One-Dimensional 13C NMR Is a Simple and Highly Quantitative Method for Enantiodiscrimination


Abstract
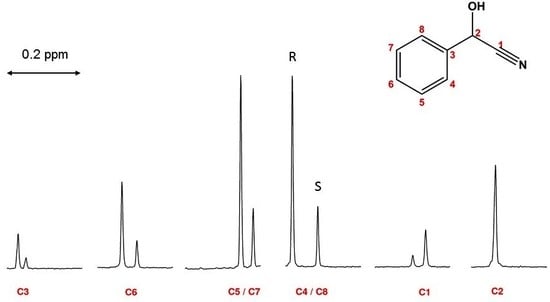
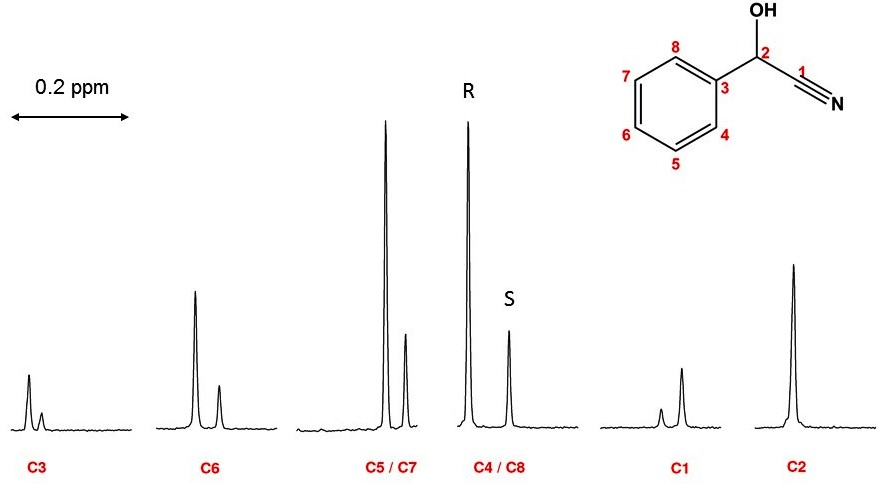
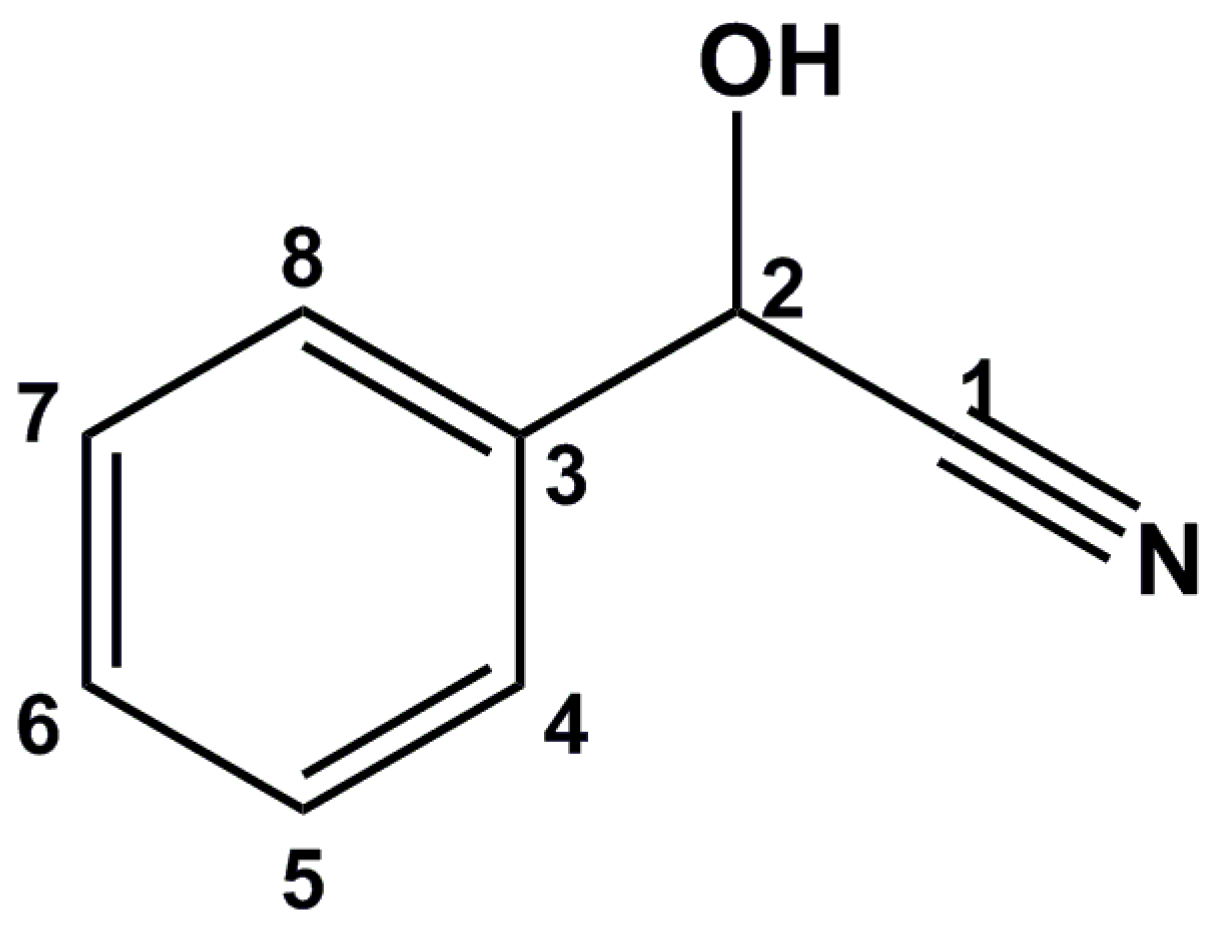
1. Introduction
2. Results
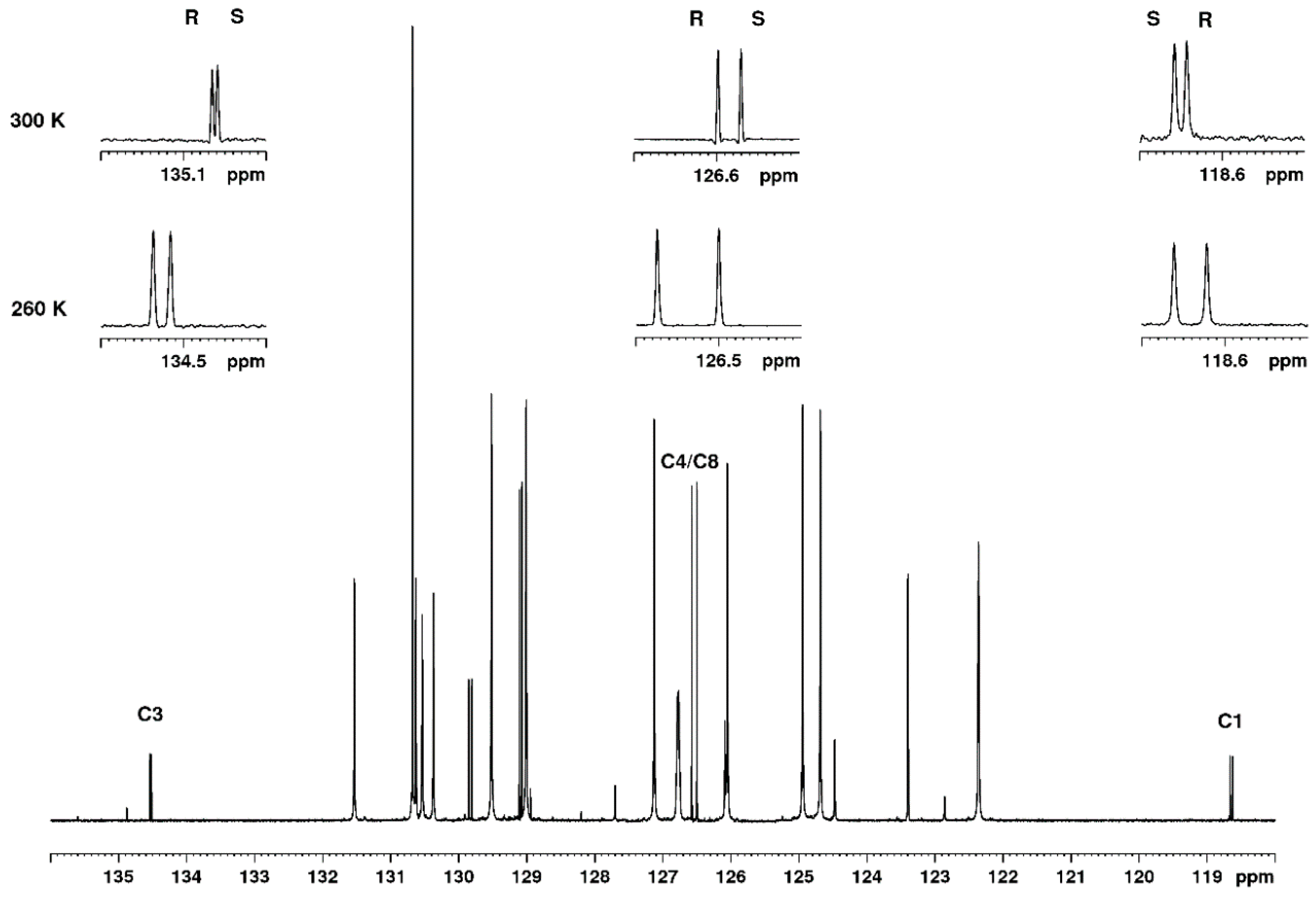
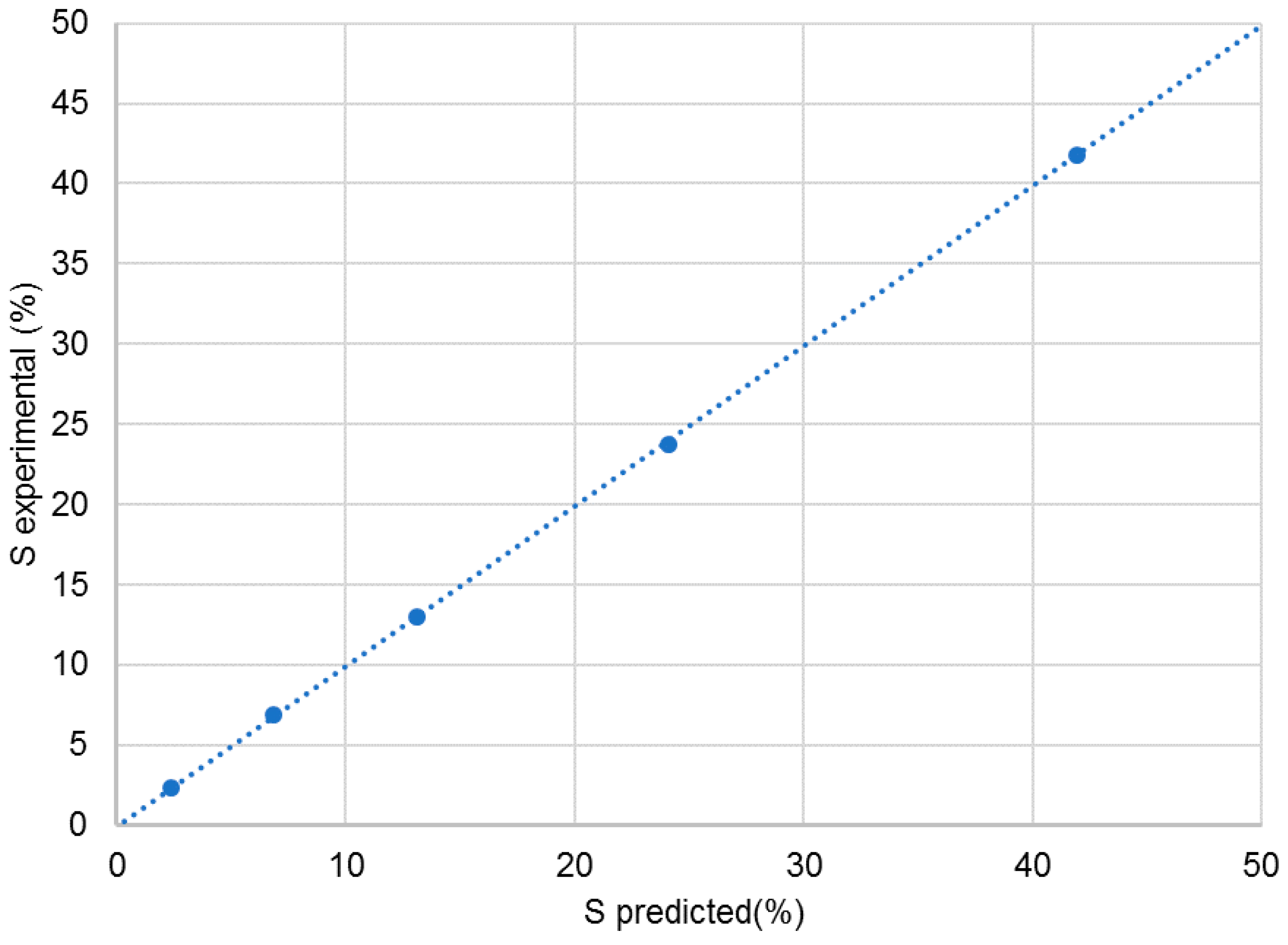
2.1. Linearity and Quantitative Trueness of Enantiodiscrimination by 1D 13C NMR
2.2. Limit of Detection (LOD)
2.3. Repeatability
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wenzel, T.J. Discrimination of Chiral Compounds Using NMR Spectroscopy; Wiley Interscience: Hoboken, NJ, USA, 2007. [Google Scholar]
- Uccello-Barretta, G.; Balzano, F. Chiral NMR solvating additives for differentiation of enantiomers. In Differentiation of Enantiomers II. Topics in Current Chemistry; Schurig, V., Ed.; Springer: Cham, Switzerland, 2013; Volume 341, pp. 69–131. [Google Scholar]
- Pirkle, W.H.; Sikkenga, D.L.; Pavlin, M.S. Nuclear Magnetic Resonance Determination of Enantiomeric Composition and Absolute Configuration of γ-Lactones Using Chiral 2,2,2-Trifluoro-1-(9-anthryl)ethanol. J. Org. Chem. 1977, 42, 384–387. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Sikkenga, D.L. The use of chiral solvating agents for nuclear magnetic resonance determination of enantiomeric purity and absolute configuration of lactones. Consequences of three-point interactions. J. Org. Chem. 1977, 42, 1370–1374. [Google Scholar] [CrossRef]
- Schripsema, J.; De Rudder, K.E.E.; Van Vliet, T.B.; Lankhorst, P.P.; De Vroom, E.; Kijne, J.W.; Van Brussel, A.A.N. Bacteriocin small of Rhizobium leguminosarum belongs to the class of N-acyl-l-homoserine lactone molecules, known as autoinducers and as quorum sensing co-transcription factors. J. Bacteriol. 1996, 178, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, A.; Virgili, A. The use of perdeuterio 2,2,2-trifluoro-1-(9-anthryl) ethanol as chiral solvating agent. Chiral induction observed on 1H and 13C NMR. Enantiomer 2001, 6, 235–243. [Google Scholar]
- Latypov, S.; Franck, X.; Jullian, J.C.; Hocquemiller, R.; Figadère, B. NMR determination of absolute configuration of butenolides of annonaceous type. Chem. Eur. J. 2002, 8, 5662–5666. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Chisholm, C.D. Using NMR spectroscopic methods to determine enantiomeric purity and assign absolute stereochemistry. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 1–63. [Google Scholar] [CrossRef] [PubMed]
- Weisman, G. Nuclear Magnetic Resonance analysis using chiral solvating agents. In Asymmetric Synthesis 1; Morrison, J.D., Ed.; Academic Press: New York, NY, USA, 1983; pp. 153–196. [Google Scholar]
- Pérez-Trujillo, M.; Monteagudo, E.; Parella, T. 13C NMR spectroscopy for the differentiation of enantiomers using chiral solvating agents. Anal. Chem. 2013, 85, 10887–10894. [Google Scholar] [CrossRef] [PubMed]
- Pavia, A.A.; Lacombe, J. 13C NMR spectroscopy, a useful tool to determine the enantiomeric purity of synthetic threonine-containing glycopeptides. Spectra of diastereomeric α- and β-d-galactopyranosyl-l- and -d-threonine and -l- and -d-allothreonine. J. Org. Chem. 1983, 48, 2564–2568. [Google Scholar] [CrossRef]
- Bremser, W.; Vogel, G. Determination of the enantiomeric purity of tocopherol by 13C NMR spectroscopy. Org. Magn. Reson. 1980, 14, 155–156. [Google Scholar] [CrossRef]
- Lankhorst, P.P.; Netscher, T.; Duchateau, A.L.L. A simple 13C NMR method for the discrimination of complex mixtures of stereoisomers: All eight stereoisomers of α-tocopherol resolved. Chirality 2015, 27, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.S. Recent advances in multinuclear NMR spectroscopy for chiral recognition of organic compounds. Molecules 2017, 22, 247. [Google Scholar] [CrossRef] [PubMed]
- Castañar, L.; Pérez-Trujillo, M.; Nolis, P.; Monteagudo, E.; Virgili, A.; Parella, T. Enantiodifferentiation through frequency-selective pure-shift 1H Nuclear Magnetic Resonance. ChemPhysChem 2014, 15, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Trujillo, M.; Castañar, L.; Monteagudo, E.; Kuhn, L.T.; Nolis, P.; Virgili, A.; Thomas Williamson, R.; Parella, T. Simultaneous 1H and 13C enantiodifferentiation from highly-resolved pure shift HSQC spectra. Chem. Commun. 2014, 50, 10214–10217. [Google Scholar] [CrossRef] [PubMed]
- Hickel, A.; Gradnig, G.; Schall, M.; Zangger, K.; Griengl, H. Determination of enantiomeric purity of mandelonitrile with derivatized cyclodextrins in water. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1997, 53, 451–455. [Google Scholar] [CrossRef]
- Hanefeld, U.; Straathof, A.J.J.; Heijnen, J.J. Enzymatic formation and esterification of (S)-mandelonitrile. J. Mol. Catal. B Enzym. 2001, 11, 213–218. [Google Scholar] [CrossRef]
- Kato, Y.; Yoshida, S.; Xie, S.-X.; Asano, Y. Aldoxime dehydratase co-existing with nitrile hydratase and amidase in the iron-type nitrile hydratase-producer Rhodococcus sp. N-771. J. Biosci. Bioeng. 2004, 97, 250–259. [Google Scholar] [CrossRef]
- Ueatrongchit, T.; Komeda, H.; Asano, Y.; H-Kittikun, A. Parameters influencing asymmetric synthesis of (R)-mandelonitrile by a novel (R)-hydroxynitrile lyase from Eriobotrya japonica. J. Mol. Catal. B Enzym. 2009, 56, 208–214. [Google Scholar] [CrossRef]
- Nanda, S.; Kato, Y.; Asano, Y. A new (R)-hydroxynitrile lyase from Prunus mume: Asymmetric synthesis of cyanohydrins. Tetrahedron Lett. 2005, 61, 10908–10916. [Google Scholar] [CrossRef]
- Asano, Y.; Tamura, K.; Doi, N.; Ueatrongchit, T.; H-Kittikun, A.; Ohmiya, T. Screening for New Hydroxynitrilases from Plants. Biosci. Biotechnol. Biochem. 2005, 69, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Wu, J.; Xu, G.; Yang, L. Kinetic study of lipase catalyzed asymmetric transesterification of mandelonitrile in solvent-free system. Chem. Eng. J. 2008, 138, 258–263. [Google Scholar] [CrossRef]
- Pawar, S.V.; Yadav, G.D. Enantioselective enzymatic hydrolysis of rac-mandelonitrile to R-mandelamide by nitrile hydratase immobilized on poly(vinyl alcohol)/chitosan-glutaraldehyde support. Ind. Eng. Chem. Res. 2014, 53, 7986–7991. [Google Scholar] [CrossRef]
- Ribeiro, S.S.; De Oliveira, J.R.; Porto, A. Lipase-catalyzed kinetic resolution of (±)-mandelonitrile under conventional condition and microwave irradiation. J. Braz. Chem. Soc. 2012, 23, 1395–1399. [Google Scholar] [CrossRef]
- Andexer, J.; Von Langermann, J.; Mell, A.; Bocola, M.; Kragl, U.; Eggert, T.; Pohl, M. An R-selective hydroxynitrile lyase from Arabidopsis thaliana with an α/β-hydrolase fold. Angew. Chem. Int. Ed. 2007, 46, 8679–8681. [Google Scholar] [CrossRef] [PubMed]
- Baleizão, C.; Gigante, B.; Garcia, H.; Corma, A. Ionic liquids as green solvents for the asymmetric synthesis of cyanohydrins catalysed by VO(salen) complexes. Green Chem. 2002, 4, 272–274. [Google Scholar] [CrossRef]
- Thomas, J.C.; Aggio, B.B.; Marques de Oliveira, A.R.; Piovan, L. High-Throughput Preparation of Optically Active Cyanohydrins Mediated by Lipases. Eur. J. Org. Chem. 2016, 2016, 5964–5970. [Google Scholar] [CrossRef]
- Veum, L.; Hanefeld, U.; Pierre, A. The first encapsulation of hydroxynitrile lyase from Hevea brasiliensis in a sol-gel matrix. Tetrahedron 2004, 60, 10419–10425. [Google Scholar] [CrossRef]
- Queseda-Moreno, M.M.; Virgili, A.; Monteagudo, E.; Claramunt, R.M.; Aviles-Moreno, J.R.; Lopez-Gonzalez, J.J.; Alkorta, I.; Elguero, J. A vibrational circular dichroism (VCD) methodology for the measurement of enantiomeric excess in chiral compounds in the solid phase and for the complementary use of NMR and VCD techniques in solution: The camphor case. Analyst 2018, 143, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Otte, D.A.L.; Borchmann, D.E.; Lin, C.; Weck, M.; Woerpel, K.A. 13C NMR spectroscopy for the quantitative determination of compound ratios and polymer end groups. Org. Lett. 2014, 16, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- Lankhorst, P.P.; Poot, M.M.; De Lange, M.P.A. Quantitative determination of lovastatin and dihydrolovastatin by means of 1H NMR spectroscopy. Pharmacop. Forum 1996, 22, 2414–2422. [Google Scholar]
- Holzgrabe, U. Quantitative NMR spectroscopy in pharmaceutical applications. Prog. Nucl. Magn. Reson. Spectrosc. 2010, 57, 229–240. [Google Scholar] [CrossRef] [PubMed]
- International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use. Validation of Analytical Procedures: Text and Methodology; ICH-Q2B; ICH Expert Working Group: Geneva, Switzerland, 1996. [Google Scholar]
- Monteagudo, E.; Virgili, A.; Parella, T.; Pérez-Trujillo, M. Chiral Recognition by Dissolution DNP NMR Spectroscopy of 13C-Labeled dl-Methionine. Anal. Chem. 2017, 89, 4939–4944. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | B | C |
|---|---|---|
| % S by Weight | %S Predicted (after Correction) | % S by Experiment |
| 0 | 2.4 | 2.4 |
| 4.3 | 6.8 | 6.9 |
| 10.5 | 13.1 | 13.0 |
| 21.7 | 24.1 | 23.7 |
| 40.8 | 41.9 | 41.8 |
| 50 | 50.1 | 50.1 |
| Replicate nr | S Enantiomer (%) | S Enantiomer (%) |
|---|---|---|
| 1 | 23.7 | 6.9 |
| 2 | 23.8 | 6.8 |
| 3 | 23.8 | 6.7 |
| 4 | 23.9 | 6.7 |
| 5 | 23.9 | 6.7 |
| 6 | 23.8 | 6.8 |
| Average | 23.8 | 6.8 |
| Standard deviation | 0.1 | 0.1 |
| Relative standard deviation (%) | 0.3 | 1.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lankhorst, P.P.; Van Rijn, J.H.J.; Duchateau, A.L.L. One-Dimensional 13C NMR Is a Simple and Highly Quantitative Method for Enantiodiscrimination. Molecules 2018, 23, 1785. https://doi.org/10.3390/molecules23071785
Lankhorst PP, Van Rijn JHJ, Duchateau ALL. One-Dimensional 13C NMR Is a Simple and Highly Quantitative Method for Enantiodiscrimination. Molecules. 2018; 23(7):1785. https://doi.org/10.3390/molecules23071785
Chicago/Turabian StyleLankhorst, Peter P., Jozef H. J. Van Rijn, and Alexander L. L. Duchateau. 2018. "One-Dimensional 13C NMR Is a Simple and Highly Quantitative Method for Enantiodiscrimination" Molecules 23, no. 7: 1785. https://doi.org/10.3390/molecules23071785
APA StyleLankhorst, P. P., Van Rijn, J. H. J., & Duchateau, A. L. L. (2018). One-Dimensional 13C NMR Is a Simple and Highly Quantitative Method for Enantiodiscrimination. Molecules, 23(7), 1785. https://doi.org/10.3390/molecules23071785