Abstract
According to the precepts that C-10 amino-artemisinins display optimum biological activities for the artemisinin drug class, and that attachment of a sugar enhances specificity of drug delivery, polarity and solubility so as to attenuate toxicity, we assessed the effects of attaching sugars to N-4 of the dihydroartemisinin (DHA)-piperazine derivative prepared in one step from DHA and piperazine. N-Glycosylated DHA-piperazine derivatives were obtained according to the Kotchetkov reaction by heating the DHA-piperazine with the sugar in a polar solvent. Structure of the D-glucose derivative is secured by X-ray crystallography. The D-galactose, L-rhamnose and D-xylose derivatives displayed IC50 values of 0.58–0.87 nM against different strains of Plasmodium falciparum (Pf) and selectivity indices (SI) >195, on average, with respect to the mouse fibroblast WEHI-164 cell line. These activities are higher than those of the amino-artemisinin, artemisone (IC50 0.9–1.1 nM). Notably, the D-glucose, D-maltose and D-ribose derivatives were the most active against the myelogenous leukemia K562 cell line with IC50 values of 0.78–0.87 µM and SI > 380 with respect to the human dermal fibroblasts (HDF). In comparison, artemisone has an IC50 of 0.26 µM, and a SI of 88 with the same cell lines. Overall, the N-glycosylated DHA-piperazine derivatives display antimalarial activities that are greatly superior to O-glycosides previously obtained from DHA.
1. Introduction
The derivatives 2–4 of artemisinin 1 (Figure 1) are the most widely used drugs for treatment of malaria caused mainly by the apicomplexan parasite Plasmodium falciparum (Pf). For treatment of non-severe malaria, combinations with longer half-life drugs known as artemisinin combination therapies (ACTs) are used [1]. Because of the peroxide group, artemisinins are thermally and chemically fragile, and show variable pharmacokinetic profiles and low bioavailability [2]. In particular, DHA 2 is labile due to an unzipping process driven by the unprotected hemiacetal hydroxyl group, it rearranges irreversibly under physiological conditions into the peroxyhemiacetal 5 that decomposes via a Kornblum-de la Mare process to the dead-end compound deoxyartemisinin 6 (Figure 1) [3]. The peroxyhemiacetal 5 is observed in plasma from patients treated with artesunate [4]. In this respect, it is noted that the antimalarial activity of DHA rapidly attenuates (half-life ~ 3 h) when incubated in human plasma or serum [5]. The instability also engenders problems during formulation and storage [6]. Again because of the hydroxyl group, DHA undergoes facile Phase I metabolism [7,8]. Artesunate 4 is rapidly hydrolyzed to DHA in vivo [9]. But because of the incipient protective effect of the ester group against first-pass metabolism, artesunate is a better source of DHA in plasma than DHA itself [10]. The facile metabolism of artemether 3 involving oxidative dealkylation of the methyl group, predominantly by CYP3A4, is reflected in the detection of DHA in subjects administered with artemether [11]. These artemisinins, especially DHA, display neurotoxicity in vitro and in vivo [12,13,14].
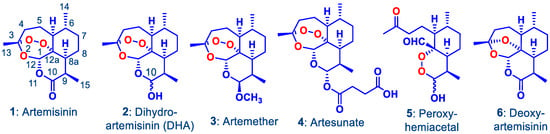
Figure 1.
Artemisinin 1 and its clinically-used derivatives—the hemiacetal dihydroartemisinin (DHA) 2, the lactol ether artemether 3, and the hemiester artesunate 4. The latter two are rapidly converted into DHA via metabolism or facile hydrolysis respectively. DHA rearranges irreversibly under physiological conditions into the peroxyhemiacetal 5 that in turn rearranges to the inert deoxyartemisinin 6.
However, resistance by malaria parasites to these derivatives, and to DHA in particular, now enormously complicates the task of global malaria control [15,16]. Thus, the development of new artemisinins that do not provide DHA and thereby supplant the current artemisinin derivatives is an urgent task. According to proposals based on the ADMET paradigm, [17] we prepared amino-artemisinin derivatives wherein the nitrogen atom of the amino group replaces the exocyclic oxygen atom attached to C-10 of the current clinical derivatives (Figure 2) [18,19,20,21]. All are significantly more active than the current clinical artemisinins against Pf. Whilst enhanced activity may be ascribed to improved pharmacokinetic parameters, this is better rationalized in terms of the conceptual model for mode of action that involves oxidation of reduced flavin cofactors important for modulating oxidative stress in the malaria parasite [22]. From a drug development perspective, the most advanced of the aminoartemisinins is artemisone 7 that is nine-fold more active than artesunate 4 against Pf in vitro and at least three-fold in in vivo [20,23,24]. Artemisone is not metabolized to DHA 2 but provides metabolites bearing unsaturation in the S,S-dioxothiomorpholino ring, and/or hydroxyl groups at C-5 or C-7 that also have potent antimalarial activity [20,25,26]. Unlike the current artemisinins, artemisone displays no clinically relevant autoinduction of metabolizing enzymes. [20,25,27,28] In comparison to DHA, [29] artemisone possesses a longer elimination half-life (~3 h vs. ~1 h), lower plasma clearance (~3.5 L/h/kg vs. ~5.4 L/h/kg) and a larger volume of distribution (14.5 L/kg vs. 7.7 L/kg), [27,28] and displays negligible neurotoxicity in vitro and in vivo [14]. Overall, artemisone is the only artemisinin derivative developed outside China that has progressed into a clinical trial against malaria. Artemisone has now been registered with the US FDA as an orphan drug planned for use in treatment of severe malaria via intravenous administration [30].
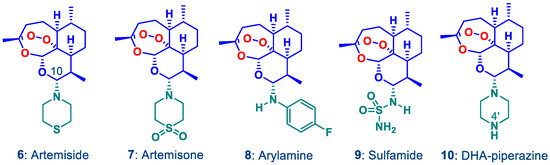
Figure 2.
Amino-artemisinins bearing alkylamino, arylamino and sulfonylamino groups attached to C-10 of the artemisinin nucleus wherein the oxygen atom at C-10 in the current artemisinins (Figure 1) is replaced by the nitrogen atom. In terms of cost of goods, compounds 8–10 are the most economically obtained from DHA. The intent of the current work is to develop new compounds by attaching polar groups to N-4 in the piperazine derivative 10.
Artemisinins have also been tested for antitumour activities in vitro on cancer cell lines, in in vivo studies with animal models [31,32,33,34], and in pilot clinical trials [34,35]. However, as clinical treatment will require protracted treatment regimens, the DHA prodrug and the neurotoxicity problems essentially will, or should, proscribe use of the current clinical artemisinins. Artemisone 7 has antitumour properties, as well, and in combination with known anticancer agents, it possesses activities in vitro in the nanomolar to low micromolar range [36,37]. Combinations of artemisone with the anticancer drugs oxaliplatin, gemcitabine and thalidomide have a markedly potentiating effect on the action of each drug against human colorectal HCT116, colorectal adenocarcinoma SW480 and breast cancer MCF7 cells, an effect that is significantly greater than artemisinin itself. [36]. When artemisone is encapsulated into niosomes and solid lipid nanoparticles, the formulations display highly selective cytotoxicity towards melanoma A375 cells with negligible toxicity towards normal skin cells [38]. This thus allows for topical delivery of artemisone for treatment of melanoma [39].
Thus, 10-amino-artemisinins are desirable compounds, given their generally enhanced efficacies against malaria, superior metabolic profiles in the case of artemisone 7, and promising activities against cancer. Of the various synthetic routes developed, a phase transfer approach involving DHA and a primary aromatic amine was the most expeditious in leading directly and with complete stereoselectivity to C-10 α-arylamino derivatives such as compound 8 [18]. For the others generally requiring more basic alkyl amine nucleophiles, phase transfer methods did not work, and N-glycosylation technology involving activation of the hydroxyl group in DHA by conversion into the ß-halide under anhydrous conditions had to be used. Treatment of the ß-halide in situ with the appropriate 2° amine nucleophile worked well in providing stereoselectively the α-alkylamino derivatives such as the α-thiomorpholino derivative artemiside 6 and its sulfone counterpart artemisone 7, the sulfamide 9 and related compounds [19,20,21,40]. Of the various compounds earmarked for further development, the piperazine derivative 10 is attractive. Piperazine is a readily accessible, cheap synthon, and the free 2° amino group in the product allows for attachment of additional ligands to modulate drug properties. Thus, we have attached ferrocene containing groups to imprint redox-active behavior [41] and ligands based on cholesterol so as to enhance uptake through membranes such as the mycolic acid layer associated with Mycobacterium tuberculosis [42].
Here we use the piperazine derivative 10 as a template for preparing N-glycosylated artemisinin derivatives. Because of stereochemical issues involving the chiral, enantiomerically pure sugars, we had to secure configuration of the piperazine group at C-10 in 10. Thereupon, the efficacies of the N-glycosylated derivatives would be evaluated in vitro against malaria and selected normal or immortalized cell lines.
For malaria, it is not yet clear if the parasite possesses proteins with lectin-like activity (see below) capable of binding a sugar [43] and thus, it is uncertain how N-glycosylated artemisinins may express antimalarial activities. However, attachment of sugars to the hydroxyl group of DHA involving activation of DHA as the trimethylsilyl ether (cf. Scheme 1 below), treatment with the partially protected acetylated sugar under Lewis acid catalysis, and eventual hydrolysis of the acetylated sugar in the DHA precursor provided the DHA O-glycosides such as the O-glucosyl 11 and O-galactosyl 12 derivatives [44]. These were substantially less active than artemisinin 1 in vitro and essentially inactive in vivo (Figure 3). The more lipophilic peracetylated derivatives, that would be less capable of protein binding, however, were more active. The observation in general supports the theses that more polar artemisinin derivatives tend to be less active, but also less neurotoxic [20,45]. We are unaware of other studies involving assessment of antimalarial activities of glycosylated artemisinin derivatives.
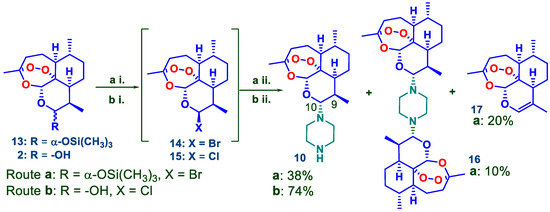
Scheme 1.
Preparation of DHA-piperazine derivative 10. Route a i. DHA α-TMS ether 13 (1.40 mmol.), TMSBr (1.43 mmol., 1.02 equiv.), CH2Cl2, 0 °C, N2, 30 min; a ii. Solution from a i added to piperazine (5.61 mmol, 4.0 equiv.) in CH2Cl2, 0 °C—room temperature, 12 h, 10 38%; 16 10%; 17 20%. Route b i. DHA 2 (3.52 mmol.), (COCl)2 (3.6 mmol., 1.02 equiv.), DMSO (0.35 mmol, 0.1 equiv.), toluene, N2, room temperature, 30 min; b ii. Solution from b i added to piperazine (14 mmol, 4 equiv.), CH2Cl2, 12 h; 10 74%.
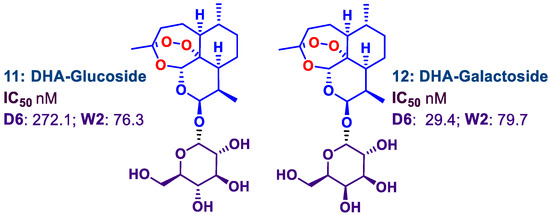
Figure 3.
O-Glycosylated DHA derivatives and antimalarial activities against Plasmodium falciparum (Pf) D6 and W2 strains in vitro; cf. artemisinin 1: IC50 D6 10.4 nM; W2 2.4 nM.
In cancer pathogenesis, it is established that cell surface proteins called lectins, a family of carbohydrate-binding proteins, contribute to neoplastic transformation, tumor cell survival, angiogenesis and tumor metastasis [46]. Lectins have binding sites for galactose, N-acetyl glucosamine and others [47,48]. The feasibility of using carbohydrate ligands to target protein receptors at such binding sites, termed ‘glycotargeting’, is long established [49]. Because of the carbohydrate tag, the drug delivery system can be recognized, bound and internalized by endogenous lectin receptors at a tumour cell surface [50]. The technique has been extended to guide nanoparticles bearing fucose ligands and encapsulating an anticancer drug to lectin cell surface receptors on cancer cells [51]. Thus, through tagging artemisinins with ligands that overall may enhance specificity of drug delivery, we assess here the effects of attaching sugars known to bind to lectins to the piperazine unit of the DHA-piperazine derivative 10.
2. Results and Discussion
2.1. Synthetic Chemistry: Preparation of DHA-Piperazine Derivative 10
The protocol for preparing artemiside 6 from the α-trimethylsilyl ether 13 of DHA and thiomorpholine [20,40] was applied to 13 by using piperazine, the caveat here being that with this difunctional nucleophile, formation of the bis-DHA piperazine derivative becomes a possibility. In the event, treatment of 13 firstly with trimethylsilyl bromide (TMSBr) in dichloromethane to generate the ß-bromide 14 in situ and then with anhydrous piperazine in the presence of triethylamine as base gave the polar DHA piperazine 10 (20%), the highly crystalline disubstituted piperazine 16 (10%) and the glycal 17 (20%). The last is a product common to these amination reactions; its formation is due to competing E1/E2 pathways involving the ß-bromide 14 as discussed elsewhere. [40] The bis-DHA piperazine 16 was prepared in higher yields (to 43%) by decreasing the amount of piperazine added to the solution of the ß-bromide 14. Alternatively, yields of DHA piperazine 10 were increased (to 38%) by conducting inverse addition of the solution of the ß-bromide generated in situ to an excess of piperazine with omission of the triethylamine base (Route a, Scheme 1).
The foregoing method is unsatisfactory, and thus the newer process for optimization of the preparation of artemiside 6 and artemisone 7 was applied here. This involves use of the oxalyl chloride-catalytic dimethyl sulfoxide (DMSO) system to convert DHA into the ß-chloride 15 in toluene followed by treatment with the appropriate amine nucleophile. The products are obtained in good yields in scalable reactions that could be run in relatively concentrated solutions (to 1.2 M DHA) [40]. The role of the DMSO is crucial: in its absence in toluene, very little of the product is obtained. The mechanism likely involves activation of DMSO by oxalyl chloride according to the Swern oxidation to form a chlorosulfonium salt. This reacts with DHA to provide a sulfurane intermediate that collapses to the ß-chloride 15 with regeneration of DMSO [40]. Oxidation of the hydroxyl group in DHA as such does not interfere. In the event, inverse addition of the ß-chloride 15 solution in toluene to an excess of piperazine in dichloromethane now gave the DHA piperazine 10 in 74% yield. Alternatively, addition of a solution of piperazine (1 equiv.) in dichloromethane to the toluene solution of the ß-chloride 15 gave the bis-DHA piperazine 16 (35%) as the predominant product.
The stereochemistry of the piperazine group in 10 was demonstrated by 1H NMR spectroscopy. The signal due to H-10 in the artemisinin nucleus at δ 3.95 ppm displays a coupling of 10.4 Hz, corresponding to a trans-diaxial relationship between H-10 and H-9 [52]. Therefore, the piperazine is attached via an α-equatorial bond to C-10 embedded in a chair-like pyran ring. This stereochemistry corresponds to that of all 10-aminoartemisinin derivatives prepared thus far [18,19,20]. However, it was not possible to obtain crystals of 10 suitable for X-ray crystallography, and for this purpose a single crystal of the dimer 16 was used. The structural determination clearly indicated attachment of the piperazine by equatorial bonds from N to C-10 in each of the artemisinin units; the piperazine and the pyran rings of the artemisinins are in the chair conformation (Figure 4 and Supplementary Materials).
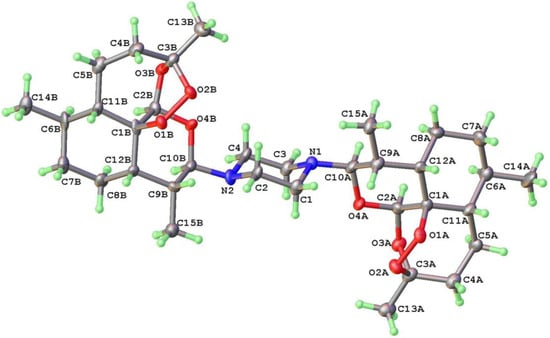
Figure 4.
ORTEP plot from X-ray crystallographic structural determination of the 1,4-bis-DHA piperazine 16 indicating diaxial arrangement of H-9 and H-10 attached to each of the pyran rings in the respective artemisinin units, the chair piperazine and α-equatorial C-10 to N bonds. Numbering in the artemisinin A and B units is as for artemisinin in Figure 1.
2.2. N-Glycosylation of DHA-Piperazine 10
The Kotchetkov aminoglycosylation reaction [53] involves condensation of amines with aldoses in aqueous alcohol containing inorganic salts [54]. This strikingly effective reaction works well for 2° amines such as indoles with glucose [55] and of relevance to the current case, for substituted piperazines with glucose or galactose [56,57,58]. Whilst attempted N-glucosylation of the DHA piperazine 10 with D-glucose by heating in methanol under reflux resulted in some decomposition, by lowering the reaction temperature to 60 °C, formation of the N-glucosylated product 18, a stable white crystalline solid, took place in acceptable yield (Scheme 2).
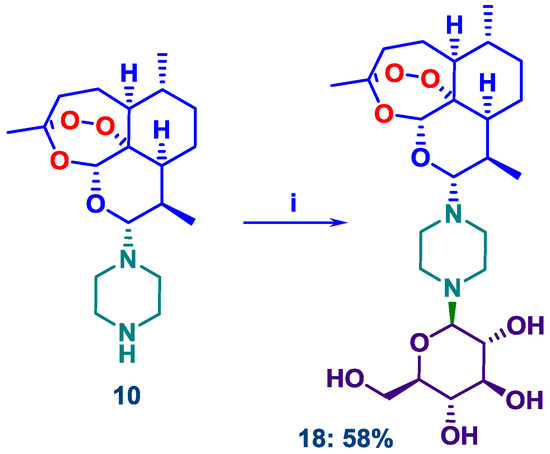
Scheme 2.
Preparation of the N-glucosylated DHA piperazine 18. i. 10, D-glucose (3.0 equiv.), 60 °C, MeOH, 24–48 h; 58%.
In the 1H NMR spectrum recorded in methanol-d4, the doublet signal at δ 4.10 ppm with J 10,9 = 10.4 Hz due to H-10 indicates that the equatorial α-configuration of the piperazine linked to the artemisinin nucleus in 10 is retained in 18. Two multiplets at δ 2.65 and 3.01 ppm are due to the protons on the piperazine linker. The signals at δ 3.19–3.85 ppm correspond to the C-H protons on the glucose moiety. Signals due to the three methyl groups and other protons in the artemisinin nucleus are readily recognized. Details are given in the Experimental Section. The structure was confirmed by X-ray crystallography, wherein crystallization to provide the single crystal incorporated water (Figure 5 and Supplementary Materials). The piperazine ring as for the disubstituted piperazine 16 above is in the chair conformation in which the artemisinin and glucosyl units are attached by equatorial bonds. Although it was initially anticipated that glucose, as a reducing sugar, might affect the artemisinin by reduction, the peroxide remained intact. In any event, the reactions were run without any attempt to exclude air in order to maintain an oxidizing environment.
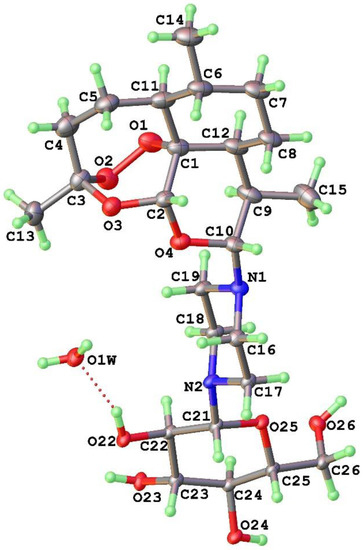
Figure 5.
ORTEP plot from X-ray crystallographic structural determination of the N-glucosylated DHA piperazine 18 indicating co-crystallization with water, the diaxial arrangement of H-9 and H-10, the chair piperazine and α-equatorial artemisinin C-10 to N-1 bond, and equatorial glucosyl C-21 to N2 bond. Numbering is as for the artemisinin nucleus in Figure 1.
The α-configuration of the amine linked to the glucose is assured by the lack of a kinetic anomeric effect exerted on the incoming amine nucleophile in the Kotchetkov reaction. Under general acid catalysis by the solvent, the anomeric hydroxyl group of the glucose is protonated to provide the oxonium intermediate (Scheme 3) that reacts with the DHA piperazine 10 via an SN1 reaction to yield the N-glucosylated DHA piperazine 18. By using this protocol, the other N-glycosylated derivatives 19–26 were prepared from DHA piperazine 10 and the corresponding aldoses; the sugars used and yields are summarized in Chart 1. Three furanose derivatives 24–26 were also able to be obtained. The disaccharide maltose was also successfully connected to the DHA-piperazine to provide the derivative 21 although in poor yield; this may have been due to the poor solubility of the sugar in methanol. The Kochetkov reaction only worked well for aldoses, in general, no reaction took place for ketose sugars such as fructose and sucrose.
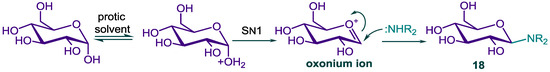
Scheme 3.
Kochetkov reaction of DHA piperazine 10 with D-glucose via the SN1pathway (NHR2 = DHA piperazine 10).
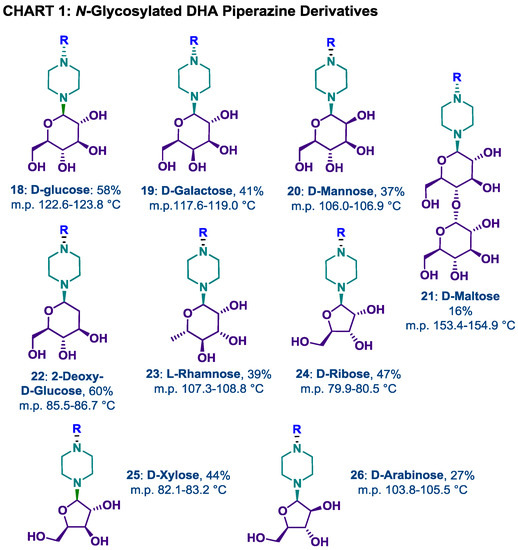
Chart 1.
N-glycosylated DHA piperazine derivatives prepared by the Kotchetkov reaction from the DHA-piperazine derivative 10 and the corresponding aldose according to Scheme 2 (R = 10-dihydroartemisinyl); isolated yields are given.
2.3. Antimalarial Activities against Different P. falciparum Strains and Cytotoxicities
The N-glycosylated DHA piperazine derivatives together with DHA 2, artemisone 7 and chloroquine as comparator drugs were screened in vitro against chloroquine (CQ)-sensitive Pf D10 and CQ-resistant Pf W-2 strains [59] by measuring effect on parasite lactate dehydrogenase (pLDH) according to modification of the method of Makler [60]. Antimalarial activities expressed as 50% inhibitory concentrations (IC50) [61] are presented in Table 1. Most of the compounds were notably active against both CQ-sensitive and -resistant Pf. The galactose 19, rhamnose 23 and xylose 25 derivatives with IC50 values of less than 1 nM were the most active against both strains, with activities comparable to those of artemisone 7 and some 4–5 fold superior to those of DHA (Table 1, column 2).

Table 1.
In vitro screening of the N-glycosylated DHA piperazine derivatives against P. falciparum CQ-sensitive D10 and CQ-resistant W2 strains, and cytotoxicity on the mouse fibroblast cell line WEHI-164.
The derivatives were submitted to comparative cytotoxicity screens on the mouse fibrosarcoma cell line WEHI-164 (Table 1) using the MTT assay as previously described [62]. All showed a selectivity index (SI) with respect to antimalarial activities of >100, indicating that the sugar insertion in the structure did not increase cytotoxicity. Indeed, the mannose and the rhamnose derivatives 20 and 23 had selectivity indices twofold greater than those of DHA or artemisone. Interestingly, the most active compounds against WEHI-164 was the parent DHA piperazine derivative 10 and the xylose derivative 25. Selected compounds were carried forward for screening against the immortalized myelogenous leukemia, K562, human microvascular endothelial cells (HMEC-1) and fresh human fibroblasts HDF (Table 2) using the MTT assay, as above. [62] Notably, the glucose 18, maltose 21 and the ribose derivative 24 were the most active against the highly proliferating K562 cell line compared to the endothelial cells or human fibroblasts, although some fourfold less active than artemisone 7 that elicits impressive activity in this screen. All the compounds were significantly less toxic to human fibroblasts than DHA 2 or artemisone 7. The natural product, camptothecin, a topoisomerase inhibitor and anticancer agent, is more active than artemisone 7, but also significantly more cytotoxic against HDF (SI < 100).
Table 2.
Cytotoxicities of N-glycosylated DHA piperazine derivatives against the myelogenous leukemia cell line K562, human dermal endothelial cells HMEC-1, and fresh human dermal fibroblasts HDF.
Overall, the glycosylated piperazine derivatives that are are easily prepared display promising activities against the malaria parasite coupled with very good selectivities. The galactose 19, rhamnose 23 and xylose 25 piperazine derivatives are the most potent among all of the N-glycosylated derivatives. The enhancement of antimalarial activities over those of DHA contrasts notably with the attenuating effect exerted by attachment of glycosides directly to the hydroxyl group of DHA itself (cf. Figure 3). The trend evidently also supports the contention that C-10 substituted amino-artemisinins are optimum substrates. [22] Thus, further studies evaluating activities of the best compounds in vivo are mandated. Clearly also, given the evidently enhancing effect that the glycoside moiety exerts on aqueous solubilities, we have here a compound class, one or more members of which have the potential to supplant use of artesunate, in particular in the preparation of formulations required for intravenous administration for severe/cerebral malaria. Currently, a dual pack comprising solid artesunate and sterile aqueous sodium bicarbonate that is mixed and diluted with aqueous dextrose immediately prior to injection is used; because artesunate is so easily hydrolyzed to DHA, the emphasis is on immediate injection. The compounds here, like artemisone, are likely to be stable at neutral pH. Thus, results from the next phases of the programme involving target profile evaluation and assessment of pharmacokinetic properties will be reported in due course. Finally, the activities of the N-glycosylated piperazine derivatives, and especially of artemisone 7 itself against the myelogenous leukemia cell line K562 are indicative of the viability of the aminoartemisinins at large for further development for antitumour therapy.
3. Conclusions
The use of sugars to enhance the selectivity of drugs is currently of great importance. New glycosylated derivatives of artemisinin were conceived on the basis of the incipient concept of carbohydrate-lectin interactions. Appending sugar groups to the artemisinin nucleus should also enhance the properties such as aqueous solubility, and especially render the compounds selectively cytotoxic. Mindful of the problem of attrition of activities observed on converting DHA into O-glycosylated DHA derivatives, we focused on preparation of N-glycosylated piperazine derivatives that were screened against different strains of P. falciparum. The compounds, in contrast to the O-glycosylated DHA derivatives, are very active, which vindicates the overall project. Should these compounds turn out to be water soluble, then there is the potential to use these in intravenous formulations for treatment of severe malaria.
4. Materials and Methods
4.1. General
Dihydroartemisinin was obtained either from the Kunming Pharmaceutical Corporation, Kunming, China; or from Haphacen, Hanoi College of Pharmacy, Vietnam. Other chemicals were purchased from commercial sources and used without further purification. Methanol (AR grade) was used as received. Dichloromethane was dried and distilled from calcium hydride. Toluene was dried over sodium and distilled from sodium benzophenone ketyl prior to use. Ethyl acetate and hexane for column chromatography were distilled from calcium chloride. Triethylamine was dried over calcium hydride and stored over sodium hydroxide pellets. TLC was performed with Merck (Darmstadt, Germany) Kieselgel 60 F254 plates and visualized either with ultra violet light (254 nm) or by heating after treatment with ammonium molydbate in 10% concentrated sulfuric acid. Column chromatography was performed with Merck silica gel 60 (0.04–0.063 mm, Darmstadt, Germany).
Melting points were recorded on a Leica (Wetzlar, Germany) Microscope Heating Stage 350 and are corrected. 1H and 13C NMR spectral data, unless otherwise stated, were obtained from samples in CDCl3 or CD3OD. 1H NMR spectrum was recorded on Bruker-400 spectrometer (Bruker AG, Karlsruhe, Germany) operating at 400 MHz and 13C NMR spectrum was recorded on Bruker-300 spectrometer (Billerica, MA, USA) operating at 75.4 MHz for 13C. Chemical shifts were reported in ppm relative to internal standard tetramethylsilane (0.3% v/v) as 0.0 ppm for 1H and CDCl3 as 77.0 ppm for 13C. Coupling constant was recorded in Hz. Abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Infrared spectra were recorded either on a Perkin (Peterborough, UK) Elmer Spectrum One spectrometer. Single crystal X-ray structure measurements were carried out on a Bruker (Billerica, MA, USA). Smart-APEX CCD four-circle diffractometer. All computations in the structure determination and refinement were performed on Silicon Graphics Indy computer using programs of the Siemens SHELXTL PLUS (version 5) (Munich, Germany) package (see Supplementary Materials).
4.2. Synthesis of Glycoside Derivatives: 10α-(1-Piperazino)-10-deoxo-10-dihydroartemisinin 10 and (bis-11′,4-piperazino)-10α-deoxo-10-dihydroartemisinin 16
Dimethyl sulfoxide (50 µL, 0.704 mmol) and then oxalyl chloride (0.4 mL, 4.73 mmol) were added to a suspension of dihydroartemisinin 2 (2.0 g, 7.04 mmol) in toluene (10 mL) under nitrogen which was then stirred for 30 min, during which time the DHA dissolved to form a clear amber solution. This was then added via cannula to a solution of piperazine (1.8 g, 21.21 mmol) in dichloromethane (20 mL). The resulting mixture was stirred for another 12 h at ambient temperature, and then diluted with more dichloromethane (40 mL). This was then filtered through a pad of Celite, and the filtrate was washed with water (3 × 30 mL) and brine (30 mL). The organic layer was dried (MgSO4) and filtered, and the filtrate was evaporated under reduced pressure to leave a residue that was submitted to chromatography over silica gel. Elution with dichloromethane-methanol-triethylamine (10:1:0.1) gave the product 10 as a cream white solid (1.8 g, 74%), m.p. 99–100 °C, [α]D22 + 12.58° (c 0.54, CHCl3). 1H NMR δ 0.81 (d, J = 7.0 Hz, 3H, H-14), 0.95 (d, J = 6.2 Hz, 3H, H-15), 1.16–1.35 (m, 2 H), 1.39 (s, 3H, H-13), 1.42–1.58 (m, 3H), 1.59-1.74 (m, 3H), 1.83–1.92 (m, 1 H), 1.96–.2.03 (m, 1H), 2.29–2.42 (m, 1H), 2.54–2.65 (m, 4H, piperazine), 2.79–2.89 (m, 4H, piperazine), 2.93 (s, 1H, NH), 2.96–3.0 (m, 1H), 3.97 (d, J = 10.3 Hz, 1H, H-10), 5.27 (s, 1H, H-12); IR (film) νmax 3327, 2927, 2872, 2346, 1714, 1645, 1450, 1376, 1319, 1260, 1198, 1161, 1056, 982, 928, 880, 852, 804, 733, 664 cm−1. MS (CI, CH4) m/z (%) 221.1 (8), 260.1 (11), 383.2 (12), 412.3 (3), 427.3 (22), 441.3 (100), 442.3 (25), 469.3 (19), 487.3 (8). HRMS (ESI): m/z calcd. for C19H33N2O4 353.2440 [M + H]+, found 353.2482.
Dimethyl sulfoxide (37.5 µL, 0.1 equiv) was added into a stirred suspension of DHA (1.5 g, 5.275 mmol) in toluene (15 mL) at room temperature under nitrogen. Oxalyl chloride (0.53 mL, 1.15 equiv) was slowly added into the suspension which during the course of the addition became a pale amber solution. This was stirred for 1 h, and then treated dropwise with a solution of piperazine (0.45 g, 1 equiv.) in dichloromethane (10 mL); the resulting mixture was stirred overnight. This was then quenched with saturated aqueous NaHCO3 (40 mL), and extracted with ethyl acetate (4 × 30 mL). The organic layers were combined, and the combined layer was washed with brine (40 mL) and dried (MgSO4). After filtration, the filtrate was evaporated under reduced pressure to leave a residue that was submitted to chromatography over silica gel. Elution with hexane-ethyl acetate 4:1 gave the product 16 (568 mg, 35%) as a white crystalline solid. Crystallization from ethyl acetate-hexane gave colourless plates, m.p. 149–150 °C, [α]D22 2.31° (c 0.58, CHCl3). 1H NMR δ 0.79 (d, J = 7.0 Hz, 6H, 14-H), 0.94 (d, J = 5.9 Hz, 6H, H-15), 1.21–1.34 (m, 6H), 1.40 (s, 6H, H-13), 1.45–1.57 (m, 4H), 1.58–1.60 (m, 4 H), 1.64–1.76 (m, 4 H), 1.83–1.91 (m, 2 H), 1.94–2.04 (m, 2 H), 2.29–2.41 (m, 2 H), 2.58–2.60 (m, 4H, piperazine), 2.99–3.02 (m, 4 H, piperazine), 3.99 (d, J = 10 Hz, 2H, H-10,), 5.27 (s, 2H, H-12). IR (film) νmax 2927, 2871, 1651, 1522, 1454, 1376, 1327, 1304, 1279, 1252, 1226, 1207, 1180, 1163, 1130, 1101, 1056, 1041, 1024, 979, 943, 926, 880, 847, 827, 734 cm–1. MS (CI, CH4) m/z (%) 221.1 (4), 306.2 (8), 346.2 (4), 455.3 (3), 469.3 (14), 513.4 (36), 527.4 (100), 530.5 (11), 557.5 (3), 573.4 (2). HRMS (ESI) m/z: calcd for C34H55N2O8 619.3958 [M + H] +, found 619.3929; Calcd. for C34H54N2O8 C 65.99, H 8.80, N 4.52; found C 66.37, H 8.93, N 4.18%.
4.3. Synthesis of Glycoside Derivatives
4.3.1. D-Glucose
10α-(Piperazinyl)-10-deoxyartemisinin 10 (654 mg, 1.85 mmol) was stirred with D-glucose (1.0 g, 5.55 mmol, 3.0 equiv.) in methanol (20 mL) at 60 °C for 24 h; an open system was used, that is, no precautions were taken to exclude air. The solvent was then evaporated under reduced pressure, and the residue was dissolved in ethyl acetate (40 mL). The solution was washed with saturated aqueous NaHCO3 (3 × 25 mL), and then brine (25 mL). The organic layer was separated and dried (MgSO4), and filtered. The filtrate was evaporated under reduced pressure, and the residue was submitted to chromatography on silica gel. Elution with ethyl acetate-methanol (20:1) gave the glucose derivative 18 (549 mg, 58%). The product was recrystallized from dichloromethane/ hexane to afford 18 as fine white needles, m.p. 122.6–123.8 °C. 1H NMR (400 MHz, CD3OD): δ 0.81 (d, J = 7.2 Hz, 3H, H-14), 0.99 (d, J = 6.4 Hz, 3H, H-15), 0.94–1.57 (m, 3H), 1.37 (s, 3H, H-13), 1.40–1.95 (m, 7H), 2.22–2.32 (m, 1H), 2.46–2.64 (m, 8H), 2.63–2.68 (m, 1H), 2.88–3.54 (m, 7H), 3.6–3.63 (m, 2H), 3.99 (d, J = 10.0 Hz, 1H, H-10), 4.43–4.49 (m, 2H), 5.41 (s, 1H, H-12) ppm. 13C NMR (75.4 MHz, CDCl3): δ 13.4, 20.2, 21.6, 24.6, 25.9, 28.5, 34.2, 36.3, 37.2, 45.8, 46.9, 47.5, 51.7, 62.1, 68.9, 70.4, 77.5, 80.2, 90.5, 91.6, 94.2, 103.8 ppm. IR (film) νmax = 551, 617, 832, 879, 924, 941, 982, 1020, 1039, 1058, 1075, 1099, 1115, 1130, 1208, 1277, 1299, 1328, 1379, 1455, 1648, 2868, 2924, 3417 cm–1; HRMS (ESI) calcd. for C25H43N2O9 515.2969 [M + H]+, found 515.2976.
4.3.2. D-Galactose
A solution of 10α-(piperazinyl)-10-deoxyartemisinin 10 (498 mg, 1.41 mmol) and D-galactose (760 mg, 4.2 mmol) in methanol (15 mL) at 60 °C was stirred for 36 h. After workup according to the foregoing procedure, the crude product was submitted to chromatography with dichloromethane-methanol (15:1) to afford the galactose derivative 19 (294 mg, 41%) as a white microcrystalline solid, m.p. 117.6–119.0 °C. 1H NMR (400 MHz, CDCl3): δ 0.69 (d, J = 8.0 Hz, 3H, H-14), 0.87 (d, J = 6.4 Hz, 3H, H-15), 0.94–1.57 (m, 3H), 1.25 (s, 3H, H-13), 1.40–2.20 (m, 8H), 2.38–2.51 (m, 7H), 2.46–2.64 (m, 8H), 2.79–2.83 (m, 4H), 2.88–3.54 (m, 7H), 3.12–3.87 (m, 7H), 3.99 (d, J = 10.0 Hz, 1H, H-10), 4.43–4.49 (m, 2H), 5.29 (s, 1H, H-12) ppm. 13C NMR (75.4 MHz, CDCl3): δ 11.7, 14.1, 15.3, 20.6, 21.0, 22.3, 25.4, 26.6, 27.6, 28.2, 29.0, 29.2, 30.4, 32.2, 34.9, 35.8, 37.0, 38.1, 41.0, 41.9, 45.7, 46.4, 47.7, 48.7, 52.4, 52.5, 53.1, 57.2, 62.3, 63.2, 67.6, 70.2, 75.5, 76.5, 80.9, 81.0, 91.2, 91.6, 92.3, 95.7, 104.7 ppm. IR (film) νmax = 510, 551, 660, 693, 746, 766, 834, 851, 880, 924, 940, 957, 981, 10101, 1061, 1101, 1134, 1177, 1208, 1279, 1383, 1419, 1455, 1633, 1706, 2870, 2922, 3395 cm−1; HRMS (ESI): calcd. For C25H43N2O9 515.2969 [M + H]+, found 515.2987.
4.3.3. D-Mannose
A solution of 10α-(piperazinyl)-10-deoxyartemisinin 10 (629 mg, 1.78 mmol) and D-mannose (960 mg, 5.34 mmol) in methanol (20 mL) at 60 °C was stirred for 36 h. After workup according to the procedure in i. above, the crude product was submitted to chromatography with dichloromethane-methanol (15:1) to afford the mannose derivative 20 (340 mg, 37%) as a fine white powder, m.p. 106.0–106.9 °C. 1H NMR (400 MHz, CDCl3) δ 0.79 (d, J = 7.2 Hz, 3H, H-14), 0.97 (d, J = 7.6 Hz, 3H, H-15), 0.83–1.38 (m, 6H), 1.42 (s, 3H, H-13), 1.44–2.16 (m, 7H), 2.30–3.08 (m, 12H), 3.50 (d, J = 9.2 Hz, 1H, H-10), 3.65–4.01 (m, 7H), 5.28 (s, 1H, H-12) ppm. 13C NMR (75.4 MHz, CDCl3) δ 13.5, 14.2, 20.4, 21.7, 22.7, 24.8, 26.2, 28.6, 31.0, 31.7, 34.4, 36.4, 37.5, 45.9, 47.1, 51.8, 54.7, 61.3, 62.7, 69.4, 70.99, 71.7, 75.6, 80.1, 80.4, 83.7, 90.6, 91.8, 96.7, 102.0, 104.0 ppm. IR (film) νmax = 572, 642, 658, 734, 756, 803, 826, 879, 927, 982, 1040, 1380, 1448, 1631, 1708, 2929, 3433 cm−1; HRMS (ESI): calcd. for C25H43N2O9 515.2969 [M + H]+, found 515.2980.
4.3.4. D-Maltose
According to the general procedure, the reaction was started with 10α-(piperazinyl)-10-deoxyartemisinin 10 (421 mg, 1.19 mmol) and maltose (1.2 g, 3.6 mmol) in methanol (20 mL) at 60 °C for 48 h. The solvent was removed directly from the reaction mixture by evaporation under reduced pressure, and the residue was submitted directly to column chromatography. Elution with dichloromethane-methanol (4:1) gave the maltose derivative 21 (131 mg, 16%), that slowly precipitated as a fine white microcrystalline powder from ethyl acetate-methanol (5:1), m.p. 153.4–154.9 °C. 1H NMR (400 MHz, CD3OD): δ 0.70 (d, J = 6.4 Hz, 3H, H-14), 0.92 (d, J = 6.0 Hz, 3H, H-15), 0.91–1.67 (m, 7H), 1.45 (s, 3H, H-13), 1.84–2.33 (m, 8H), 2.90–4.35 (m, 14H), 4.01 (d, J = 10.2 Hz, 1H, H-10), 5.64 (s, 1H, H-12) ppm. 13C NMR (75.4 MHz, CD3OD): δ 13.5, 21.7, 26.4, 27.5, 29.9, 31.1, 32.6, 35.3, 38.4, 40.4, 42.0, 46.8, 52.7, 57.3, 61.1, 63.2, 63.5, 68.5, 75.7, 77.7, 79.2, 80.3, 85.2, 90.8, 91.7, 96.0, 100.9, 102.3, 173.4 ppm. IR (film) νmax = 824, 836, 879, 925, 989, 1015, 1054, 1112, 1131, 1147, 1178, 1210, 1277, 1305, 1328, 1383, 1455, 1640, 2868, 2924, 2949, 3392 cm−1; HRMS (ESI) calcd. for C31H53N2O14 677.3497 [M + H]+, found 677.3475.
4.3.5. 2-Deoxy-D-glucose
A solution of 10α-(piperazinyl)-10-deoxyartemisinin 10 (447 mg, 1.27 mmol) and 2-deoxy-D-glucose (625 mg, 3.81 mmol) in methanol (15 mL) at 60 °C was stirred for 24 h. After workup according to the procedure in i. above, the crude product was submitted to chromatography with dichloromethane-methanol (20:1) to afford the 2-deoxyglucose derivative 22 (377 mg, 60%) as a fine white powder, m.p. 85.0–86.7 °C. 1H NMR (400 MHz, CDCl3): δ 0.80 (d, J = 6.8 Hz, 3H, H-14), 0.95 (d, J = 5.6 Hz, 3H, H-15), 0.85–1.05 (m, 1H), 1.38 (s, 3H, H-13), 1.23–1.56 (m, 7H), 1.68–2.67 (m, 12H), 2.91–4.03 (m, 8H), 5.27 (s, 1H, H-12) ppm. 13C NMR (75.4 MHz, CDCl3): δ 13.5, 20.4, 21.7, 24.8, 26.0, 28.7, 29.8, 35.7, 36.4, 37.5, 45.9, 47.0, 48.2, 62.4, 72.6, 72.7, 77.6, 80.4, 90.6, 91.4, 91.7, 104.1 ppm. IR (film) vmax = 825, 879, 984, 1054, 1208, 1378, 1449, 1632, 1708, 2871, 2927, 3414 cm−1; HRMS (ESI) calcd. for C25H43N2O8 499.3019 [M+H]+, found 499.3012.
4.3.6. L-Rhamnose
A solution of 10α-(piperazinyl)-10-deoxyartemisinin 10 (337 mg, 0.96 mmol) and L-rhamnose (473 mg, 2.88 mmol) in methanol (10 mL) at 60 °C was stirred for 24 h. After workup according to the procedure in i. above, the crude product was submitted to chromatography with dichloromethane-methanol (15:1) to afford the rhamnose derivative 23 (185 mg, 38.5%) that slowly precipitated from dichloromethane-hexane as a white powder, m.p. 107.3–108.8 °C. 1H NMR (400 MHz, CDCl3): δ 0.81 (d, J = 6.8 Hz, 3H, H-14), 0.98 (d, J = 6.0 Hz, 3H, H-15), 1.04–1.29 (m, 7H), 1.36 (s, 3H, H-13), 1.42–2.49 (m, 15H), 2.85–4.80 (m, 11H), 4.01 (d, 1H, J = 10.2 Hz, H-10), 5.41 (s, 1H, H-12) ppm. 13C NMR (75.4 MHz, CDCl3): δ 13.6, 17.8, 20.4, 21.8, 24.9, 26.1, 28.7, 34.4, 36.5, 37.5, 46.0, 46.7, 48.8, 49.3, 51.9, 69.0, 73.1, 74.0, 75.2, 80.4, 90.6, 91.8, 93.0, 104.1 ppm. IR (film) νmax = 826, 879, 925, 982, 1015, 1058, 1208, 1280, 1277, 1454, 1633, 2870, 2927, 3416 cm−1; HRMS (ESI), calcd. for C25H43N2O8499.3019, m/z [M + H]+ found 499.3002.
4.3.7. D-Ribose
A solution of 10α-(piperazinyl)-10-deoxyartemisinin 10 (377 mg, 1.07 mmol) and D-ribose (480 mg, 3.21 mmol) in methanol (10 mL) at 60 °C was stirred for 24 h. After workup according to the procedure in i. above, the crude product was submitted to chromatography with dichloromethane-methanol (20:1) to afford the ribose derivative 24 (247 mg, 47%) that slowly precipitated from dichloromethane-hexane as a white powder, m.p. 79.9–80.5 °C. 1H NMR (400 MHz, CDCl3): δ 0.78 (d, J = 7.2 Hz, 3H, H-14), 0.95 (d, J = 6.0 Hz, 3H, H-15), 0.83–1.38 (m, 5H), 1.41 (s, 3H, H-13), 1.44–2.02 (m, 7H), 2.21–2.98 (m, 12H), 3.32–4.41 (m, 9H), 5.28 (s, 1H, H-12) ppm. 13C NMR (75.4 MHz, CDCl3): δ 13.5, 20.4, 21.7, 24.8, 26.1, 28.6, 34.4, 36.4, 37.5, 45.9, 51.8, 54.4, 54.7, 62.1, 69.9, 71.5, 72.7, 73.8, 80.4, 80.4, 90.5, 91.7, 91.8, 100.4, 104.0, 104.1 ppm. IR (film) νmax = 617, 826, 858, 879, 926, 1041, 1130, 1207, 1280, 1378, 1454, 1632, 1720, 2871, 2927, 3424 cm−1; HRMS (ESI): calcd. for C24H41N2O8 485.2863 [M + H]+, found 485.2832.
4.3.8. D-Xylose
A solution of 10α-(piperazinyl)-10-deoxyartemisinin 10 (421 mg, 1.19 mmol) and D-xylose (536 mg, 3.57 mmol) in methanol (10 mL) at 60 °C was stirred for 24 h. After workup according to the procedure in i. above, the crude product was submitted to chromatography with dichloromethane-methanol (15:1) to afford the xylose derivative 25 (258 mg, 44%), that slowly precipitated from dichloromethane-hexane as a white powder, m.p. 82.1–83.2 °C. 1H NMR (400 MHz, CDCl3): δ 0.78 (d, J = 6.8 Hz, 3H, H-14), 0.94 (d, J = 6.4 Hz, 3H, H-15), 0.79–1.07 (m, 2H), 1.41 (s, 3H, H-13), 1.18–1.55 (m, 6H), 1.63–2.05 (m, 4H), 2.30–2.38 (td, J = 14.4, 3.6 Hz, 1H), 2.54–2.66 (m, 5H), 2.91–3.97 (m, 12H), 3.78 (d, J = 10.4 Hz, 1H, H-10), 5.27 (s, 1H, H-12) ppm. 13C NMR (75.4 MHz, CDCl3): δ 13.5, 20.4, 21.8, 24.8, 26.1, 28.6, 28.7, 34.4, 36.5, 37.5, 45.8, 46.0, 47.3, 47.9, 51.9, 67.7, 69.0, 70.0, 78.2, 80.3, 80.4, 90.8, 91.8, 95.3, 104.1, 104.2 ppm. IR (film) νmax = 826, 879, 925, 979, 1040, 1129, 1207, 1281, 1378, 1455, 1634, 1722, 2871, 2928, 3420 cm−1; HRMS (ESI): calcd. for C24H41N2O8 485.2863 [M + H]+, found 485.2817.
4.3.9. D-Arabinose
A solution of 10α-(piperazinyl)-10-deoxyartemisinin 10 (314 mg, 0.80 mmol) and D-arabinose (400 mg, 2.67 mmol) in methanol (10 mL) at 60 °C was stirred for 24 h. After workup according to the procedure in i. above, the crude product was submitted to chromatography with dichloromethane-methanol (15:1) to afford the arabinose derivative 26 (114 mg, 27%), that slowly precipitated from dichloromethane-hexane as a white powder, m.p. 103.8–105.5 °C. 1H NMR (400 MHz, CDCl3): δ 0.79 (d, J = 6.8 Hz, 3H, H-14), 0.94 (d, J = 6.0 Hz, 3H, H-15), 0.79–1.07 (m, 1H), 1.40 (s, 3H, H-13), 1.13–2.24 (m, 9H), 2.30–3.00 (m, 11H), 3.49–4.16 (m, 8H), 5.28 (s, 1H, H-12) ppm. 13C NMR (75.4 MHz, CDCl3): δ 13.5, 20.4, 21.7, 24.8, 26.1, 28.6, 31.0, 34.4, 36.4, 37.4, 45.8, 47.3, 48.0, 51.8, 54.4, 54.7, 62.1, 67.2, 68.2, 69.8, 71.5, 73.8, 74.6, 80.4, 90.5, 91.7, 95.4, 100.5, 104.0, 104.4 ppm. IR (film) νmax = 825, 879, 925, 978, 1053, 1085, 1131, 1206, 1378, 1454, 1632, 1715, 2870, 2926, 3424 cm−1; HRMS (ESI): calcd. for C24H41N2O8 485.2863 [M + H]+, found 485.2817.
4.4. Biological Assays
All the reagents unless indicated otherwise were purchased from Sigma (Sigma Italia, Milan, Italy).
4.4.1. Cultures of Pf and In Vitro Antimalarial Assays
The Pf strains D10 (CQ-sensitive), and W-2 (CQ-resistant) were cultured in vitro as described by Trager and Jensen with minor modifications [60]. Parasites were maintained at 5% hematocrit (human type A-positive red blood cells) in RPMI 1640 (EuroClone, Celbio) medium with the addition of 10% heat inactivate human serum, 20 mM Hepes and 2 mM glutamine (Euroclone). The cultures were maintained at 37 °C in a standard gas mixture consisting of 1–3% O2, 5% CO2, and 92–94% N2. Compounds were dissolved in either water or DMSO and then diluted with medium to achieve the required concentrations (final DMSO concentration <1%, which is non-toxic to the parasite). Asynchronous cultures of Pf with parasitaemia of 1–1.5% and 1% final hematocrit were aliquoted into 96-well flat-bottom microplates (COSTAR) with serial dilutions of test compounds and incubated for 72 h at 37 °C. Parasite growth was determined spectrophotometrically (OD650) by measuring the activity of the parasite lactate dehydrogenase (pLDH) according to a modification of the method of Makler in control and drug-treated cultures [61]. The antimalarial activity is expressed as 50% inhibitory concentrations (IC50); each IC50 value is the mean and standard deviation of at least three separate experiments performed in duplicate [62].
4.4.2. Cell Cytotoxicity Assays
A long term microvascular endothelial cell line (HMEC-1) immortalized by SV 40 large T antigen was kindly provided by Dr. Francisco J. Candal, Center for Disease Control, Atlanta, GA, USA. Cells were maintained in MCDB 131 medium (Invitrogen, Milan, Italy) supplemented with 10% fetal calf serum (HyClone, Celbio, Milan, Italy), 10 ng/mL of epidermal growth factor (Chemicon), 1 µg/mL of hydrocortisone, 2 mM glutamine, 100 units/ml of penicillin, 100 µg/mL of streptomycin and 20 mM Hepes buffer (EuroClone). K562, the human myelogenous leukemia cell line and WEHI 164 murine fibrosarcoma line were cultured in RPMI 1640 supplemented with 2 mM glutamine, 100 units/ml of penicillin, 100 µg/mL of streptomycin and 10% fetal calf serum. Fibroblasts from skin biopsies were maintained in DMEM (EuroClone, Pero, Italy) supplemented with 10% fetal calf serum (HyClone, Logan, UT, USA), 2 mM glutamine, 100 U/mL of penicillin and 100 µg/mL of streptomycin.
For the cytotoxicity assays, HMEC-1 and WEHI 164 cells were seeded in 96 well flat bottom tissue culture clusters at 104 cells/well; HDF were seeded at 1.5 × 104 cells/well and let to adhere for 24 h before the drug treatment; K562 were seeded in round bottom clusters at 1.5 × 104 cells/well. Cells were then treated with serial dilutions of test compounds and cell proliferation evaluated using the MTT assay as previously described [62]. Plates were incubated for 72 h at 37 °C in 5% CO2, then 20 µL of a 5 mg/ml solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) in PBS were added for an additional 3 h at 37 °C. The plates were then centrifuged, the supernatants discarded and the dark blue formazan crystals dissolved using 100 µL of lysing buffer consisting of 20% (w/v) of a solution of SDS (sodium dodecyl sulfate), 40% of N,N-dimethyl formamide (Merck) in distilled water adjusted with 80% acetic acid to pH 4.7. The plates were then read on a microplate reader (Molecular Devices Co., Menlo Park, CA, USA) at a test wavelength of 550 nm and a reference wavelength of 650 nm. The results are expressed as IC50 which is the dose of compound necessary to inhibit cell growth by 50%. All the tests were performed in triplicate at least three times.
4.4.3. IC50 Calculation
The results of the antimalarial and cytotoxicity assays were expressed as the percent viability compared to the untreated controls, calculated with the following formula: 100([OD of treated sample blank]/[OD of untreated sample blank]) (OD, optical density). As a blank, uninfected RBCs were used for the antimalarial assays or media without cells for the cytotoxicity assays. The percent viability was plotted as a function of drug concentrations, and the curve fitting was obtained by nonlinear regression analysis using a four parameter logistic method (software Gen5 1.10 provided with the Synergy4 plate reader [Biotek]). The IC50 was extrapolated as the dose that induced a 50% inhibition of parasites or cell viability.
Supplementary Materials
The Supplementary Materials are available online S1: HRMS spectra of N-glycosylated derivatives 18–26, Tables S1–S6: X-Ray structural tables for compound 16; Tables S7–S13: X-Ray structural tables for compound 18.
Author Contributions
R.K.H. and Y.W conceived the project, Y.W. synthesized and characterized the compounds, H.N.W. optimized the synthetic pathways, I.D.W. conducted the X-ray crystallography, N.B. and S.P. designed and performed the biological evaluation in vitro against P. falciparum and the toxicity assays, N.B., S.P., P.M., D.T., analyzed the biological data, N.B., S.P., P.M., D.T., H.N.W. and R.K.H. wrote the paper. All the co-authors reviewed and approved the manuscript.
Funding
The work at the University of Milan was supported by the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (PRIN) Project 20154JRJPP_004. Funds from the COST Action CM1307, entitled “Targeted Chemotherapy towards Diseases Caused by Endoparasites” were attributed for covering the costs to publish the special issue of Molecules “COST Action CM1307—Proceedings in Medicinal and Natural Product Chemistry” in open access. The authors acknowledge AVIS-Milano for providing fresh human blood for parasite cultures.
Acknowledgments
The South African Medical Research Council (MRC) with funds from the National Treasury under its Economic Competitiveness and Support Package, North-West University and the South African National Research Foundation are thanked for financial support to RKH (NRF UIDs 90682 and 98934). Work at HKUST was originally supported through the HKSAR University Grants Committee Areas of Excellence Fund, and the HKSAR Research Grants Council Projects No. AoE P/10-01/01-02-I, AOE/P-10/01-2-II, and the University Grants Council Grants No. HKUST 6493/06M and 600507 (R.K.H.)
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Geneva 2016 World Malaria Report; WHO: Geneva, Switzerland, 2016; ISBN 978-92-4-151171-1. [Google Scholar]
- Navaratnam, V.; Mansor, S.M.; Sit, N.W.; Grace, J.; Li, Q.; Olliaro, P. Pharmacokinetics of artemisinin-type compounds. Clin. Pharmacokinet. 2000, 39, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Chan, H.W.; Lung, C.M.; Ng, N.C.; Wong, H.N.; Shek, L.Y.; Williams, I.D.; Gomes, M.F.; Cartwright, A. Artesunate and dihydroartemisinin (DHA): Unusual decomposition products formed under mild conditions and comments on the fitness of DHA as an antimalarial drug. ChemMedChem 2007, 2, 1448–1463. [Google Scholar] [CrossRef] [PubMed]
- Olliaro, P.L.; Nair, N.K.; Haynes, R.K.; Tang, M.M.K.; Cheu, C.K.W.; Zanolari, B.; Decosterd, L.A.; Navaratnam, V. Unpublished work.
- Parapini, S.; Olliaro, P.; Navaratnam, V.; Taramelli, D.; Basilico, N. Stability of the antimalarial drug dihydroartemisinin under physiologically-relevant conditions: Implications for clinical treatment, pharmacokinetic and in vitro assays. Antimicrob. Agents Chemother. 2015, 59, 4046–4052. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.H. The pharmaceutical death-ride of dihydroartemisinin. Malar. J. 2010, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.M.; Scheinmann, F.; Stachulski, A.V.; Maggs, J.L.; Park, B.K. Efficient preparations of the ß-glucuronides of dihydroartemisinin and structural confirmation of the human glucuronide metabolite. J. Med. Chem. 2001, 44, 1467–1470. [Google Scholar] [CrossRef] [PubMed]
- Ilett, K.F.; Ethell, B.T.; Maggs, J.L.; Davis, T.M.E.; Batty, K.T.; Burchell, B.; Binh, T.Q.; Thu, L.T.A.; Hung, N.C.; Pirmohamed, M.; et al. Glucuronidation of dihydro-artemisinin in vivo and by human liver microsomes and expressed UDP-glucuronosyltransferases. Drug Metab. Dispos. 2002, 30, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.A.; Duparc, S.; Borghini-Fuhrer, I.; Jung, D.; Shin, C.-S.; Fleckenstein, L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar. J. 2011, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Batty, K.T.; Ilett, K.F.; Powell, S.M.; Martin, J.; Davis, T.M.E. Relative bioavailability of artesunate and dihydroartemisinin: Investigations in the isolated perfused rat liver and in healthy Caucasian volunteers. Am. J. Trop. Med. Hyg. 2002, 66, 130–136. [Google Scholar] [CrossRef]
- Van Agtmael, M.A.; Gupta, V.; van der Wösten, T.H.; Rutten, J.-P.B.; van Boxtel, C.J. Grapefruit juice increases the bioavailability of artemether. Eur. J. Clin. Pharmacol. 1999, 55, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Wesche, D.L.; DeCoster, M.A.; Tortella, F.C.; Brewer, T.G. Neurotoxicity of artemisinin analogs in vitro. Antimicrob. Agents Chemother. 1994, 38, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, G.; Roehrdanz, E.; Haynes, R.K.; Kahl, R. Neurotoxic mode of action of artemisinin. Antimicrob. Agents. Chemother. 2002, 46, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, G.; Temerowski, M.; Haynes, R.K.; Fugmann, B. Identification of non-neurotoxic artemisinin derivatives in vivo and in vitro. Res. Adv. Antimicrob. Agents Chemother. 2003, 3, 35–47. [Google Scholar]
- Paloque, L.; Ramadani, A.P.; Mercereau-Puijalon, O.; Augereau, J.M.; Benoit-Vical, F. Plasmodium falciparum: Multifaceted resistance to artemisinins. Malar. J. 2016, 15, 149. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Culleton, R.; Zhang, M.; Ramaprasad, A.; von Seidlein, L.; Zhou, H.; Zhu, G.; Tang, J.; Liu, Y.; Wang, W.; et al. Emergence of indigenous artemisinin-resistant Plasmodium falciparum in Africa. N. Engl. J. Med. 2017, 376, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K. From artemisinin to new artemisinin antimalarials: Biosynthesis, extraction, old and new derivatives, stereochemistry and medicinal chemistry requirements. Curr. Top. Med. Chem. 2006, 6, 509–537. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Chan, H.-W.; Ho, W.-Y.; Ko, C.K.F.; Gerena, L.; Kyle, D.E.; Peters, W.; Robinson, B.L. Convenient access both to highly antimalaria active 10-arylaminoartemisinins, and to 10-alkyl ethers including artemether, arteether, and artelinate. ChemBioChem 2005, 6, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Ho, W.-Y.; Chan, H.-W.; Fugmann, B.; Stetter, J.; Croft, S.L.; Vivas, L.; Peters, W.; Robinson, B.L. Highly antimalaria-active artemisinin derivatives: Biological activity does not correlate with chemical reactivity. Angew. Chem. Int. Ed. 2004, 43, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Fugmann, B.; Stetter, J.; Rieckmann, K.; Heilmann, H.D.; Chan, H.W.; Cheung, M.K.; Lam, W.L.; Wong, H.N.; Croft, S.L.; et al. Artemisone-a highly active antimalarial drug of the artemisinin class. Angew. Chem. Int. Ed. 2006, 45, 2082–2088. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K. Strategies in the development and chemical modification of the new artemisinin antimalarial artemisone. Synform 2008, 3, A34–A36. [Google Scholar]
- Haynes, R.K.; Wong, H.N.; Wu, Y.; Wu, W.K.; Cheu, K.W.; Williams, I.D.; Krishna, S.; Slavic, K.; Gravett, A.M.; Liu, W.M. Methylene homologues of artemisone: An unexpected structure–activity relationship and a possible implication for the design of C10-substituted artemisinins. ChemMedChem 2016, 11, 1469–1479. [Google Scholar]
- Ramharter, M.; Burkhardt, D.; Nemeth, J.; Adegnika, A.A.; Kremsner, P.G. In vitro activity of artemisone compared with artesunate against Plasmodium falciparum. Am. J. Trop. Med. Hyg. 2006, 75, 637–639. [Google Scholar] [PubMed]
- Vivas, L.; Rattray, L.; Stewart, L.B.; Robinson, B.; Fugmann, B.; Haynes, R.K.; Peters, W.; Croft, S.L. Antimalarial efficacy and drug interactions of the novel semi-synthetic endoperoxide artemisone in vitro and in vivo. J. Antimicrob. Chemother 2007, 59, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Schmeer, K.; Breyer, M.; Hucke, K.; Gaefke, D.; Kern, A.; Schmuck, G. Artemifone, a New Anti-Malarial Artemisinin Derivative: Metabolism In Vitro in Liver Microsomes and Hepatocyte Sandwich Cultures of Different Species Including Man. Presentation PO63. In Proceedings of the XVI International Congress for Tropical Medicine and Malaria: Medicine and Health in the Tropics, Marseille, France, 11–15 September 2005; Abstract Book, O-063. p. 144. [Google Scholar]
- Krudsood, S.; Wilairatana, P.; Chalermrut, K.; Leowattana, W.; Voith, B.; Hampel, B.; Looareesuwan, S. Artemifone, a New Anti-Malarial Artemisinin Derivative: Open Pilot Trial to Investigate the Antiparasitic Activity of Bay 44-9585 in Patients with Uncomplicated P. falciparum Malaria. In Proceedings of the XVI International Congress for Tropical Medicine and Malaria: Medicine and Health in the Tropics, Marseille, France, 11–15 September 2005; Abstract Book, P054. p. 142. [Google Scholar]
- Nagelschmitz, J.; Voith, B.; Römer, A. Artemifone, A New Anti- Malaria Artemisinin Derivative- Safety And Pharmacokinetics In Healthy Male Subjects. In Proceedings of the XVI International Congress for Tropical Medicine and Malaria: Medicine and Health in the Tropics, Marseille, France, 11–15 September 2005; Abstract Book, P047. p. 142. [Google Scholar]
- Nagelschmitz, J.; Voith, B.; Wensing, G.; Roemer, A.; Fugmann, B.; Haynes, R.K.; Kotecka, B.M.; Rieckmann, K.H.; Edstein, M.D. First assessment in humans of the safety, tolerability, pharmacokinetics, and ex vivo pharmacodynamic antimalarial activity of the new artemisinin derivative artemisone Antimicrob. Agents Chemother. 2008, 52, 3085–3091. [Google Scholar] [CrossRef] [PubMed]
- Na-Bangchang, K.; Krudsood, S.; Silachamroon, U.; Molunto, P.; Tasanor, O.; Chalermrut, K.; Tangpukdee, N.; Matangkasombut, O.; Kano, S.; Looareesuwan, S. The pharmacokinetics of oral dihydroartemisinin and artesunate in healthy Thai volunteers. Southeast Asian J. Trop. Med. Public Health 2004, 35, 575–582. [Google Scholar] [PubMed]
- http://www.artemis-therapeutics.com/news/ (accessed on 30 June 2018).
- Efferth, T. Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells. Curr. Drug Targets 2006, 7, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Lai, H.; Singh, N.P.; Sasaki, T. Anticancer properties of artemisinin derivatives and their targeted delivery by transferrin conjugation. Int. J. Pharm. 2008, 354, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Ortiz, M.P.; Wei, M.Q. Antitumor activity of artemisinin and its derivatives: From a well-known antimalarial agent to a potential anticancer drug. J. Biomed. Biotechnol. 2012, 247597. [Google Scholar] [CrossRef] [PubMed]
- Das, A.K. Anticancer effect of antimalarial artemisinin compounds. Ann. Med. Health Sci. Res. 2015, 5, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Ganapathi, S.; Ster, I.C.; Saeed, M.E.M.; Cowan, M.; Finlayson, C.; Kovacsevics, H.; Jansen, H.; Kremsner, P.G.; Efferth, T.; et al. A randomised, double blind, placebo-controlled pilot study of oral artesunate therapy for colorectal cancer. EBioMedicine 2015, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Gravett, A.M.; Liu, W.M.; Krishna, S.; Chan, W.-C.; Haynes, R.K.; Wilson, N.L.; Dalgleish, A.G. In vitro study of the anti-cancer effects of artemisone alone or in combination with other chemotherapeutic agents. Cancer Chemother. Pharmacol. 2011, 67, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Van Huijsduijnen, R.H.; Guy, R.K.; Chibale, K.; Haynes, R.K.; Peitz, I.; Kelter, G.; Phillips, M.A.; Vennerstrom, J.L.; Yuthavong, Y.; Wells, T.N.C. Anticancer properties of distinct antimalarial drug classes. PLoS ONE 2013, 8, e82962. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, A.; Mazumder, A.; du Plessis, L.; du Preez, J.L.; Haynes, R.K.; du Plessis, J. In vitro anti-cancer effects of artemisone nano-vesicular formulations on melanoma cells. Nanomedicine 2015, 11, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, A.; Mazumder, A.; Fox, L.T.; Brümmer, A.; Gerber, M.; du Preez, J.L.; Haynes, R.K.; du Plessis, J. In vitro Skin Permeation of Artemisone and its nano-vesicular formulations. Int. J. Pharm. 2016, 503, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; Chan, D.H.W.; Lee, K.W.; Tin, W.S.; Wong, H.N.; Haynes, R.K. Evaluation and optimization of synthetic routes from dihydroartemisinin to the alkylamino-artemisinins artemiside and artemisone: A test of N-glycosylation methodologies on a lipophilic peroxide. Tetrahedron 2018. [Google Scholar] [CrossRef]
- De Lange, C.; Coertzen, D.; Smit, F.J.; Wentzel, J.F.; Wong, H.N.; Birkholtz, L.-M.; Haynes, R.K.; N’Da, D.D. Synthesis, in vitro antimalarial activities and cytotoxicities of amino-artemisinin-ferrocene derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Morake, M.; Coertzen, D.; Ngwane, A.; Wentzel, J.F.; Wong, H.N.; Smit, F.J.; Birkholtz, L.-M.; Pietersen, R.-D.; Baker, B.; Wiid, I.; et al. Preliminary evaluation of artemisinin–cholesterol conjugates as potential drugs for the treatment of intractable forms of malaria and tuberculosis. ChemMedChem 2018, 13, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Kupferschmid, M.; AquinoGil, M.O.; Shams-Eldin, H.; Schmidt, J.; Yamakawa, N.; Krzewinski, F.; Schwarz, R.T.; Lefebvre, T. Identification of O-GlcNAcylated proteins in Plasmodium falciparum. Malar. J. 2017, 16, 485. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, L.-Q.; Andersen, S.L.; Klayman, D.L. Antimalarial activity of new dihydroartemisinin derivatives. 5. Sugar analogues. J. Med. Chem. 1992, 35, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.K.; Karle, J.M. Stereoelectronic properties of antimalarial artemisinin analogues in relation to neurotoxicity. Chem. Res. Toxicol. 1999, 12, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Rabinovich, G.A. Galectins as modulators of tumor progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.S.; Ashraf, G.M.; Banu, N. Galectins-Potential targets for cancer therapy. Cancer Lett. 2007, 253, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Sörme, P.; Arnoux, P.; Kahl-Knutsson, B.; Leffler, H.; Rini, J.M.; Nilsson, U.J. Structural and thermodynamic studies on galectin-3 in complex with synthetic inhibitors: Carbohydrate-protein affinity enhancements through fine–tuning of an arginine-arene interaction. J. Am. Chem. Soc. 2005, 127, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.C.; Kornfeld, S. Hepatic uptake of proteins coupled to fetuin glycopeptide. Biochem. Biophys. Res. Commun. 1971, 45, 622–629. [Google Scholar] [CrossRef]
- Witczak, Z.J. Carbohydrate therapeutics: New developments and strategies. ACS Symp. Ser. 2006, 932, 25–46. [Google Scholar]
- Garg, N.K.; Singh, B.; Jain, A.; Nirbhavane, P.; Sharma, R.; Tyagi, R.K.; Kushwah, V.; Jain, S.; Katare, O.P. Fucose decorated solid-lipid nanocarriers mediate efficient delivery of methotrexate in breast cancer therapeutics. Colloids Surf. B Biointerfaces 2016, 146, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Chan, H.W.; Cheung, M.K.; Lam, W.L.; Soo, M.K.; Tsang, H.W.; Voerste, A.; Williams, I.D. C-10 Ester and ether derivatives of dihydroartemisinin: 10-α artesunate, preparation of authentic 10-β artesunate, and of other esters and ether derivatives bearing potential aromatic intercalating groups at C-10. Eur. J. Org. Chem. 2002, 2002, 113–132. [Google Scholar] [CrossRef]
- Likhosherstov, L.M.; Novikova, O.S.; Dervitskaya, V.A.; Kochetkov, N.K. A new simple synthesis of amino sugar β-D-glycosylamines. Carbohydr. Res. 1986, 146, C1–C5. [Google Scholar] [CrossRef]
- Oscarson, S. Conjugation of Monosaccharides-Synthesis of Glycosidic Linkages in Glycosides, Oligosaccharides and Polysaccharides. In Carbohydrates; Finch, P., Ed.; Springer: Dordrecht, The Netherlands, 1999; Chapter 4; pp. 150–186. ISBN 978-90-481-4033-6. [Google Scholar] [CrossRef]
- Chisholm, J.D.; Vrankem, D.L.V. Glycosylation of 2,2-indolylindolines. J. Org. Chem. 1995, 60, 6672–6673. [Google Scholar] [CrossRef]
- Jung, M.E.; Yang, E.C.; Vu, B.T.; Kiankarimi, M.; Spyrou, E.; Kaunitz, J. Glycosylation of fluoro-quinolones through direct and oxygenated polymethylene linkages as a sugar-mediated active transport system for antimicrobials. J. Med. Chem. 1999, 42, 3899–3909. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, J.D.; Van Vranken, D.L. Regio-controlled synthesis of the antitumor antibiotic AT2433-A1. J. Org. Chem. 2000, 65, 7541–7553. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, M.; Nishimura, T.; Kawamoto, H.; Nakano, M.; Honma, T.; Yoshinari, T.; Arakawa, H.; Suda, H.; Morishima, H.; Nishimura, S. Synthesis and biological activities of NB-506 analogues modified at the glucose group. Bioorg. Med. Chem. Lett. 2000, 10, 419–422. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Makler, M.T.; Ries, J.M.; Williams, J.A.; Bancroft, J.E.; Piper, R.C.; Gibbins, B.L.; Hinrichs, D.J. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 1993, 48, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Sparatore, A.; Basilico, N.; Parapini, S.; Romeo, S.; Novelli, F.; Sparatore, F.; Taramelli, D. 4-Aminoquinoline quinolizidinyl- and quinolizidinylalkyl-derivatives with antimalarial activity. Bioorg. Med. Chem. 2005, 13, 5338–5345. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, S.; Gelati, M.; Basilico, N.; Parati, E.A.; Haynes, R.K.; Taramelli, D. Differential effects on angiogenesis of two antimalarial compounds, dihydroartemisinin and artemisone: Implications for embryotoxicity. Toxicology 2007, 241, 66–74. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).