagitannin with a diarylheptanoid moiety, from Alnus hirsuta var. Microphylla. Phytochemistry 1992, 31, 967–970. [Google Scholar]
- Amagata, T. Misaligned Structures: Case Examples from the Past Decade. In Comprehensive Natural Products II, Chemistry and Biology; Liu, H.W., Ed.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 2, pp. 581–621. [Google Scholar]
- Suyama, T.L.; Gerwick, W.H.; McPhail, K.L. Survey of marine natural product structure revisions: A synergy of spectroscopy and chemical synthesis. Bioorg. Med. Chem. 2011, 19, 6675–6701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridolin. Chebulinic acid. Chem. Zentralbl. 1884, 641. [Google Scholar]
- Partridge, S.M. Aniline Hydrogen Phthalate as a Spraying Reagent for Chromatography of Sugars. Nature 1949, 164, 443. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.T.; Lademann, R. Corilagin, ein weiterer kristallisierter gerbstoff aus dividivi. Liebigs Ann. Chem. 1951, 571, 232–237. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Schmidt, D.M.; Herok, J. Die konstitution und konfiguration des corilagins XIX. Mitteilung uber natürliche gerbstoffe. Liebigs Ann. Chem. 1954, 587, 67–74. (In German) [Google Scholar] [CrossRef]
- Mislow, K.; Glass, M.A.W.; O’brien, R.E.; Rutkin, P.; Steinberg, D.H.; Weiss, J.; Djerassi, C. Configuration, conformation and rotatory dispersion of optically active biaryls. J. Am. Chem. Soc. 1962, 84, 1455–1478. [Google Scholar] [CrossRef]
- Okuda, T.; Yoshida, T.; Hatano, T. Equilibrated stereostructures of hydrated geraniin and mallotusinic acid. Tetrahedron Lett. 1980, 21, 2561–2564. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Blinn, F.; Lademann, R. Über die bindung der ellagsäure in corilagin und chebulagsäure. XII. Mitteilung über natürliche gerbstoffe. Liebigs Ann. Chem. 1952, 576, 75–84. (In German) [Google Scholar] [CrossRef]
- Reeves, R.E.; Goebel, W.F. Chemoimmunological studies on the soluble specific substance of pneumococcus V. The structure of the type III polysaccharide. J. Biol. Chem. 1941, 139, 511–519. [Google Scholar]
- For β-isomer: Adams, M.H.; Reeves, R.E.; Goebel, W.F. The synthesis of 2,4-dimethyl-β-methylglucoside. J. Biol. Chem. 1941, 140, 653–661. [Google Scholar]
- Schmidt, O.T.; Herok, J. α-Glucogallin XVIII. Mitteilung über natürliche gerbstoffe. Liebigs Ann. Chem. 1954, 587, 63–66. (In German) [Google Scholar] [CrossRef]
- Jochims, J.C.; Taigel, G.; Schmidt, O.T. Uber natürliche gerbstoffe, XLI. Protonenresonanz-spektren und konformationsbestimmung einiger natürlicher gerbstoffe. Liebigs Ann. Chem. 1968, 717, 169–185. (In German) [Google Scholar]
- Schmidt, O.T.; Demmler, K. Racemische und optisch aktive 2,3,4,2′,3′,4′-hexaoxydiphenyl-6,6′-dicarbonsaure. XVII. Mitteilung uber natürliche gerbstoffe. Liebigs Ann. Chem. 1954, 586, 179–193. (In German) [Google Scholar] [CrossRef]
- Hatano, T.; Yoshida, T.; Shingu, T.; Okuda, T. 13C Nuclear magnetic resonance spectra of hydrolyzable tannins. III. Tannins having 1C4 glucose and C-glucosidic linkage. Chem. Pharm. Bull. 1988, 36, 3849–3856. [Google Scholar]
- Seikel, M.K.; Hills, W.E. Hydrolysable tannins of Eucalyptus delegatensis wood. Phytochemistry 1970, 9, 1115–1128. [Google Scholar]
- Okuda, T.; Yoshida, T.; Nayeshiro, H. Geraniin, a new ellagitannin from Geranium thunbergii. Tetrahedron Lett. 1976, 3721–3722. [Google Scholar] [CrossRef]
- Luger, P.; Weber, M.; Kashino, S.; Amakura, Y.; Yoshida, T.; Okuda, T.; Beurskens, G.; Dauter, Z. Structure of the tannin geraniin based on conventional X-ray data at 295 K and on synchrotron data at 293 and 120 K. Acta Cryst. 1998, B54, 687–694. [Google Scholar] [CrossRef]
- Yamada, H.; Nagao, K.; Dokei, K.; Kasai, Y.; Michihata, N. Total synthesis of (–)-corilagin. J. Am. Chem. Soc. 2008, 130, 7566–7567. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nonaka, G.-I.; Nishioka, I. Tannins and related compounds. XLI. Isolation and characterization of novel ellagitannins, punicacorteins A, B, C and D, and punigluconin from the bark of Punica granatum L. Chem. Pharm. Bull. 1986, 34, 656–663. [Google Scholar] [CrossRef]
- Tanaka, T.; Tong, H.-H.; Xu, Y.-M.; Ishimaru, K.; Nonaka, G.-I.; Nishioka, I. Tannins and related compounds. CXVII. Isolation and characterization of three new ellagitannins, lagerstannins A, B, and C, having a gluconic acid core, from Lagerstroemia speciosa (L.) PERS. Chem. Pharm. Bull. 1992, 40, 2975–2980. [Google Scholar] [CrossRef]
- Okuda, T.; Yoshida, T.; Ashida, M. Casuarictin and casuarinin, two new ellagitannins from Casuarina stricta. Heterocycles 1981, 16, 1681–1685. [Google Scholar]
- Nonaka, G.-I.; Ishimatsu, M.; Ageta, A.; Nishioka, I. Tannins and related Compounds. LXXVI. Isolation and characterization of cercidinins A and B and cuspinin, unusual 2,3-(R)-hexahydroxydiphenoyl glucoses from Cercidiphyllum japonicum and Castanopsis cuspidate var. sieboldii. Chem. Pharm. Bull. 1989, 37, 50–53. [Google Scholar]
- Khanbabaee, K.; Lötzerich, K. Synthesis of enantiomerically pure unusual ellagitannins 1,4,6-tri-O-galloyl 2,3-(R)-hexahydroxydiphenoyl-β-d-glucoside. The proposed chemical structures for cercidinin A and B must be revised. J. Org. Chem. 1998, 63, 8723–8728. [Google Scholar] [CrossRef]
- Tanaka, T.; Nonaka, G.-I.; Ishimatsu, M.; Nishioka, I.; Kouno, I. Revised structure of cercidinin A, a novel ellagitannin having (R)-hexahydroxydiphenoyl esters at the 3,4-postions of glucopyranose. Chem. Pharm. Bull. 2001, 49, 486–487. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Ohara, K.; Ogura, T. Total synthesis of cercidinin A. Eur. J. Org. Chem. 2013, 7872–7875. [Google Scholar] [CrossRef]
- Ikeya, K.; Taguchi, H.; Yoshioka, I.; Kobayashi, H. The constituents of Schisandra chinensis BAILL. I. Isolation and structure determination of five new lignans, gomisin A, B, C, F, and G, and the absolute structure of schizandrin. Chem. Pharm. Bull. 1979, 27, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nonaka, G.-I.; Nishioka, I. Tannins and related compounds. Part 28. Revision of the structures of sanguiins H-6, H-2, and H-3, and isolation and characterization of sanguiin H-11, a novel tetrameric hydrolysable tannin, and seven related tannins, from Sanguisorba officinalis. J. Chem. Res. (S) 1985, 176–177. [Google Scholar]
- Yoshida, T.; Chen, X.; Hatano, T.; Fukushima, M.; Okuda, T. Tannins and related polyphenols of rosaceous medicinal plants. IV. Roxbin A and B from Rosa roxburghii Fruits. Chem. Pharm. Bull. 1987, 35, 1817–1822. [Google Scholar] [PubMed]
- Yamaguchi, S.; Ashikaga, Y.; Nishii, K.; Yamada, H. Total synthesis of the proposed structure of roxbin B; the nonidentical outcome. Org. Lett. 2012, 14, 5928–5931. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Hirokane, T.; Yoshida, T.; Tanaka, T.; Hatano, T.; Ito, H.; Nonaka, G.-I.; Yamada, H. Roxbin B is cuspinin: Structural revision and total synthesis. J. Org. Chem. 2013, 78, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Al-Shari, S.M.K.; Layden, K.; Haslam, E. The metabolism of gallic acid and hexahydroxydiphenic acid in plant. Esters of (S)- hexahydroxydiphenic acid with d-glucopyranose(4C1). J. Chem. Perkin. Trans. 1982, 1, 2525–2534. [Google Scholar] [CrossRef]
- Okuda, T.; Yoshida, T.; Nayeshiro, H. Constituents of Geranium thunbergii SIEB. et Zucc. IV. Ellagitannins. (2). Structure of geraniin. Chem. Pharm. Bull. 1977, 25, 1862–1869. [Google Scholar]
- Okuda, T.; Nayeshiro, H.; Seno, K. Structure of geraniin in the equilibrium state. Tetrahedron Lett. 1977, 18, 4421–4424. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Voigt, H.; Puff, W.; Köster, R. Benzyläther der ellagsäure und hexaoxy-diphensäure XVI. Mitteilung über natürliche gerbstoffe. Liebigs Ann. Chem. 1954, 586, 165–178. (In German) [Google Scholar]
- Pfeffer, P.E.; Valentine, K.M.; Parrish, F.W. Deuterium-induced differential isotope shift 13C NMR. 1. Resonance reassignments of mono- and disaccharides. J. Am. Chem. Soc. 1979, 101, 1265–1274. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Schulz, J.; Wurmb, R. Uber natürliche gerbstoffe, XXXVI. Terchebin. Liebigs Ann. Chem. 1967, 706, 169–179. (In German) [Google Scholar] [CrossRef]
- Okuda, T.; Hatano, T.; Nitta, H.; Fujii, R. Hydrolysable tannins having enantiomeric dehydrohexahydroxydiphenoyl group: Revised structure of terchebin and structure of granatin B. Tetrahedron Lett. 1980, 21, 4361–4364. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Klinger, G. Über natürliche gerbstoffe XXVIII. Synthese der 1.3.6-trigalloyl-glucose. Liebigs Ann. Chem. 1957, 609, 199–208. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Schanz, R.; Eckert, R.; Wurmb, R. Über natürliche gerbstoffe, XXXIV. Brevilagin 1. Liebigs Ann. Chem. 1967, 706, 131–153. (In German) [Google Scholar] [CrossRef]
- Tillmans, J.; Hirsch, P.; Hirsch, W. Uber die anwendung von 2,6-dichlorphenol-indophenol als reduktionsindicator bei der untersuchung yon lebensmitteln. Z. Unters. Lebensm. 1928, 56, 272–292. (In German) [Google Scholar] [CrossRef]
- Schmidt, O.T.; Hensler, R.H.; Stephan, P. Über natürliche gerbstoffe XXVI. Neochebulagsäure Liebigs Ann. Chem. 1957, 609, 186–191. (In German) [Google Scholar]
- Schmidt, O.T.; Demmler, K.; Bittermann, H. Über natürliche gerbstoffe XXVII. Neochebulinsäure und 1.3.6-trigalloyl-glucose. Liebigs Ann. Chem. 1957, 609, 192–199. (In German) [Google Scholar] [CrossRef]
- Fürstenwerth, H.; Schildknecht, H. Isoterchebin, der gerbe Farbestoff des Zistrosenwürgers Cytinus hypocistis (Rafflessiaceae, Schmarotzerblumengewächse). Liebigs Ann. Chem 1976, 112–123. (In German) [Google Scholar] [CrossRef]
- Okuda, T.; Hatano, T.; Yasui, T. Revised structure of isoterchebin, isolated from Cornus Officinalis. Heterocycles 1981, 16, 1321–1324. [Google Scholar] [CrossRef]
- Nonaka, G.I.; Matsumoto, Y.; Nishioka, I.; Nishizawa, M.; Yamagishi, T. Trapain, a new hydrolysable tannin from Trapa japonica Flerov. Chem. Pharm. Bull. 1981, 29, 1184–1187. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Bernauer, K. Brevifolin and Brevifolin-carbosäure XXI. Mitteilung über natürliche Gerbstoffe. Libigs Ann. Chem. 1954, 588, 211–230. (In German) [Google Scholar] [CrossRef]
- Schmidt, O.T.; Wurmb, R.; Schulz, J. Hexahydroxy-diphensäure (ellagsäure), brevifolin-carbosäure und chebulsäure als Umwandlungsprodukte der brevilagin und des terchebins. Libigs Ann. Chem. 1967, 706, 180–186. (In German) [Google Scholar] [CrossRef]
- Nonaka, G.I.; Harada, M.; Nishioka, I. Eugeniin, a new ellagitannin from cloves. Chem. Pharm. Bull. 1980, 28, 685–687. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Schulz, J.; Fiesser, H. Über natürliche gerbstoffe XXVIII. Die gerbstoffe der myrobalanen. Liebigs Ann. Chem. 1967, 706, 187–197. (In German) [Google Scholar] [CrossRef]
- Haslam, E.; Uddin, M. Gallotannins. Part XV. Some observations on the structures of chebulinic acid and its derivatives. J. Chem. Soc. (C) 1967, 2381–2384. [Google Scholar]
- Yoshida, T.; Fujii, R.; Okuda, T. Revised structures of chebulinic acid and chebulagic acid. Chem. Pharm. Bull. 1980, 28, 3713–3715. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Mayer, W. Die Konstitution der spaltsaure C14H12O11 aus chebulin- und chebulagsäure. Liebigs Ann. Chem. 1951, 571, 1–15. [Google Scholar]
- Schmidt, O.T.; Schmidt, D.M. Die umwandlung von chebulagsäure in corilagin XIV. Mitteilung über natürliche gerbstoffe. Liebigs Ann. Chem. 1952, 578, 25–30. [Google Scholar] [CrossRef]
- Schilling, G.; Schweiger, R.; Weis, G.; Mayer, W.; Weiβ, J.; Siegel, R. Die relative konfiguration der chebulsäure. Liebigs Ann. Chem. 1981, 1981, 603–609. [Google Scholar] [CrossRef]
- Yoshida, T.; Okuda, T.; Koga, T.; Toh, N. Absolute configurations of chebulic, chebulinic and chebulagic acid. Chem. Pharm. Bull. 1982, 30, 2655–2658. [Google Scholar] [CrossRef]
- Hay, J.E.; Haynes, L.J. Bergenin, a C-glycopyranosyl derivative of 4-O-methylgallic acid. J. Chem. Soc. 1958, 2231–2238. [Google Scholar] [CrossRef]
- Ito, T.; Harada, N.; Nakanishi, K. Application of the aromatic chirality method to the bergenin system. Agric. Biol. Chem. 1971, 35, 797–798. [Google Scholar] [CrossRef]
- Mayer, W.; Gabler, W.; Riester, A.; Korger, H. Die Isolierung von Castalagin, Vescalagin, Castalin und Vescalin. Liebigs Ann. Chem. 1967, 707, 177–181. [Google Scholar] [CrossRef]
- Mayer, W.; Einwiller, A.; Jochims, J.C. Die Struktur des Castalins. Liebigs Ann. Chem. 1967, 707, 182–189. (In German) [Google Scholar] [CrossRef]
- Mayer, W.; Kuhlmann, F.; Schilling, G. Die Struktur des Vescalins. Liebigs Ann. Chem. 1971, 747, 51–59. (In German) [Google Scholar]
- Mayer, W.; Seitz, H.; Jochims, J.C. Die Struktur des Castalagins. Liebigs Ann. Chem. 1969, 721, 186–193. (In German) [Google Scholar] [CrossRef]
- Mayer, W.; Seitz, H.; Jochims, J.C.; Schauerte, K.; Schilling, G. Struktur des Vescalagins. Liebigs Ann. Chem. 1971, 751, 60–68. (In German) [Google Scholar] [CrossRef]
- Okuda, T.; Yoshida, T.; Ashida, M.; Yazaki, K. Casuarinin, stachyurin and strictinin, new ellagitannins from Casuarina stricta and Stachyurus praecox. Chem. Pharm. Bull. 1982, 30, 766–769. [Google Scholar] [CrossRef]
- Nonaka, G.I.; Ishimaru, K.; Watanabe, M.; Nishioka, I.; Yamauchi, T.; Wan, A.S.C. Tannins and Related Compounds. LI. Elucidation of the Stereochemistry of the Triphenoyl Moiety in Castalagin and Vescalagin, from Eugenia grandis. Chem. Pharm. Bull. 1987, 35, 217–220. [Google Scholar] [CrossRef]
- Nonaka, G.I.; Sakai, T.; Tanaka, T.; Mihashi, K.; Nishioka, I. Tannins and Related Compounds. XCVII. Structure Revision of C-Glycosidic Ellagitannins, Castalagin, Vescalagin, Casuarinin and Stachyurin, and Related Hydrolyzable Tannins. Chem. Pharm. Bull. 1990, 38, 2151–2156. [Google Scholar] [CrossRef]
- Matsuo, Y.; Wakamatsu, H.; Omar, M.; Tanaka, T. Reinvestigation of the Stereochemistry of the C-Glycosidic Ellagitannins, Vescalagin and Castalagin. Org. Lett. 2015, 17, 46–49. [Google Scholar] [PubMed]
- Richieu, A.; Peixoto, P.; Pouységu, L.; Denis, D.; Quideau, S. Bio-inspired Total Synthesis of (–)-Vescalin, A Nonahydroxytriphenoylated C-Glucosidic Ellagitannin. Angew. Chem. Int. Ed. 2017, 56, 13833–13837. [Google Scholar]
- Tanaka, T.; Nonaka, G.I.; Nishioka, I. Tannins and Related Compounds. XLII. Isolation and Characterization of Four New Hydrolyzable Tannins, Terflavins A and B, Tergallagin and Tercatain from the Leaves of Terminalia catappa L. Chem. Pharm. Bull. 1986, 34, 1039–1049. [Google Scholar] [CrossRef]
- Hervé du Penhoat, C.L.M.; Michon, V.M.F.; Peng, S.; Viriot, C.; Scalbert, A.; Gage, D. Structural elucidation of new dimeric ellagitannins from Quercus robur L. roburins A–E. J. Chem. Soc. Perkin Trans. 1 1991, 1653–1660. [Google Scholar] [CrossRef]
- Tanaka, T.; Ueda, N.; Shinohara, H.; Nonaka, G.I.; Fujioka, T.; Mihashi, K.; Kouno, I. C-glycosidic ellagitannin metabolites in the heartwood of Japanese chestnut tree (Castanea crenate SIEB. et ZUCC.). Chem. Pharm. Bull. 1996, 44, 2236–2242. [Google Scholar] [CrossRef]
- Lin, T.C.; Tanaka, T.; Nonaka, G.I.; Nishioka, I.; Young, T.J. Tannins and related compounds. CVIII. Isolation and characterization of novel complex tannins (flavono-ellagitannins), anogeissinin and anogeissusins A and B, from Anogeissus acuminata (ROXB ex DC.) GUILL. et PERR. var. lanceolata WALL. ex CLARKE. Chem. Pharm. Bull. 1991, 39, 1144–1147. [Google Scholar] [CrossRef]
- Nonaka, G.I.; Ishimaru, K.; Mihashi, K.; Iwase, Y.; Ageta, M.; Nishioka, I. Tannins and related compounds. LXIII. Isolation and characterization of mongolicains A and B, novel tannins from Quercus and Castanopsis species. Chem. Pharm. Bull. 1988, 36, 857–869. [Google Scholar] [CrossRef]
- Mayer, W.; Görner, A.; Andrä, K. Punicalagin und Punicalin, zwei Gerbstoffe aus den Schalen der Granatäpfel. Liebigs Ann. Chem. 1978, 1977, 1976–1986. [Google Scholar] [CrossRef]
- Tanaka, T.; Nonaka, G.-I.; Nishioka, I. Tannins and Related Compounds. XL. Revision of the Structures of Punicalin and Punicalagin, and Isolation and Characterization of 2-O-Galloylpunicalin from the Bark of Punica granatum L. Chem. Pharm. Bull. 1986, 34, 650–655. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Würtele, L.; Harréus, A. Pedunculagin, eine 2.3;4.6-Di-[(–)-hexahydroxy-diphenoyl]glucose aus Knoppern. Liebigs Ann. Chem. 1965, 690, 150–162. [Google Scholar] [CrossRef]
- Schilling, G.; Schick, H. Zur Struktur des Punicalagins und Punicalins. Liebigs Ann. Chem. 1985, 1985, 2240–2245. [Google Scholar] [CrossRef]
- Nonaka, G.I.; Tanaka, T.; Nishioka, I. Tannins and Related Compounds. Part 3. A New Phenolic Acid, Sanguisorbic Acid Dilactone, and Three New Ellagitannins, Sanguiins H-1, H-2, and H-3, from Sanguisorba officinalis. J. Chem. Soc., Perkin Trans. 1982, 1, 1067–1073. [Google Scholar] [CrossRef]
- Nonaka, G.I.; Tanaka, T.; Nishioka, I. A dimeric hydrolyzable tannin, sanguiin H-6 from Sanguisorba officinalis L. Chem. Pharm. Bull. 1982, 27, 2255–2257. [Google Scholar]
- Bacon, R.G.R.; Stewart, O.J. Metal Ions and Complexes in Organic Reactions. Part IV. Copper-promoted Preparations of Diaryl Ethers and Competing Hydrogen-transfer Processes. J. Chem. Soc. 1965, 4953–4961. [Google Scholar]
- Yoshida, T.; Memon, M.U.; Okuda, T. Alnusiin, a novel ellagitannin from Alnus sieboldiana fruits. Heterocycles 1981, 16, 1085–1088. [Google Scholar]
- Yoshida, T.; Yazaki, K.; Memon, M.U.; Maruyama, I.; Kurokawa, K.; Okuda, T. Bicornin, a new hydrolysable tannin from Trapa bicornis, and revised structure of alnusiin. Heterocycles 1989, 29, 861–864. [Google Scholar]
- Mayer, W.; Bilzer, W.; Schilling, G. Castavaloninsäure, isolierung und strukturermittlung. Liebigs Ann. Chem. 1976, 1976, 876–881. [Google Scholar]
- Yoshida, T.; Yazaki, K.; Memon, M.U.; Maruyama, I.; Kurokawa, K.; Shingu, T.; Okuda, T. Structures of alnusiin and bicornin, new hydrolysable tannins having a monolactonized tergalloyl group. Chem. Pharm. Bull. 1989, 37, 2655–2660. [Google Scholar] [CrossRef]
- Ishimatsu, M.; Tanaka, T.; Nonaka, G.I.; Nishioka, I. Alnusnins A and B, from the leaves of Alnus sieboldiana. Phytochemistry 1989, 28, 3179–3184. [Google Scholar] [CrossRef]
- Tanaka, T.; Kirihara, S.; Nonaka, G.I.; Nishioka, I. Tannins and related compounds CXXIV. Five new ellagitannins, platycaryanins A, B, C, and D, and platycariin, and new complex tannin, strobilanin, from the fruits and bark of Platycarya strobilacea SIEV et al ZUCC., and biomimetic synthesis of C-glycoside ellagitannins from glucopyranose-based ellagitannins. Chem. Pharm. Bull. 1993, 41, 1708–1716. [Google Scholar]
- Eerdunbayaer, L.B.; Nozaki, A.; Takahashi, E.; Okamoto, K.; Ito, H.; Hatano, T. Hydrolyzable tannins isolated from Syzygium aromaticum: structure of a new C-glucosidic ellagitannin and spectral features of tannins with a tergalloyl group. Heterocycles 2012, 85, 365–381. [Google Scholar]
- Yoshida, T.; Ikeda, Y.; Ohbayashi, H.; Ishihara, K.; Ohwashi, W.; Shingu, T.; Okuda, T. Dimeric ellagitannins in plants of melastomataceae. Chem. Pharm. Bull. 1986, 34, 2676–2679. [Google Scholar] [CrossRef]
- Yoshida, T.; Haba, K.; Arata, R.; Okuda, T. Structures of new hydrolysable tannin oligomers from Melastomataceae plants. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu 1987, 29, 676–683. [Google Scholar]
- Yoshida, T.; Haba, K.; Arata, R.; Okuda, T. Presented at 108th Annual Meeting of the Pharmaceutical Society of Japan, Hiroshima, Japan, 4 April 1988; p. 339.
- Yoshida, T.; Ohwashi, W.; Haba, K.; Ohbayashi, H.; Ishihara, K.; Okano, Y.; Shingu, T.; Okuda, T. Tannins and Related Polyphenols of Melastomaceous Plants. II. Nobotanins B, C, and E, Hydrolyzable Tannin Dimer and Trimers from Tibouchina semidecandra COGN. Chem. Pharm. Bull. 1991, 39, 2264–2270. [Google Scholar] [CrossRef]
- Yoshida, T.; Haba, K.; Nakata, F.; Okano, Y.; Shingu, T.; Okuda, T. Tannins and Related Polyphenols of Melastomaceous Plants. III. Nobotanins G, H, and I, Dimeric Hydrolyzable Tannins from Heterocentron roseum. Chem. Pharm. Bull. 1992, 40, 66–71. [Google Scholar] [CrossRef]
- Yoshida, T.; Haba, K.; Arata, R.; Nakata, F.; Shingu, T.; Okuda, T. Tannins and Related Polyphenols of Melastomaceous Plants. VII. Nobotanins J and K, Trimeric Hydrolyzable Tannins from Heterocentron roseum. Chem. Pharm. Bull. 1995, 43, 1101–1106. [Google Scholar] [CrossRef]
- Okuda, T.; Yoshida, T.; Hatano, T.; Yazaki, K.; Ashida, M. Ellagitannins of the Casuarinaceae, Stachyuraceae and Myrtaceae. Phytochemistry 1982, 21, 2871–2874. [Google Scholar] [CrossRef]
- Okuda, T.; Hatano, T.; Yazaki, K. Praecoxin B, C, D and E, novel ellagitannins from Stachyurus praecox. Chem. Pharm. Bull. 1983, 31, 333–336. [Google Scholar] [CrossRef]
- Hatano, T.; Kira, R.; Yasuhara, T.; Okuda, T. Tannins of Hamamelidaceous Plants. III. Isorugosins A, B and D, New Ellagitannins from Liquidambar formosana. Chem. Pharm. Bull. 1988, 36, 3920–3927. [Google Scholar] [CrossRef]
- Hatano, T.; Ogawa, N.; Yasuhara, T.; Okuda, T. Tannins of Rosaceous Plants. VIII. Hydrolyzable Tannin Monomers Having a Valoneoyl Group from Flower Petals of Rosa rugosa THUNB. Chem. Pharm. Bull. 1990, 38, 3308–3313. [Google Scholar] [CrossRef]
- Yoshida, T.; Hatano, T.; Namba, O.; Atallah, A.F.; Chu, T.; Okonogi, A.; Kuwajima, T.; Okuda, T. Determination of orientation of acyl group in hydrolyzable tannin oligomers. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu 1991, 33, 401–408. [Google Scholar]
- Yoshida, T.; Nakata, F.; Hosotani, K.; Nitta, A.; Okuda, T. Dimeric hydrolysable tannins from Melastoma malabathricum. Phytochemistry 1992, 31, 2829–2833. [Google Scholar]
- Hatano, T.; Namba, O.; Chen, L.; Yasuhara, T.; Yazaki, K.; Yoshida, T.; Okuda, T. Hydrolyzable tannins having a depsidone-forming valoneoyl group. Heterocycles 1990, 31, 1221–1224. [Google Scholar]
- Yoshida, T.; Namba, O.; Chen, L.; Liu, Y.; Okuda, T. Ellagitannin Monomers and Oligomers from Euphorbia prostrata AIT. and Oligomers from Loropetalum chinense OLIV. Chem. Pharm. Bull. 1990, 38, 3296–3302. [Google Scholar] [CrossRef]
- Elyashberg, M.; Williams, A.J.; Blinov, K. Structural revisions of natural products by Computer-Assisted Structure Elucidation (CASE) systems. Nat. Prod. Rep. 2010, 27, 1296–1328. [Google Scholar] [PubMed]
- Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature 2013, 495, 461–466. [Google Scholar] [CrossRef] [PubMed]
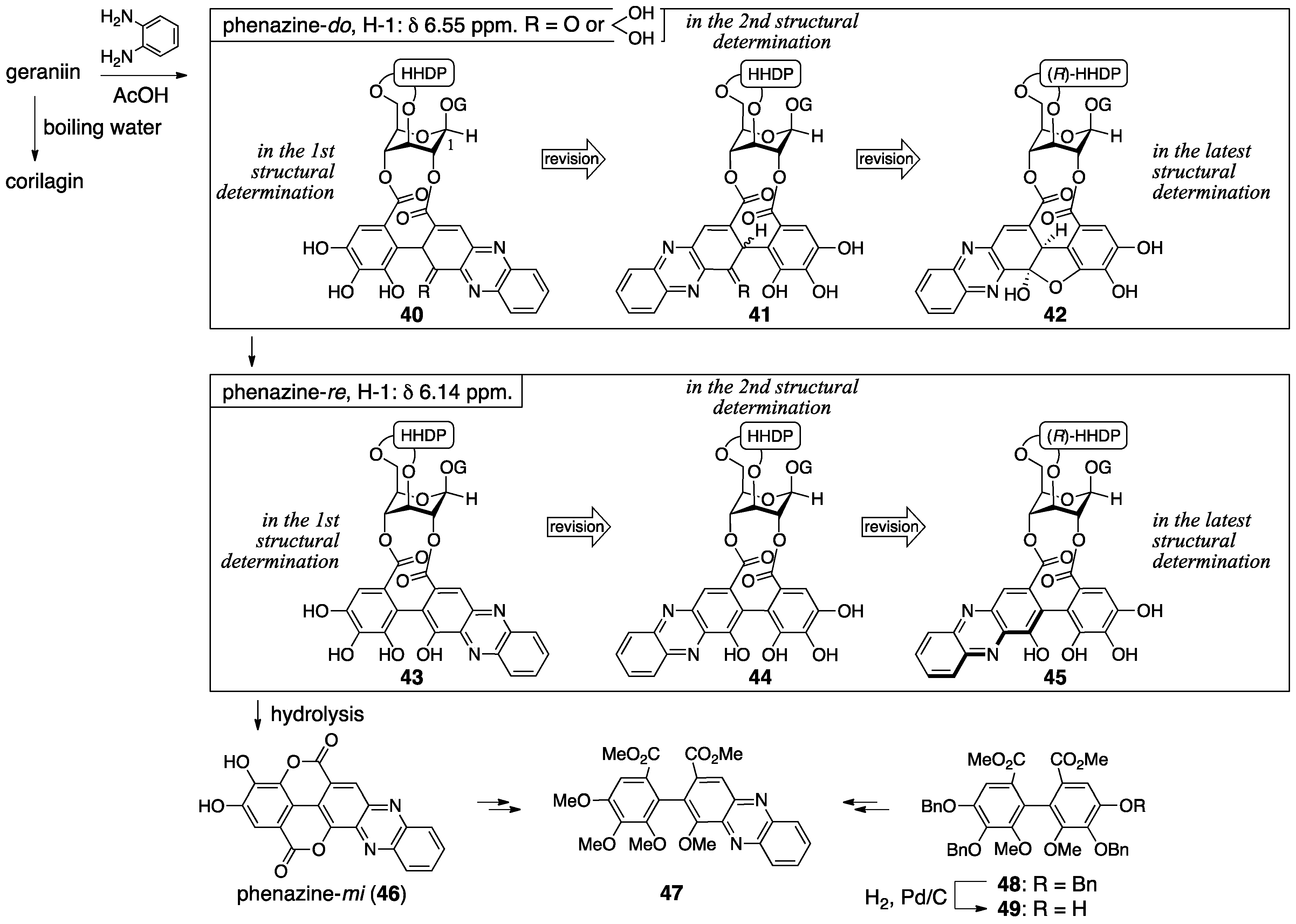



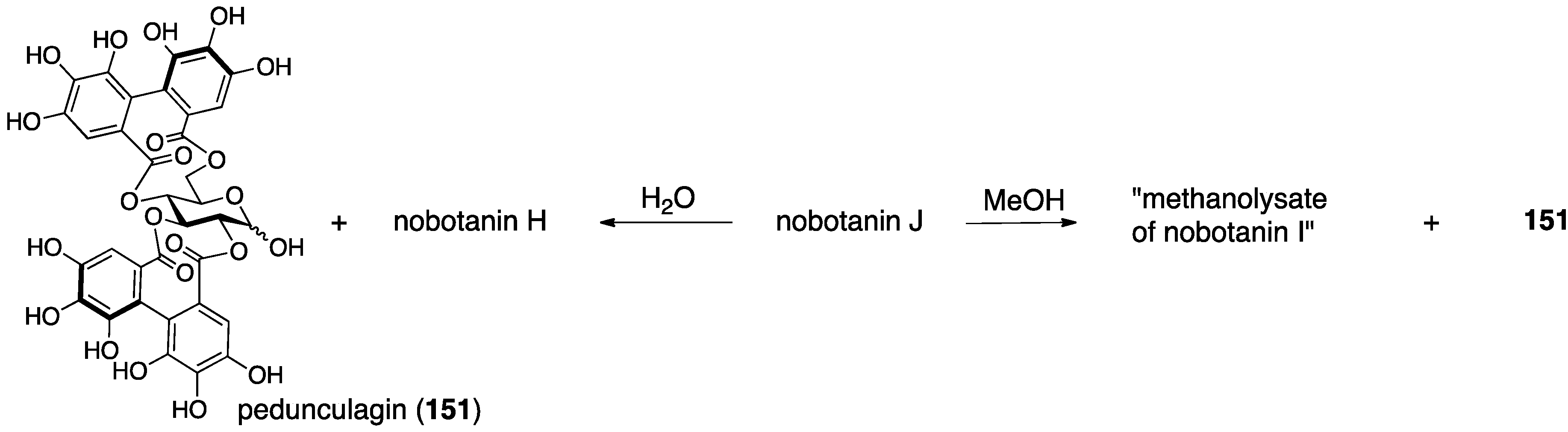

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
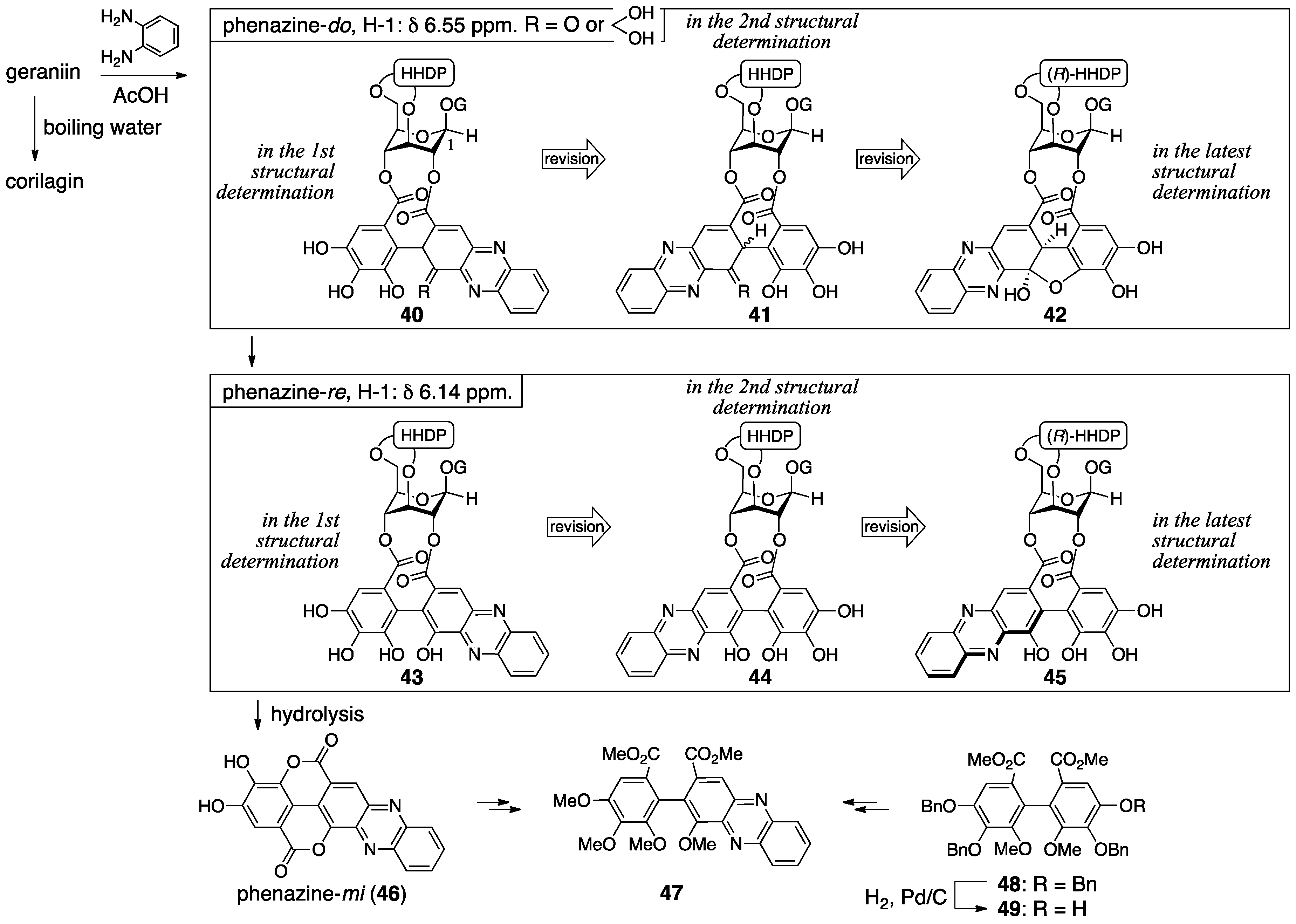{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
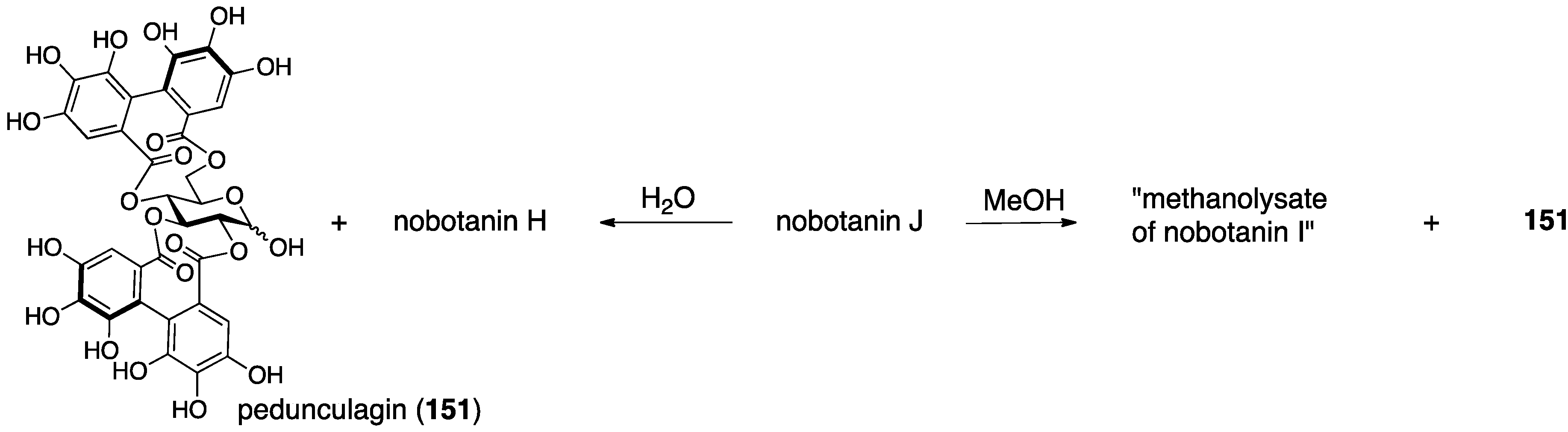{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section | Compound Name | Cause(s) for Wrong Structure | How the Error Was Realized | Methods Used for Latest Structural Confirmation |
|---|---|---|---|---|
| 4.1.1. | corilagin | Prediction based on similarity of CD/ORD spectra | unclear | Identification with a known fragment |
| Total synthesis and identification | ||||
| 4.1.2. | punigluconin | Misassignment of NMR data | Structural determination of analogous compounds | NMR studies |
| 4.1.3. | cercidinins A and B | Prediction based on similarity of NMR data with analogous compounds | Total synthesis | NMR studies with long-range methods |
| Total synthesis and identification | ||||
| 4.1.4. | roxbin B | Prediction based on similarity of CD/ORD spectra | Total synthesis | Identification of NMR data to a known compound |
| Total synthesis and identification | ||||
| 4.2.1 | geraniin | Misinterpretation of NMR data | Contradiction between NMR data and reported structure | NMR studies |
| Single-crystal X-ray diffraction | ||||
| 4.2.2. | terchebin | Prediction based on analogous compounds Incorrect experimental results | Contradiction between NMR data and reported structure | Structural determination from the beginning |
| 4.2.3. | isoterchebin | Incorrect experimental results | Identification of the reported structure to the other compound | Structural determination from the beginning involving identification |
| 4.3. | chebulinic acid and chebulagic acid | Unreasonable structure determination under lack of evidence | Contradiction between NMR data and reported structure | Structural determination from the beginning |
| Single-crystal X-ray diffraction of a fragment | ||||
| 4.4.1. | castalin, vescalin, castalagin, vescalagin, casuarinin, and stachyurin | Misinterpretation of NMR data | unclear | NMR studies |
| Prediction based on similarity of CD/ORD spectra | Chemical calculation | |||
| Total synthesis and identification | ||||
| 4.4.2. | punicalin and punicalagin | Use of molecular model for the final basis | Structural determination of analogous compounds | Structural determination from the beginning involving identification |
| 4.5.1 | sanguiin H-2, H-3, and H-6 | Prediction based on similarity of CD/ORD spectra | Structural determination of analogous compounds | Structural determination from the beginning |
| Synthesis of a fragment and identification | ||||
| 4.5.2. | alnusiin | Use of the additivity of the substituent effect in the 13C-NMR spectrum | unclear | NMR studies with long-range methods |
| 4.5.3 | alnusnins A and B | Incorrect experimental results | Structural determination of analogous compounds | Correct mass spectra |
| 4.5.4. | nobotanins B, C, E, G, H, I, J, and K | Prediction based on wrong structures | Structural determination of analogous compounds | NMR studies with long-range methods |
| Use of molecular model for the final basis |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, H.; Wakamori, S.; Hirokane, T.; Ikeuchi, K.; Matsumoto, S. Structural Revisions in Natural Ellagitannins. Molecules 2018, 23, 1901. https://doi.org/10.3390/molecules23081901
Yamada H, Wakamori S, Hirokane T, Ikeuchi K, Matsumoto S. Structural Revisions in Natural Ellagitannins. Molecules. 2018; 23(8):1901. https://doi.org/10.3390/molecules23081901
Chicago/Turabian StyleYamada, Hidetoshi, Shinnosuke Wakamori, Tsukasa Hirokane, Kazutada Ikeuchi, and Shintaro Matsumoto. 2018. "Structural Revisions in Natural Ellagitannins" Molecules 23, no. 8: 1901. https://doi.org/10.3390/molecules23081901
APA StyleYamada, H., Wakamori, S., Hirokane, T., Ikeuchi, K., & Matsumoto, S. (2018). Structural Revisions in Natural Ellagitannins. Molecules, 23(8), 1901. https://doi.org/10.3390/molecules23081901