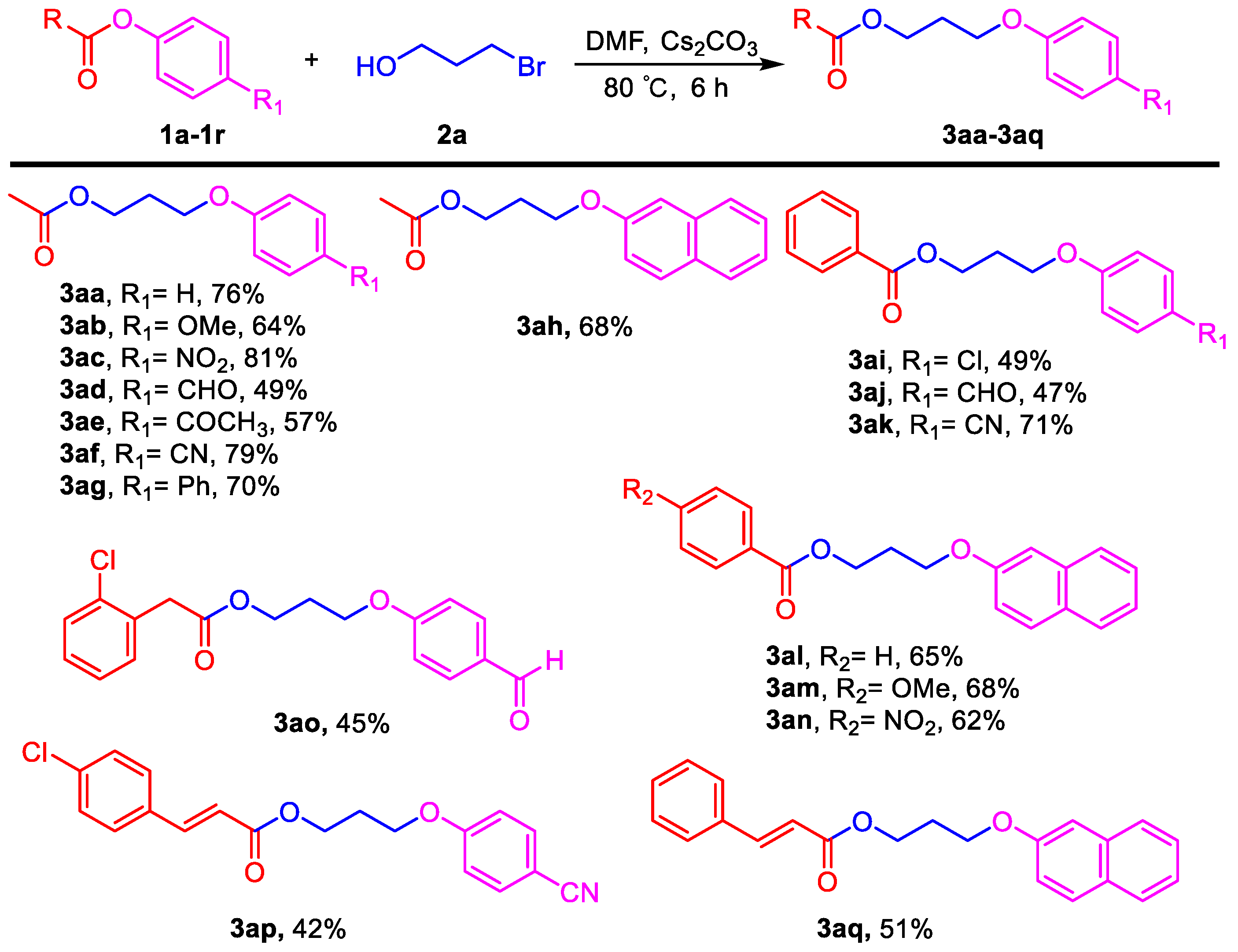
3.2. General Procedure for the Aryloxyalkyl Esters from Phenolic Esters (3aa–3aq)
Phenyl esters 1a–1c (0.5 mmol) were loaded into a flask (10 mL). DMF (2 mL) and Cs2CO3 (488 mg, 1.5 mmol, 3.0 equiv) were then added, which was followed by the addition of 3-bromoethanol (2a, 0.525 mmol, 1.05 equiv). Then the reaction mixture was stirred at 80 °C for 6 h under an Ar atmosphere. After completion of the reaction, as confirmed by TLC, the reaction mixture was cooled down to room temperature and 10 mL of CH2Cl2 (DCM) and 10 mL of water were added. After separation of the dichloromethane layer from the water, the aqueous phase was extracted with CH2Cl2 (2 × 5 mL) again. The combined organic layers were then dried over anhydrous Na2SO4, filtered, and concentrated under vacuum to yield the crude product. The crude product was purified by silica gel column chromatography to obtain the desired pure compound.
3-Phenoxypropyl acetate (3aa). Compound 3aa (CAS: 58883-98-0) was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 200:1) to provide the pure compound (73 mg, 76%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.33 (t, J = 8.2 Hz, 2H), 6.93–6.99 (m, 1H), 6.93 (d, J = 8.6 Hz, 2H), 4.31 (t, J = 6.1 Hz, 2H), 4.07 (t, J = 6.1 Hz, 2H), 2.13–2.19 (m, 2H), 2.09 (s, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 170.7, 158.2, 129.1, 120.3, 113.9, 63.6, 60.9, 28.1, 20.4; MS (ESI): m/z = 193.1 [M − H]−; Rf = 0.5 (DCM:MeOH 100:1).
3-(4-Methoxyphenoxy)propyl acetate (3ab). Compound 3ab was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 200:1) to provide the pure compound (71 mg, 64%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 6.85 (s, 4H, Ar-H), 4.27 (t, J = 6.1 Hz, 2H), 4.01 (t, J = 6.1 Hz, 2H), 3.78 (s, 3H, -OCH3), 2.08–2.14 (m, 2H), 2.07 (s, 3H, -COCH3); 13C-NMR (75 MHz, CDCl3, δ ppm): 196.8, 171.0, 162.6, 130.6, 130.4, 114.1, 64.6, 61.0, 28.4, 26.3, 20.9; HRMS (ESI): m/z calcd. for C12H17O4: 225.1121, found: 225.1119 [M + H]+; Rf = 0.5 (DCM:MeOH 100:1).
3-(4-Nitrophenoxy)propyl acetate (3ac). Compound 3ac (CAS: 99986-51-3) was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (96 mg, 81%) as a light yellow oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.17 (d, J = 8.1 Hz, 2H), 6.5 (d, J = 8.1 Hz, 2H), 4.26 (t, J = 6.1 Hz, 2H), 4.14 (t, J = 6.1 Hz, 2H), 2.12–2.20 (m, 2H), 2.05 (s, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 170.4, 163.2, 140.9, 125.3, 113.9, 64.7, 60.3, 27.8, 20.4; MS (ESI): m/z = 238.1 [M − H]−; Rf = 0.2 (DCM:MeOH 100:1).
3-(4-Formylphenoxy)propyl acetate (3ad). Compound 3ad was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 50:1) to provide the pure compound (54 mg, 49%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 9.83 (s, 1H, -CHO), 7.78 (d, J = 8.1 Hz, 2H), 6.97 (d, J = 8.1 Hz, 2H), 4.11 (t, J = 6.1 Hz, 2H), 4.04 (t, J = 6.1 Hz, 2H), 2.02 (s, 3H, -COCH3), 1.77–1.89 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 190.3, 170.6, 163.4, 131.5, 129.3, 114.1, 67.1, 63.4, 24.7, 20.4; MS (ESI): m/z = 221.1 [M − H]−; HRMS (ESI): m/z calcd. for C12H15O4: 223.0965, found: 223.0961 [M + H]+; Rf = 0.3 (DCM:MeOH 50:1).
3-(4-Acetylphenoxy)propyl acetate (3ae). Compound 3ae (CAS: 146274-20-6) was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (66 mg, 57%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.94 (d, J = 8.1 Hz, 2H), 6.95 (d, J = 8.1 Hz, 2H), 4.27 (t, J = 6.1 Hz, 2H), 4.12 (t, J = 6.1 Hz, 2H), 2.56 (s, 3H), 2.13–2.17 (m, 2H), 2.06 (s, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 196.8, 171.0, 162.7, 130.6, 130.4, 114.1, 64.6, 61.0, 28.4, 26.3, 20.9; MS (ESI): m/z = 235.1 [M + H]+; Rf = 0.3 (DCM:MeOH 100:1).
3-(4-Cyanophenoxy)propyl acetate (3af). Compound 3af was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (86 mg, 79%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.58 (d, J = 8.2 Hz, 2H), 6.96 (d, J = 8.2 Hz, 2H), 4.26 (t, J = 6.1 Hz, 2H), 4.09 (t, J = 6.1 Hz, 2H), 2.07–2.18 (m, 2H), 2.04 (s, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 161.5, 150.4, 133.4, 118.6, 114.6, 103.5, 64.2, 60.4, 27.8, 20.4; HRMS (ESI): m/z calcd. for C12H14NO4: 220.0968, found: 220.0972 [M + H]+; Rf = 0.3 (DCM:MeOH 100:1).
3-([1,1′-Biphenyl]-4-yloxy)propyl acetate (3ag). Compound 3ag (CAS: 1417800-59-9) was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (94 mg, 70%) as a light yellow solid. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.58 (d, J = 8.2 Hz, 2H), 7.47 (t, J = 6.2 Hz, 2H), 7.34–7.41 (m, 3H), 7.10 (t, J = 8.2 Hz, 1H), 7.05 (t, J = 8.2 Hz, 1H), 4.22 (t, J = 6.1 Hz, 2H), 4.09 (t, J = 6.1 Hz, 2H), 2.09–2.13 (m, 2H), 2.05 (s, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 170.5, 155.1, 137.9, 130.5, 129.0, 128.1, 127.4, 126.4, 120.7, 112.1, 64.3, 60.8, 28.1, 20.4; MS (ESI): m/z = 293.2 [M + Na]+; Rf = 0.5 (DCM:MeOH 100:1).
3-(Naphthalen-2-yloxy)propyl acetate (3ah). Compound 3ah (CAS: 1174495-88-5) was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (83 mg, 68%) as a yellow solid. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.78–7.84 (m, 3H), 7.51 (t, J = 6.2 Hz, 1H), 7.40 (t, J = 6.2 Hz, 1H), 7.18–7.23 (m, 2H), 4.36 (t, J = 6.1 Hz, 2H), 4.17 (t, J = 6.1 Hz, 2H), 2.17–2.25 (m, 2H), 2.12 (s, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 170.6, 156.3, 134.1, 129.0, 128.5, 127.2, 126.3, 125.9, 123.2, 118.4, 106.1, 63.8, 60.92, 28.1, 20.5; MS (ESI): m/z = 243.1 [M − H]−; Rf = 0.6 (DCM:MeOH 100:1).
3-(4-Chlorophenoxy)propyl benzoate (3ai). Compound 3ai was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (71 mg, 49%) as a light yellow solid. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.08 (d, J = 8.2 Hz, 2H), 7.58 (t, J = 6.2 Hz, 1H), 7.46 (t, J = 6.2 Hz, 2H), 7.24 (d, J = 8.2 Hz, 2H), 6.85 (d, J = 8.2 Hz, 2H), 4.55 (t, J = 6.1 Hz, 2H), 4.11 (t, J = 6.1 Hz, 2H), 2.23–2.31 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 165.9, 156.9, 132.5, 129.6, 129.0, 128.8, 127.9, 125.1, 115.2, 64.3, 61.2, 28.2; HRMS (ESI): m/z calcd. for C16H16ClO3: 291.0782, found: 291.0779 [M + H]+; Rf = 0.3 (DCM:MeOH 100:1).
3-(4-Formylphenoxy)propyl benzoate (3aj). Compound 3aj (CAS: 864845-56-7) was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 50:1) to provide the pure compound (67 mg, 47%) as a yellow solid. 1H-NMR (300 MHz, CDCl3, δ ppm): 9.88 (s, 1H, -CHO), 8.04 (d, J = 8.6 Hz, 2H), 7.84 (d, J = 8.2 Hz, 2H), 7.57 (t, J = 6.2 Hz, 1H), 7.42 (t, J = 8.2 Hz, 2H), 7.01 (d, J = 8.2 Hz, 2H), 4.54 (t, J = 6.1 Hz, 2H), 4.22 (t, J = 6.1 Hz, 2H), 2.26–2.33 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 190.3, 159.2, 132.5, 131.5, 129.0, 127.9, 114.2, 64.4, 61.0, 28.1; MS (ESI): m/z = 283.1 [M − H]−; Rf = 0.2 (DCM:MeOH 50:1).
3-(4-Cyanophenoxy)propyl benzoate (3ak). Compound 3ak was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (100 mg, 71%) as yellow solids. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.07 (d, J = 8.6 Hz, 2H), 7.61 (t, J = 6.2 Hz, 1H), 7.46 (t, J = 8.2 Hz, 2H), 7.24 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 8.2 Hz, 2H), 4.55 (t, J = 6.1 Hz, 2H), 4.11 (t, J = 6.1 Hz, 2H), 2.23–2.32 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 165.8, 161.5, 133.4, 132.5, 129.5, 129.0, 127.9, 118.7, 114.7, 103.4, 64.4, 61.0, 28.0; HRMS (ESI): m/z calcd. for C17H16NO3: 282.1125, found: 282.1129 [M + H]+; Rf = 0.6 (DCM:MeOH 50:1).
3-(Naphthalen-2-yloxy)propyl benzoate (3al). Compound 3al was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (99 mg, 65%) as yellow solids. 1H-NMR (300 MHz, DMSO-d6, δ ppm): 1H-NMR (300 MHz, CDCl3, δ ppm): 8.05 (d, J = 8.6 Hz, 2H), 7.78–7.85 (m, 3H), 7.60 (t, J = 6.2 Hz, 1H), 7.51 (t, J = 6.2 Hz, 1H), 7.37 (t, J = 6.2 Hz, 1H), 7.26 (d, J = 8.2 Hz, 2H), 6.93 (d, J = 8.2 Hz, 2H), 4.62 (t, J = 6.1 Hz, 2H), 4.29 (t, J = 6.1 Hz, 2H), 2.31–2.40 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 166.1, 162.4, 133.9, 131.7, 129.1, 128.2, 127.6, 127.1, 126.8, 126.2, 123.6, 122.7, 118.9, 113.2, 106.3, 63.8, 61.4, 27.9; HRMS (ESI): m/z calcd. for C20H19O3: 307.1329, found: 307.1335 [M + H]+; Rf = 0.4 (DCM:MeOH 100:1).
3-(Naphthalen-2-yloxy)propyl 4-methoxybenzoate (3am). Compound 3am was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (114 mg, 68%) as yellow solids. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.07 (d, J = 8.6 Hz, 2H), 7.76–7.83 (m, 3H), 7.49 (t, J = 6.2 Hz, 1H), 7.39 (t, J = 6.2 Hz, 1H), 7.23 (d, J = 8.2 Hz, 2H), 6.95 (d, J = 8.2 Hz, 2H), 4.60 (t, J = 6.1 Hz, 2H), 4.27 (t, J = 6.1 Hz, 2H), 3.86 (s, 3H, -OCH3), 2.36–2.42 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 165.8, 162.9, 156.3, 134.1, 131.2, 129.0, 128.5, 127.2, 126.3, 125.9, 123.2, 122.1, 118.5, 113.2, 106.1, 64.0, 61.1, 54.9, 28.3; HRMS (ESI): m/z calcd. for C21H21O4: 337.1434, found: 337.1431 [M + H]+; Rf = 0.3 (DCM:MeOH 100:1).
3-(Naphthalen-2-yloxy)propyl 4-nitrobenzoate (3an). Compound 3an was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 50:1) to provide the pure compound (108 mg, 62%) as yellow solids. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.28 (d, J = 8.6 Hz, 2H), 8.21 (d, J = 8.6 Hz, 2H), 7.72–7.80 (m, 3H), 7.46 (t, J = 6.2 Hz, 1H), 7.37 (t, J = 6.2 Hz, 1H), 7.17 (d, J = 8.2 Hz, 2H), 4.66 (t, J = 6.1 Hz, 2H), 4.28 (t, J = 6.1 Hz, 2H), 2.36–2.42 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 164.1, 156.1, 149.9, 133.9, 130.5, 130.2, 129.0, 128.5, 127.1, 126.2, 125.9, 123.2, 123.0, 118.2, 106.0, 63.7, 62.4, 28.1; HRMS (ESI): m/z calcd. for C20H18NO5: 352.1179, found: 352.1175 [M + H]+; Rf = 0.2 (DCM:MeOH 100:1).
3-(4-Formylphenoxy)propyl 2-(2-chlorophenyl)acetate (3ao). Compound 3ao was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 50:1) to provide the pure compound (74 mg, 45%) as yellow solids. 1H-NMR (300 MHz, CDCl3, δ ppm): 9.87 (s, 2H, -CHO), 7.83 (d, J = 8.6 Hz, 2H), 7.35 (t, J = 6.2 Hz, 1H), 7.26–7.29 (m, 1H), 7.19–7.23 (m, 2H), 6.96 (d, J = 8.2 Hz, 2H), 4.33 (t, J = 6.1 Hz, 2H), 4.06 (t, J = 6.1 Hz, 2H), 3.78 (s, 2H), 2.12–2.16 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 190.2, 169.9, 163.2, 131.8, 131.4, 130.9, 129.5, 129.4, 128.9, 128.2, 126.4, 114.2, 64.0, 60.9, 38.7, 27.8; HRMS (ESI): m/z calcd. for C18H18ClO4: 333.0888, found: 333.0892 [M + H]+; Rf = 0.4 (DCM:MeOH 50:1).
3-(4-Cyanophenoxy)propyl (E)-3-(4-chlorophenyl)acrylate (3ap). Compound 3ap was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 40:1) to provide the pure compound (71 mg, 42%) as yellow solids. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.63 (d, J = 15.6 Hz, 1H), 7.58 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 8.2 Hz, 2H), 6.95 (d, J = 8.1 Hz, 2H), 6.39 (d, J = 15.6 Hz, 1H), 4.40 (t, J = 6.1 Hz, 2H), 4.13 (t, J = 6.1 Hz, 2H), 2.17–2.6 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 166.5, 162.0, 143.6, 136.2, 133.9, 132.7, 129.2, 129.1, 119.2, 118.3, 115.1, 104.0, 64.8, 61.3, 28.4; HRMS (ESI): m/z calcd. for C19H17ClNO3: 342.0891, found: 342.0896 [M + H]+; Rf = 0.3 (DCM:MeOH 50:1).
3-(Naphthalen-2-yloxy)propyl cinnamate (3aq). Compound 3aq was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 50:1) to provide the pure compound (84 mg, 51%) as yellow solids. 1H-NMR (300 MHz, DMSO-d6, δ ppm): 1H-NMR (300 MHz, CDCl3, δ ppm): 7.74–7.85 (m, 4H), 7.53–7.59 (m, 2H), 7.49 (t, J = 6.2 Hz, 1H), 7.36–7.47 (m, 4H), 7.20–7.24 (m, 2H), 6.52 (d, J = 15.6 Hz, 1H), 4.53 (t, J = 6.1 Hz, 2H), 4.27 (t, J = 6.1 Hz, 2H), 2.27–2.37 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 166.5, 156.3, 144.5, 134.1, 133.8, 129.8, 128.9, 128.4, 127.6, 127.3, 127.2, 126.3, 125.9, 123.2, 118.4, 117.5, 106.1, 63.9, 61.0, 28.3; HRMS (ESI): m/z calcd. for C22H21O3: 333.1485, found: 333.1487 [M + H]+; Rf = 0.4 (DCM:MeOH 50:1).
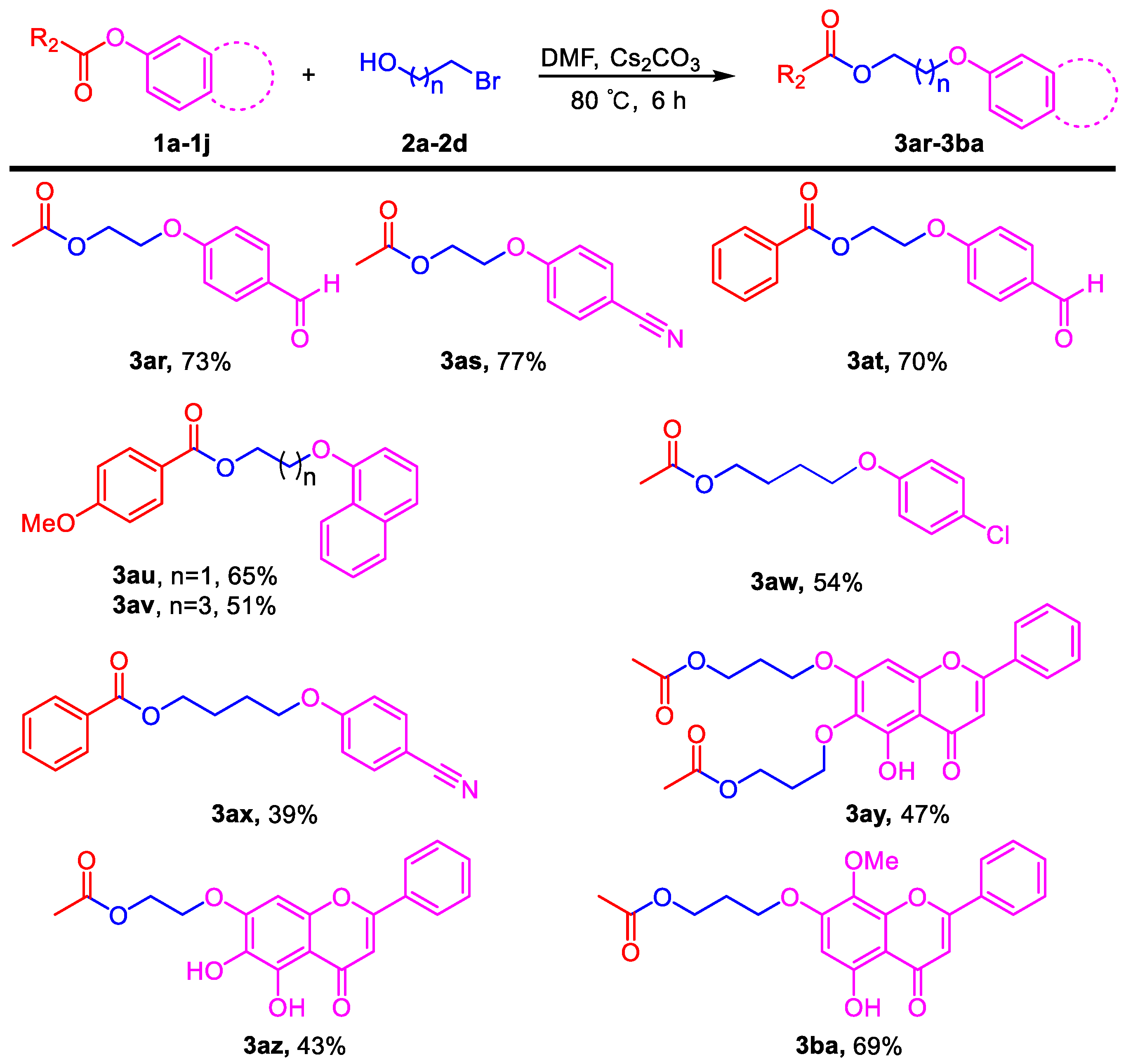
3.3. General Procedure for the Reaction of Halogenated Alcohols and Application to the Synthesis of Flavonoid Derivatives (3ar–3ba)
Phenyl esters 1a–1j (0.5 mmol) were loaded into a flask (10 mL). DMF (4 mL) and Cs2CO3 (488 mg, 1.5 mmol, 3.0 equiv) were then added, which was followed by the addition of bromohydrin (0.525 mmol, 1.05 equiv). Then, the reaction mixture was stirred at 80 °C for 6 h under an Ar atmosphere. After completion of the reaction, as confirmed by TLC, the reaction mixture was cooled down to room temperature and 10 mL of EtOAc and 10 mL of water were added. After separation of the EtOAc layer from the water, the aqueous phase was extracted with EtOAc (2 × 5 mL) again. The combined organic layers were then dried over anhydrous Na2SO4, filtered, and concentrated under vacuum to yield the crude product. The crude product was purified by silica gel column chromatography to obtain the desired pure compound.
2-(4-Formylphenoxy)ethyl acetate (3ar). Compound 3ar (CAS: 151270-72-3) was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (76 mg, 73%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 9.84 (s, 1H, -CHO), 7.79 (d, J = 8.6 Hz, 2H), 6.99 (d, J = 8.6 Hz, 2H), 4.41 (t, J = 6.1 Hz, 2H), 4.22 (t, J = 6.1 Hz, 2H), 2.06 (s, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 170.2, 162.8, 131.3, 129.6, 121.8, 114.2, 65.6, 61.8, 20.2; MS (ESI): m/z = 207.1 [M − H]−; Rf = 0.3 (DCM:MeOH 50:1).
2-(4-Cyanophenoxy)ethyl acetate (3as). Compound 3as (CAS: 150195-05-4) was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (79 mg, 77%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.56 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.6 Hz, 2H), 4.41 (t, J = 6.1 Hz, 2H), 4.20 (t, J = 6.1 Hz, 2H), 2.07 (s, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 170.3, 161.2, 133.4, 118.5, 114.7, 103.7, 65.6, 61.7, 20.3; MS (ESI): m/z = 204.1 [M − H]−; Rf = 0.3 (DCM:MeOH 100:1).
2-(4-Formylphenoxy)ethyl benzoate (3at). Compound 3at was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (94 mg, 70%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 9.91 (s, 1H, -CHO), 8.07 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 7.59 (t, J = 8.1 Hz, 1H), 7.43–7.48 (m, 2H), 7.07 (d, J = 8.1 Hz, 2H), 4.72 (t, J = 6.1 Hz, 2H), 4.41 (t, J = 6.1 Hz, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 165.9, 163.0, 132.7, 131.9, 131.5, 129.8, 129.2, 127.9, 115.8, 114.3, 65.7, 62.4; HRMS (ESI): m/z calcd. for C16H15O4: 271.0965, found: 271.0971 [M + H]+; Rf = 0.2 (DCM:MeOH 50:1).
2-(Naphthalen-1-yloxy)ethyl 4-methoxybenzoate (3au). Compound 3au was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (104 mg, 65%) as light yellow solids. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.10 (d, J = 8.1 Hz, 2H), 7.77–7.84 (m, 3H), 7.51 (t, J = 8.1 Hz, 1H), 7.40 (t, J = 8.1 Hz, 1H), 7.25 (m, 1H), 6.93 (d, J = 8.1 Hz, 2H), 4.75 (t, J = 6.1 Hz, 2H), 4.42 (t, J = 6.1 Hz, 2H), 3.85 (s, 3H, -OCH3); 13C-NMR (75 MHz, CDCl3, δ ppm): 166.3, 163.5, 156.6, 134.5, 131.8, 129.6, 129.1, 127.7, 126.8, 126.5, 123.9, 122.3, 118.9, 113.7, 106.9, 66.1, 63.2, 55.4; HRMS (ESI): m/z calcd. for C20H19O4: 323.1278, found: 323.1273 [M + H]+; Rf = 0.4 (DCM:MeOH 50:1).
4-(Naphthalen-1-yloxy)butyl 4-methoxybenzoate (3av). Compound 3av was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (89 mg, 51%) as light yellow solids. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.02 (d, J = 8.2 Hz, 2H), 7.79 (d, J = 8.1 Hz, 2H), 7.73 (d, J = 8.1 Hz, 2H), 7.46 (t, J = 8.1 Hz, 1H), 7.36 (t, J = 8.1 Hz, 1H), 7.18 (m, 1H), 6.91 (d, J = 8.2 Hz, 2H), 4.43 (t, J = 6.1 Hz, 2H), 4.18 (t, J = 6.1 Hz, 2H), 3.86 (s, 3H, -OCH3), 2.03–2.05 (m, 4H); 13C-NMR (75 MHz, CDCl3, δ ppm): 165.9, 162.8, 156.3, 147.0, 134.0, 131.0, 128.9, 127.1, 126.2, 125.8, 123.0, 122.2, 118.4, 113.0, 106.1, 66.8, 63.9, 54.9, 25.5, 25.1; HRMS (ESI): m/z calcd. for C22H23O4: 351.1591, found: 351.1587 [M + H]+; Rf = 0.4 (DCM:MeOH 50:1).
4-(4-Chlorophenoxy)butyl acetate (3aw). Compound 3aw was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (65 mg, 54%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.23 (d, J = 8.2 Hz, 2H), 6.82 (d, J = 8.2 Hz, 2H), 4.15 (t, J = 6.1 Hz, 2H), 3.95 (t, J = 6.1 Hz, 2H), 2.07 (s, 3H), 1.82–1.86 (m, 4H); 13C-NMR (75 MHz, CDCl3, δ ppm): 157.0, 128.7, 124.9, 116.1, 115.2, 67.0, 63.6, 25.2, 24.8, 20.4; HRMS (ESI): m/z calcd. for C12H16O3Cl: 243.0782, found: 243.0786 [M + H]+; Rf = 0.5 (DCM:MeOH 100:1).
5-(4-Cyanophenoxy)pentyl benzoate (3ax). Compound 3ax was prepared following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (60 mg, 39%) as a colorless oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.05 (d, J = 8.2 Hz, 2H), 7.55–7.59 (m, 3H), 7.45 (t, J = 6.3 Hz, 1H), 6.94 (d, J = 8.2 Hz, 2H), 4.38 (t, J = 6.1 Hz, 2H), 4.04 (t, J = 6.1 Hz, 2H), 1.83–1.95 (m, 4H), 1.60–1.70 (m, 2H); 13C-NMR (75 MHz, CDCl3, δ ppm): 166.6, 162.2, 133.9, 132.9, 129.5, 128.3, 119.3, 115.1, 68.0, 64.6, 28.6, 28.4, 22.6; HRMS (ESI): m/z calcd. for C19H20O3N: 310.1438, found: 310.142 [M + H]+; Rf = 0.6 (DCM:MeOH 50:1).
((5-Hydroxy-4-oxo-2-phenyl-4H-chromene-6,7-diyl)bis(oxy))bis(propane-3,1-diyl) diacetate (3ay). Compound 3ay was prepared from 6,7-diacetyl baicalein following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 50:1) to provide the pure compound (110 mg, 47%) as yellow solids. 1H-NMR (300 MHz, CDCl3, δ ppm): 12.67 (s, 1H, -OH), 7.90 (d, J = 8.1 Hz, 2H), 7.51–7.60 (m, 3H), 6.69 (s, 1H), 6.57 (s, 1H), 4.37 (t, J = 6.1 Hz, 2H), 4.33 (t, J = 6.1 Hz, 2H), 4.18 (t, J = 6.1 Hz, 2H), 4.15 (t, J = 6.1 Hz, 2H), 2.21–2.30 (m, 2H), 2.13 (m, 2H), 2.10 (s, 3H), 2.08 (s, 3H); HRMS (ESI): m/z calcd. for C25H27O9: 471.1650, found: 471.1659 [M + H]+; Rf = 0.6 (DCM:MeOH 30:1).
2-((5,6-Dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)ethyl acetate (3az). Compound 3az was prepared from 7-acetyl baicalein following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 40:1) to provide the pure compound (76 mg, 43%) as yellow solids. 1H-NMR (300 MHz, DMSO-d6, δ ppm): 12.53 (s, 1H, -OH), 8.81 (s, 1H, -OH), 8.06–8.11 (m, 2H), 7.55–7.63 (m, 3H), 7.00 (s, 1H), 6.98 (s, 1H), 4.39 (t, J = 6.1 Hz, 2H), 4.35 (t, J = 6.1 Hz, 2H), 2.07 (s, 3H); 13C-NMR (75 MHz, DMSO-d6, δ ppm): 182.2, 170.3, 163.1, 153.2, 149.5, 146.2, 131.9, 130.7, 130.2, 129.0, 126.2, 105.4, 104.6, 92.1, 66.9, 62.2, 20.6; HRMS (ESI): m/z calcd. for C19H17O7: 357.0969, found: 357.0975 [M + H]+; Rf = 0.3 (DCM:MeOH 30:1).
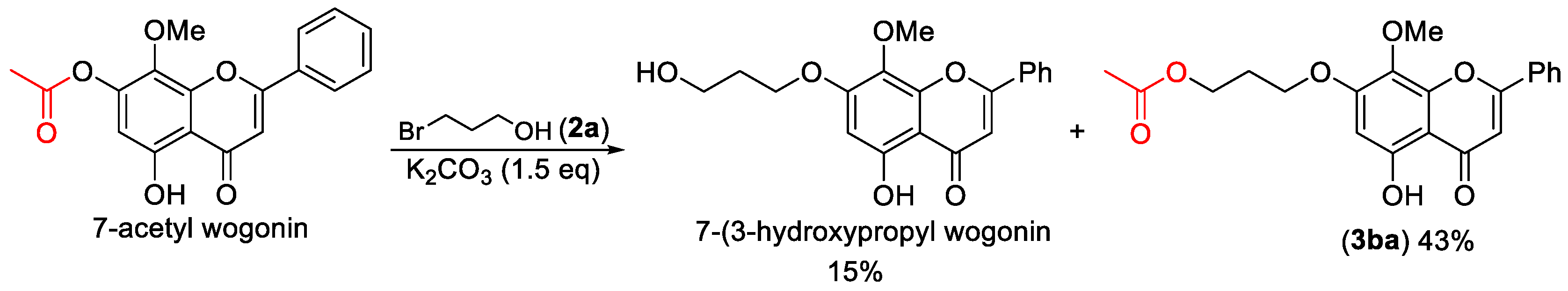
3-((5-Hydroxy-8-methoxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)propyl acetate (3ba). Compound 3ba was prepared from 7-acetyl wogonin following the general procedure described above and purified by silica gel column chromatography (DCM:MeOH 50:1) to provide the pure compound (132 mg, 69%) as yellow solids. 1H-NMR (300 MHz, CDCl3, δ ppm): 12.56 (s, 1H, -OH), 7.95 (t, J = 6.1 Hz, 2H), 7.55 (m, 3H), 6.69 (s, 1H), 6.44 (s, 1H), 4.33 (t, J = 6.1 Hz, 2H), 4.19 (t, J = 6.1 Hz, 2H), 3.94 (s, 3H, -OCH3), 2.19–2.28 (m, 2H), 2.10 (s, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 182.7, 178.0, 164.1, 163.9, 157.9, 1574, 157.3, 131.9, 131.2, 129.1, 126.3, 126.2, 105.3, 96.5, 65.5, 61.6, 60.9, 28.4, 20.9; HRMS (ESI): m/z calcd. for C21H21O7: 385.1282, found: 385.1278 [M + H]+; Rf = 0.3 (DCM:MeOH 50:1).
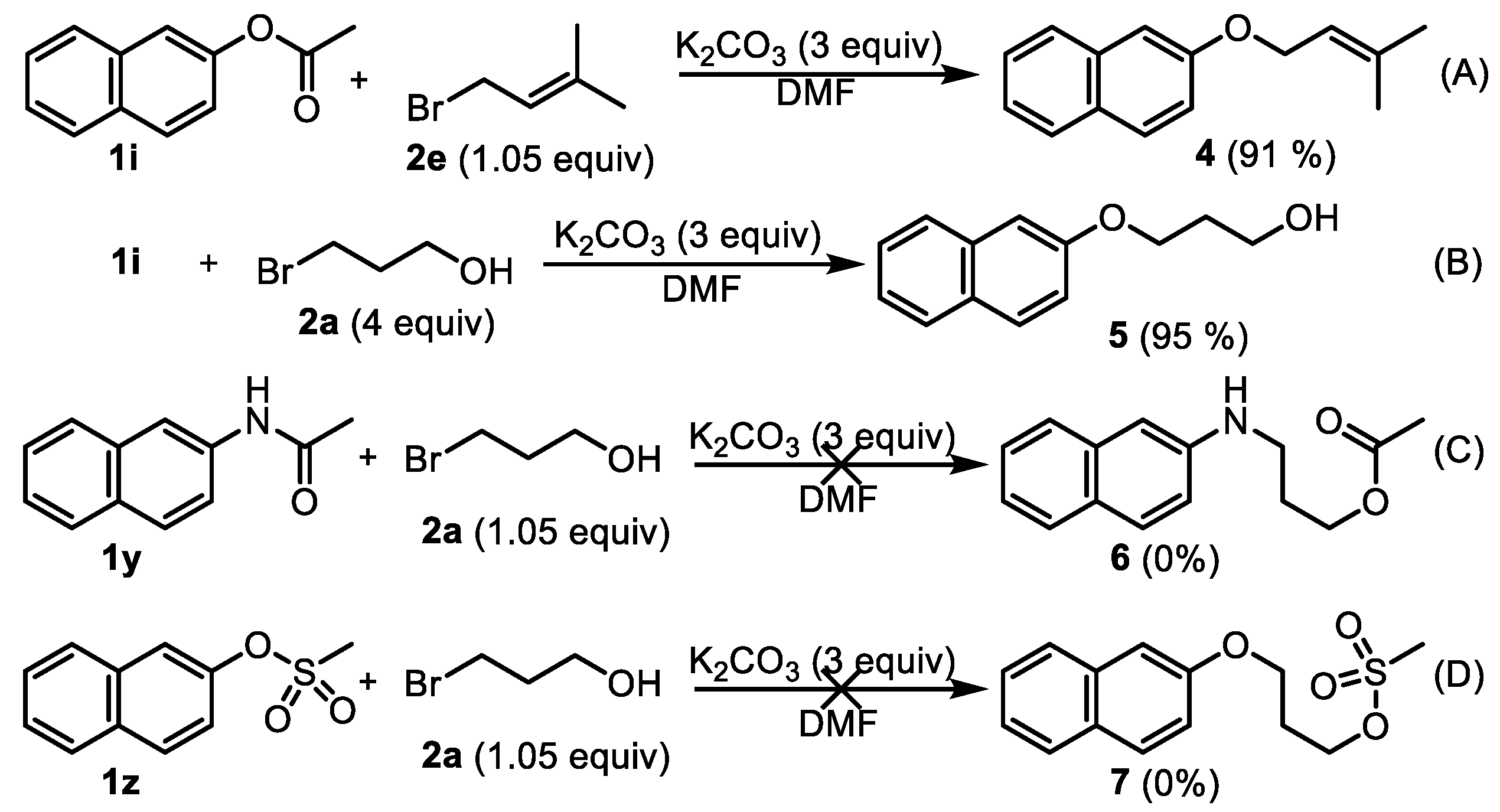
3.4. Control Experiments to Investigate the Mechanism
2-((3-Methylbut-2-en-1-yl)oxy)naphthalene (4). Phenyl esters 1i (0.5 mmol) were loaded into a flask (10 mL). DMF (2 mL) and K2CO3 (207 mg, 1.5 mmol, 3.0 equiv) were then added, which was followed by the addition of 3-methyl-2-butenyl bromide 2e (0.525 mmol, 1.05 equiv). Then, the reaction mixture was stirred at 80 °C for 6 h under an Ar atmosphere. After completion of the reaction, as confirmed by TLC, the reaction mixture was cooled down to room temperature and 10 mL of CH2Cl2 and 10 mL of water were added. After separation of the dichloromethane layer from the water, the aqueous phase was extracted with CH2Cl2 (2 × 5 mL) again. The combined organic layers were then dried over anhydrous Na2SO4, filtered, and concentrated under vacuum to yield the crude product. The crude product was purified by silica gel column chromatography with hexane/EtOAc (8:1 v/v) to afford the desired compound, 4 (CAS: 23676-18-8), as a slightly yellow solid (96 mg, 91% yield). 1H-NMR (300 MHz, CDCl3, δ ppm): 7.94–8.01 (m, 3H), 7.69 (t, J = 6.1 Hz, 1H), 7.58 (t, J = 6.1 Hz, 1H), 7.49 (dd, J = 8.1 Hz, 1.8 Hz, 1H), 7.40 (d, J = 1.8 Hz, 1H), 5.85 (t, J = 6.2 Hz, 1H, -CH=C), 4.83 (d, J = 9.1 Hz, 2H, -CH2-), 2.06 (s, 3H, -CH3), 1.99 (s, 3H, -CH3); 13C-NMR (75 MHz, CDCl3, δ ppm): 156.7, 137.6, 134.4, 129.2, 127.4, 126.5, 126.1, 123.3, 119.6, 118.9, 115.2, 106.4, 64.4, 25.6, 17.9; MS (ESI): m/z = 213.1 [M + H]+; Rf = 0.7 (DCM:MeOH 100:1).
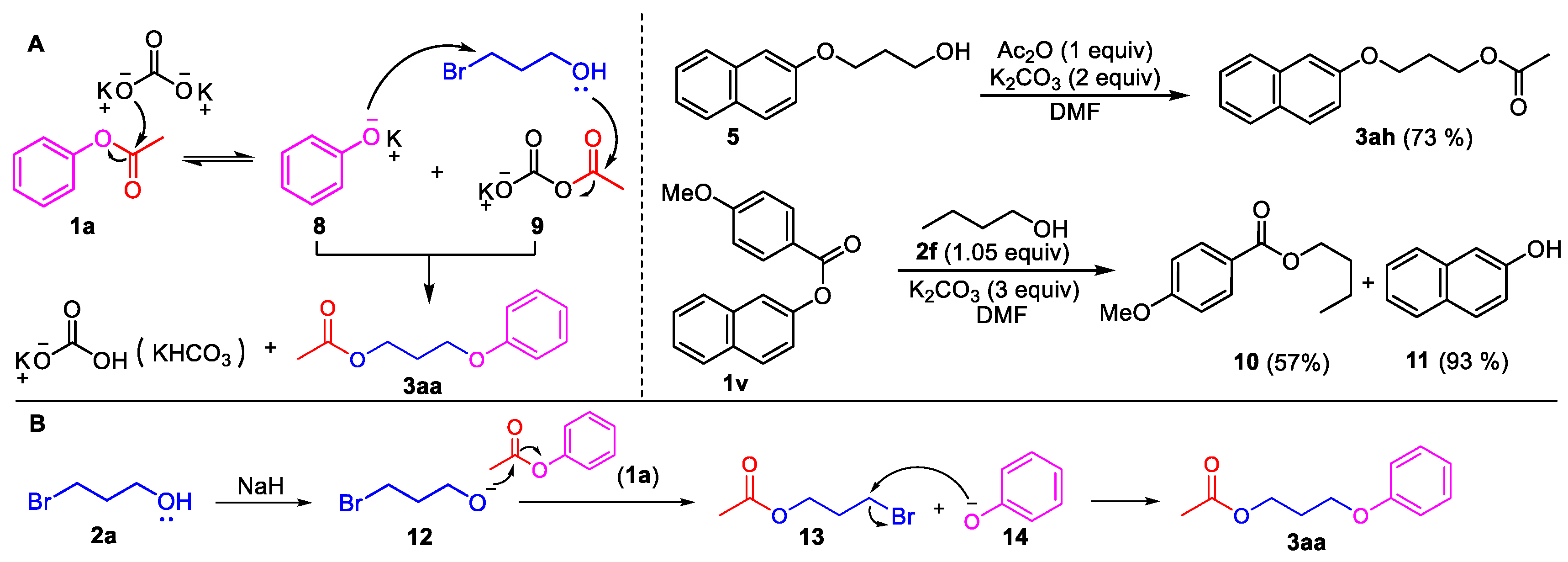
3-(Naphthalen-2-yloxy)propan-1-ol (5). Phenyl ester 1i (0.5 mmol) was loaded into a flask (10 mL). DMF (2 mL) and K2CO3 (207 mg, 1.5 mmol, 3.0 equiv) were then added, which was followed by the addition of 3-bromo-1-propanol 2a (2.0 mmol, 4 equiv). Then, the reaction mixture was stirred at 80 °C for 6 h under an Ar atmosphere. After completion of the reaction, as confirmed by TLC, the reaction mixture was cooled down to room temperature and 10 mL of CH2Cl2 and 10 mL of water were added. After separation of the dichloromethane layer from the water, the aqueous phase was extracted with CH2Cl2 (2 × 5 mL) again. The combined organic layers were then dried over anhydrous Na2SO4, filtered, and concentrated under vacuum to yield the crude product. The crude product was purified by silica gel column chromatography with hexane/EtOAc (4:1 v/v) to afford the desired compound, 5 (CAS: 7598-29-0), as a slightly yellow oil (96 mg, 95% yield). 1H-NMR (300 MHz, CDCl3, δ ppm): 7.74–7.82 (m, 3H), 7.48 (t, J = 6.2 Hz, 1H), 7.38 (t, J = 6.2 Hz, 1H), 7.19–7.24 (m, 2H), 4.63 (br s, 1H, -OH), 4.22 (t, J = 6.1 Hz, 2H), 4.05 (t, J = 6.1 Hz, 2H), 2.16–2.19 (m, 2H); MS (ESI): m/z = 203.1 [M + H]+; Rf = 0.4 (DCM:MeOH 50:1).
Butyl 4-methoxybenzoate (10). Compound 10 (CAS: 6946-35-6) was prepared from 1v and 1-butanol (2f), following the above general procedure and purified by silica gel column chromatography (DCM:MeOH 100:1) to provide the pure compound (59 mg, 57%) as a light oil. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.01 (d, J = 8.1 Hz, 1H), 6.92 (d, J = 8.1 Hz, 1H), 4.30 (t, J = 6.2 Hz, 2H), 3.85 (s, 3H, -OCH3), 1.74 (m, 2H), 1.49 (m, 2H), 0.98 (t, J = 6.2 Hz, 3H); 13C-NMR (75 MHz, CDCl3, δ ppm): 165.9, 162.7, 131.0, 122.4, 113.0, 64.0, 54.8, 30.3, 18.7, 13.2; MS (ESI): m/z = 209.1 [M + H]+; Rf = 0.8 (DCM:MeOH 100:1).
5-Hydroxy-8-methoxy-4-oxo-2-phenyl-4H-chromen-7-yl acetate (7-acetyl wogonin). 5-Acetyl wogonin (65 mg, 0.2 mmol) were loaded into a flask (25 mL). DMF (1.5 mL) and K2CO3 (82 mg, 0.6 mmol, 3.0 equiv) was added. Then, the reaction mixture was stirred at 80 °C for 6 h under an Ar atmosphere. After completion of the reaction, as confirmed by TLC, the reaction mixture was cooled down to room temperature and 5 mL of CH2Cl2 and 5 mL of water were added. After separation of the CH2Cl2 layer from the water, the aqueous phase was extracted with CH2Cl2 (2 × 3 mL) again. The combined organic layers were then dried over anhydrous Na2SO4, filtered, and concentrated under vacuum to yield the crude product. The crude product was purified by silica gel column chromatography with hexane/EtOAc (5:1 v/v) to afford 7-acyl wogonin (CAS: 95480-80-1) as slightly yellow solids (56 mg, 86% yield). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 12.49 (s, 1H, 5-OH), 7.99–8.02 (m, 2H, H-2′, H-6′), 7.63–7.69 (m, 3H, H-3′, H-4′, H-5′), 6.73 (s, 1H, H-3), 6.38 (s, 1H, H-6), 3.81 (s, 3H, 8-OCH3), 2.19 (s, 3H, -CH3); MS (ESI): m/z = 325.1 [M − H]−; Rf = 0.3 (DCM:MeOH 100:1).

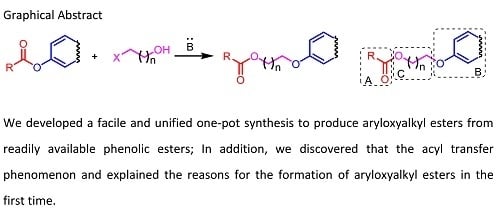
 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
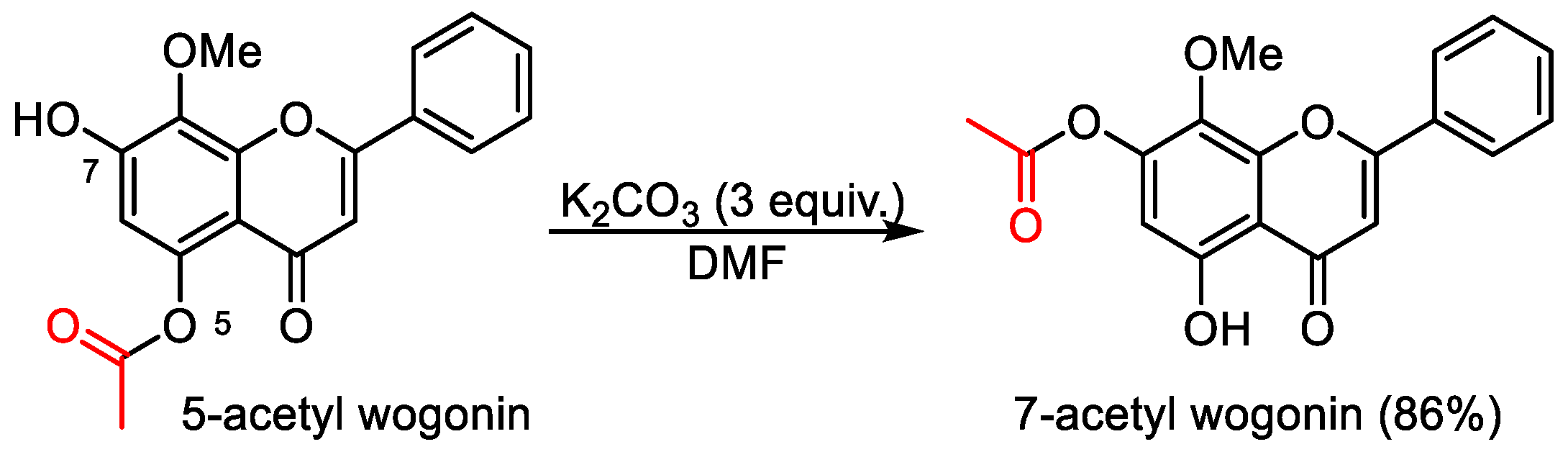{kind=link}
