Spectroscopical Investigations on the Redox Chemistry of [FeFe]-Hydrogenases in the Presence of Carbon Monoxide


 ,
, 
Abstract
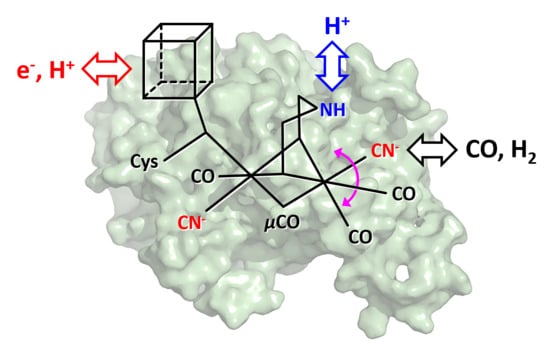
1. Introduction
2. Results
2.1. ATR-FTIR Spectro-Electrochemistry and 13CO Isotope Editing
2.2. CO binding in the Presence of H2
2.3. Assignment of H-Cluster Species by Density Functional Theory
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Fourier-Transform Infrared Spectroscopy
4.3. Density Functional Theory
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lubitz, W.; Ogata, H.; Ru, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Leung, D.Y.C.; Leung, M.K.H.; Sumathy, K. An Overview of Hydrogen Production from Biomass. Fuel Process. Technol. 2006, 87, 461–472. [Google Scholar] [CrossRef]
- Marbán, G.; Valdés-Solís, T. Towards the Hydrogen Economy? Int. J. Hydrogen Energy 2007, 32, 1625–1637. [Google Scholar] [CrossRef]
- Meher Kotay, S.; Das, D. Biohydrogen as a Renewable Energy Resource-Prospects and Potentials. Int. J. Hydrogen Energy 2008, 33, 258–263. [Google Scholar] [CrossRef]
- Fontecilla-Camps, J.C.; Volbeda, A.; Cavazza, C.; Nicolet, Y. Structure/function Relationships of [NiFe]- and [FeFe]-Hydrogenases. Chem. Rev. 2007, 107, 4273–4303. [Google Scholar] [CrossRef] [PubMed]
- Fontecilla-Camps, J.C.; Amara, P.; Cavazza, C.; Nicolet, Y.; Volbeda, A. Structure-Function Relationships of Anaerobic Gas-Processing Metalloenzymes. Nature 2009, 460, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.W.; Schut, G.J.; Boyd, E.S.; Mulder, D.W.; Shepard, E.M.; Broderick, J.B.; King, P.W.; Adams, M.W. [FeFe]- and [NiFe]-Hydrogenase Diversity, Mechanism, and Maturation. Biochim. Biophys. Acta 2015, 1853, 1350–1369. [Google Scholar] [CrossRef] [PubMed]
- Gloaguen, F.; Rauchfuss, T.B. Small Molecule Mimics of Hydrogenases: Hydrides and Redox. Chem. Soc. Rev. 2009, 38, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Schilter, D.; Camara, J.M.; Huynh, M.T.; Hammes-Schiffer, S.; Rauchfuss, T.B. Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides. Chem. Rev. 2016, 116, 8693–8749. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.R.; Berggren, G.; Bacchi, M.; Fontecave, M.; Artero, V. Mimicking Hydrogenases: From Biomimetics to Artificial Enzymes. Coord. Chem. Rev. 2014, 270–271, 127–150. [Google Scholar] [CrossRef]
- Vincent, K.A.; Parkin, A.; Armstrong, F.A. Investigating and Exploiting the Electrocatalytic Properties of Hydrogenases. Chem. Rev. 2007, 107, 4366–4413. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, F.A.; Evans, R.M.; Hexter, S.V.; Murphy, B.J.; Roessler, M.M.; Wul, P. Guiding Principles of Hydrogenase Catalysis Instigated and Clarified by Protein Film Electrochemistry. Acc. Chem. Res. 2016, 49, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Del Barrio, M.; Sensi, M.; Orain, C.; Baffert, C.; Dementin, S.; Fourmond, V.; Léger, C. Electrochemical Investigations of Hydrogenases and Other Enzymes That Produce and Use Solar Fuels. Acc. Chem. Res. 2018, 51, 769–777. [Google Scholar] [CrossRef] [PubMed]
- De Lacey, A.L.; Fernandez, V.M.; Rousset, M.; Cammack, R. Activation and Inactivation of Hydrogenase Function and the Catalytic Cycle: Spectroelectrochemical Studies. Chem. Rev. 2007, 107, 4304–4330. [Google Scholar] [CrossRef] [PubMed]
- Fourmond, V.; Greco, C.; Sybirna, K.; Baffert, C.; Wang, P.-H.; Ezanno, P.; Montefiori, M.; Bruschi, M.; Meynial-Salles, I.; Soucaille, P.; et al. The Oxidative Inactivation of FeFe Hydrogenase Reveals the Flexibility of the H-Cluster. Nat. Chem. 2014, 6, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Vignais, P.M.; Billoud, B. Occurrence, Classification, and Biological Function of Hydrogenases: An Overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef] [PubMed]
- Goldet, G.; Brandmayr, C.; Stripp, S.T.; Happe, T.; Cavazza, C.; Fontecilla-Camps, J.C.; Armstrong, F.A. Electrochemical Kinetic Investigations of the Reactions of [FeFe]-Hydrogenases with Carbon Monoxide and Oxygen: Comparing the Importance of Gas Tunnels and Active-Site Electronic/redox Effects. J. Am. Chem. Soc. 2009, 131, 14979–14989. [Google Scholar] [CrossRef] [PubMed]
- Hemschemeier, A.; Fouchard, S.; Cournac, L.; Peltier, G.; Happe, T. Hydrogen Production by Chlamydomonas Reinhardtii: An Elaborate Interplay of Electron Sources and Sinks. Planta 2008, 227, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.W.; Lanzilotta, W.N.; Lemon, B.J.; Seefeldt, L.C. X-ray Crystal Structure of the Fe-Only Hydrogenase (CpI) from Clostridium Pasteurianum to 1.8 Angstrom Resolution. Science 1998, 282, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Piras, C.; Legrand, P.; Hatchikian, C.E.; Fontecilla-Camps, J.C. Desulfovibrio Desulfuricans Iron Hydrogenase: The Structure Shows Unusual Coordination to an Active Site Fe Binuclear Center. Structure 1999, 7, 13–23. [Google Scholar] [CrossRef]
- Silakov, A.; Wenk, B.; Reijerse, E.; Lubitz, W. 14N HYSCORE Investigation of the H-Cluster of [FeFe] Hydrogenase: Evidence for a Nitrogen in the Dithiol Bridge. Phys. Chem. Chem. Phys. 2009, 11, 6553–6554. [Google Scholar] [CrossRef] [PubMed]
- Adamska-Venkatesh, A.; Krawietz, D.; Siebel, J.F.; Weber, K.; Happe, T.; Reijerse, E.; Lubitz, W. New Redox States Observed in [FeFe] Hydrogenases Reveal Redox Coupling within the H-Cluster. J. Am. Chem. Soc. 2014, 136, 11339–11346. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; King, P.W.; Chang, C.H. Proton Transport in Clostridium Pasteurianum [FeFe] Hydrogenase I: A Computational Study. J. Phys. Chem. B 2014, 118, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Pierik, A.J.; Hulstein, M.; Hagen, W.R.; Albracht, S.P. A Low-Spin Iron with CN and CO as Intrinsic Ligands Forms the Core of the Active Site in [Fe]-Hydrogenases. Eur. J. Biochem. 1998, 258, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, W.; De Lacey, A.L.; Fernandez, V.M.; Hatchikian, E.C.; Albracht, S.P.J. The Active Site of the [FeFe]-Hydrogenase from Desulfovibrio Desulfuricans. II. Redox Properties, Light Sensitivity and CO-Ligand Exchange as Observed by Infrared Spectroscopy. J. Biol. Inorg. Chem. 2006, 11, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Adamska-Venkatesh, A.; Silakov, A.; Lambertz, C.; Rüdiger, O.; Happe, T.; Reijerse, E.; Lubitz, W. Identification and Characterization of The “super-Reduced” state of the H-Cluster in [FeFe] Hydrogenase: A New Building Block for the Catalytic Cycle? Angew. Chem. Int. Ed. 2012, 51, 11458–11462. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.; Adamska-Venkatesh, A.; Pawlak, K.; Birrell, J.A.; Rüdiger, O.; Reijerse, E.J.; Lubitz, W. Proton Coupled Electronic Rearrangement within the H-Cluster as an Essential Step in the Catalytic Cycle of [FeFe] Hydrogenases. J. Am. Chem. Soc. 2017, 139, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Esselborn, J.; Muraki, N.; Klein, K.; Engelbrecht, V.; Metzler-Nolte, N.; Apfel, U.-P.; Hofmann, E.; Kurisu, G.; Happe, T. A Structural View of Synthetic Cofactor Integration into [FeFe]-Hydrogenases. Chem. Sci. 2016, 7, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Lampret, O.; Adamska-Venkatesh, A.; Konegger, H.; Wittkamp, F.; Apfel, U.-P.; Reijerse, E.J.; Lubitz, W.; Rü, O.; Happe, T.; Winkler, M. Interplay between CN− Ligands and the Secondary Coordination Sphere of the H-Cluster in [FeFe]-Hydrogenases. J. Am. Chem. Soc. 2017, 139, 18222–18230. [Google Scholar] [CrossRef] [PubMed]
- Silakov, A.; Reijerse, E.J.; Albracht, S.P.J.; Hatchikian, E.C.; Lubitz, W. The Electronic Structure of the H-Cluster in the [FeFe]-Hydrogenase from Desulfovibrio Desulfuricans: A Q-Band 57Fe-ENDOR and HYSCORE Study. J. Am. Chem. Soc. 2007, 129, 11447–11458. [Google Scholar] [CrossRef] [PubMed]
- Stripp, S.T.; Goldet, G.; Brandmayr, C.; Sanganas, O.; Vincent, K.A.; Haumann, M.; Armstrong, F.A.; Happe, T. How Oxygen Attacks [FeFe] Hydrogenases from Photosynthetic Organisms. Proc. Natl. Acad. Sci. USA 2009, 106, 17331–17336. [Google Scholar] [CrossRef] [PubMed]
- Lemon, B.J.; Peters, J.W. Binding of Exogenously Added Carbon Monoxide at the Active Site of the Iron-Only Hydrogenase (CpI) from Clostridium Pasteurianum. Biochemistry 1999, 38, 12969–12973. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.; Lemon, B.J.; Peters, J.W. Reversible Carbon Monoxide Binding and Inhibition at the Active Site of the Fe-Only Hydrogenase. Biochemistry 2000, 39, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Happe, T.; Naber, J.D. Isolation, Characterization and N-Terminal Amino Acid Sequence of Hydrogenase from the Green Alga Chlamydomonas Reinhardtii. Eur. J. Biochem. 1993, 214, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.; Mebs, S.; Duan, J.; Wittkamp, F.; Apfel, U.-P.; Heberle, J.; Haumann, M.; Stripp, S.T. Stepwise Isotope Editing of [FeFe]-Hydrogenases Exposes Cofactor Dynamics. Proc. Natl. Acad. Sci. USA 2016, 113, 8454–8459. [Google Scholar] [CrossRef] [PubMed]
- Mebs, S.; Senger, M.; Duan, J.; Wittkamp, F.; Apfel, U.-P.; Happe, T.; Winkler, M.; Stripp, S.T.; Haumann, M. Bridging Hydride at Reduced H-Cluster Species in [FeFe]-Hydrogenases Revealed by Infrared Spectroscopy, Isotope Editing, and Quantum Chemistry. J. Am. Chem. Soc. 2017, 139, 12157–12160. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; Senger, M.; Duan, J.; Esselborn, J.; Wittkamp, F.; Hofmann, E.; Apfel, U.-P.; Stripp, S.T.; Happe, T. Accumulating the Hydride State in the Catalytic Cycle of [FeFe]-Hydrogenases. Nat. Commun. 2017, 8, 16115. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.; Laun, K.; Wittkamp, F.; Duan, J.; Haumann, M.; Happe, T.; Winkler, M.; Apfel, U.-P.; Stripp, S.T. Proton-Coupled Reduction of the Catalytic [4Fe-4S] Cluster in [FeFe]-Hydrogenases. Angew. Chem. Int. Ed. 2017, 56, 16503–16506. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.; Mebs, S.; Duan, J.; Shulenina, O.; Laun, K.; Kertess, L.; Wittkamp, F.; Apfel, U.-P.; Happe, T.; Winkler, M.; et al. Protonation/reduction Dynamics at the [4Fe–4S] Cluster of the Hydrogen-Forming Cofactor in [FeFe]-Hydrogenases. Phys. Chem. Chem. Phys. 2018, 20, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Wittkamp, F.; Senger, M.; Stripp, S.T.; Apfel, U.-P. [FeFe]-Hydrogenases: Recent Developments and Future Perspectives. Chem. Commun. 2018, 54, 5934–5942. [Google Scholar] [CrossRef] [PubMed]
- Haumann, M.; Stripp, S.T. The Molecular Proceedings of Biological Hydrogen Turnover. Acc. Chem. Res. 2018. [Google Scholar] [CrossRef]
- Berggren, G.; Adamska-Venkatesh, A.; Lambertz, C.; Simmons, T.R.; Esselborn, J.; Atta, M.; Gambarelli, S.; Mouesca, J.-M.; Reijerse, E.J.; Lubitz, W.; et al. Biomimetic Assembly and Activation of [FeFe]-Hydrogenases. Nature 2013, 499, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Esselborn, J.; Lambertz, C.; Adamska-Venkatesh, A.; Simmons, T.; Berggren, G.; Noth, J.; Siebel, J.F.; Hemschemeier, A.; Artero, V.; Reijerse, E.; et al. Spontaneous Activation of [FeFe]-Hydrogenases by an Inorganic [2Fe] Active Site Mimic. Nat. Chem. Biol. 2013, 9, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Lemon, B.J.; Peters, J.W. Photochemistry at the Active Site of the Carbon Monoxide Inhibited Form of the Iron-Only Hydrogenase (CpI). J. Am. Chem. Soc. 2000, 122, 3793–3794. [Google Scholar] [CrossRef]
- Sommer, C.; Rumpel, S.; Roy, S.; Farès, C.; Artero, V.; Fontecave, M.; Reijerse, E.; Lubitz, W. Spectroscopic Investigations of a Semi-Synthetic [FeFe] Hydrogenase with Propane Di-Selenol as Bridging Ligand in the Binuclear Subsite: Comparison to the Wild Type and Propane Di-Thiol Variants. J. Biol. Inorg. Chem. 2018, 23, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Filippi, G.; Arrigoni, F.; Bertini, L.; De Gioia, L.; Zampella, G. DFT Dissection of the Reduction Step in H 2 Catalytic Production by [FeFe]-Hydrogenase-Inspired Models: Can the Bridging Hydride Become More Reactive Than the Terminal Isomer? Inorg. Chem. 2015, 54, 9529–9542. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Greco, C.; Kaukonen, M.; Fantucci, P.; Ryde, U.; De Gioia, L. Influence of the [2Fe]H Subcluster Environment on the Properties of Key Intermediates in the Catalytic Cycle of [FeFe] Hydrogenases: Hints for the Rational Design of Synthetic Catalysts. Angew. Chem. Int. Ed. 2009, 48, 3503–3506. [Google Scholar] [CrossRef] [PubMed]
- Knörzer, P.; Silakov, A.; Foster, C.E.; Armstrong, F.A.; Lubitz, W.; Happe, T. Importance of the Protein Framework for Catalytic Activity of [FeFe]-Hydrogenases. J. Biol. Chem. 2012, 287, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Adamska-Venkatesh, A.; Simmons, T.R.; Siebel, J.F.; Artero, V.; Fontecave, M.; Lubitz, W. Chlamydomonas Reinhardtii: HYSCORE and ENDOR Study of a Non-Natural H-Cluster. Phys. Chem. Chem. Phys. 2015, 17, 5421–5430. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P. Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Becke, A. Density-Functional Exchange-Energy Approximation With Correct Asymptotic-Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H-Cluster Species | CO Pattern a | Vibrational Frequency (cm−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| p µ d1 d2 | CN‒ | CO | µCO | |||||
| Hox-CO | 1 | 12 12 12 12 | 2092 | 2082 | 2012 | 1968 | 1962 | 1808 |
| 2 | 12 12 12 13 | 2092 | 2082 | 1991 | 1964 | 1942 | 1808 | |
| 3 | 12 13 13 13 | 2092 | 2082 | 1972 | 1960 | 1920 | 1768 | |
| 4 | 12 13 13 12 | 2092 | 2082 | 2006 | 1964 | 1927 | 1768 | |
| Hred´-CO | 1 | 12 12 12 12 | 2086 | 2076 | 2002 | 1967 | 1951 | 1793 |
| 2 | 12 12 12 13 | 2086 | 2076 | 1984 | 1964 | 1930 | 1793 | |
| 3 | 12 13 13 13 | 2086 | 2076 | 1964 | 1952 | 1908 | 1753 | |
| 4 | 12 13 13 12 | 2086 | 2076 | 1996 | 1954 | 1914 | 1753 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laun, K.; Mebs, S.; Duan, J.; Wittkamp, F.; Apfel, U.-P.; Happe, T.; Winkler, M.; Haumann, M.; Stripp, S.T. Spectroscopical Investigations on the Redox Chemistry of [FeFe]-Hydrogenases in the Presence of Carbon Monoxide. Molecules 2018, 23, 1669. https://doi.org/10.3390/molecules23071669
Laun K, Mebs S, Duan J, Wittkamp F, Apfel U-P, Happe T, Winkler M, Haumann M, Stripp ST. Spectroscopical Investigations on the Redox Chemistry of [FeFe]-Hydrogenases in the Presence of Carbon Monoxide. Molecules. 2018; 23(7):1669. https://doi.org/10.3390/molecules23071669
Chicago/Turabian StyleLaun, Konstantin, Stefan Mebs, Jifu Duan, Florian Wittkamp, Ulf-Peter Apfel, Thomas Happe, Martin Winkler, Michael Haumann, and Sven T. Stripp. 2018. "Spectroscopical Investigations on the Redox Chemistry of [FeFe]-Hydrogenases in the Presence of Carbon Monoxide" Molecules 23, no. 7: 1669. https://doi.org/10.3390/molecules23071669
APA StyleLaun, K., Mebs, S., Duan, J., Wittkamp, F., Apfel, U.-P., Happe, T., Winkler, M., Haumann, M., & Stripp, S. T. (2018). Spectroscopical Investigations on the Redox Chemistry of [FeFe]-Hydrogenases in the Presence of Carbon Monoxide. Molecules, 23(7), 1669. https://doi.org/10.3390/molecules23071669