NTyroSite: Computational Identification of Protein Nitrotyrosine Sites Using Sequence Evolutionary Features

Abstract
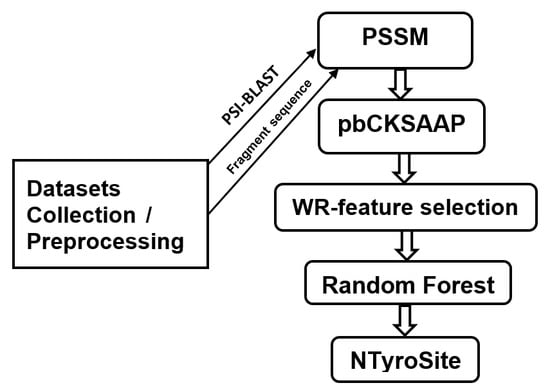
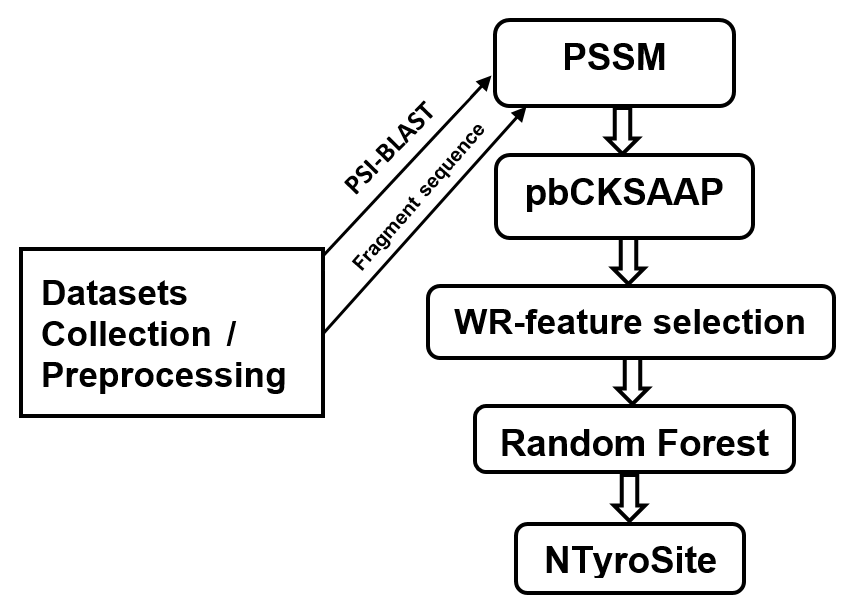
1. Introduction
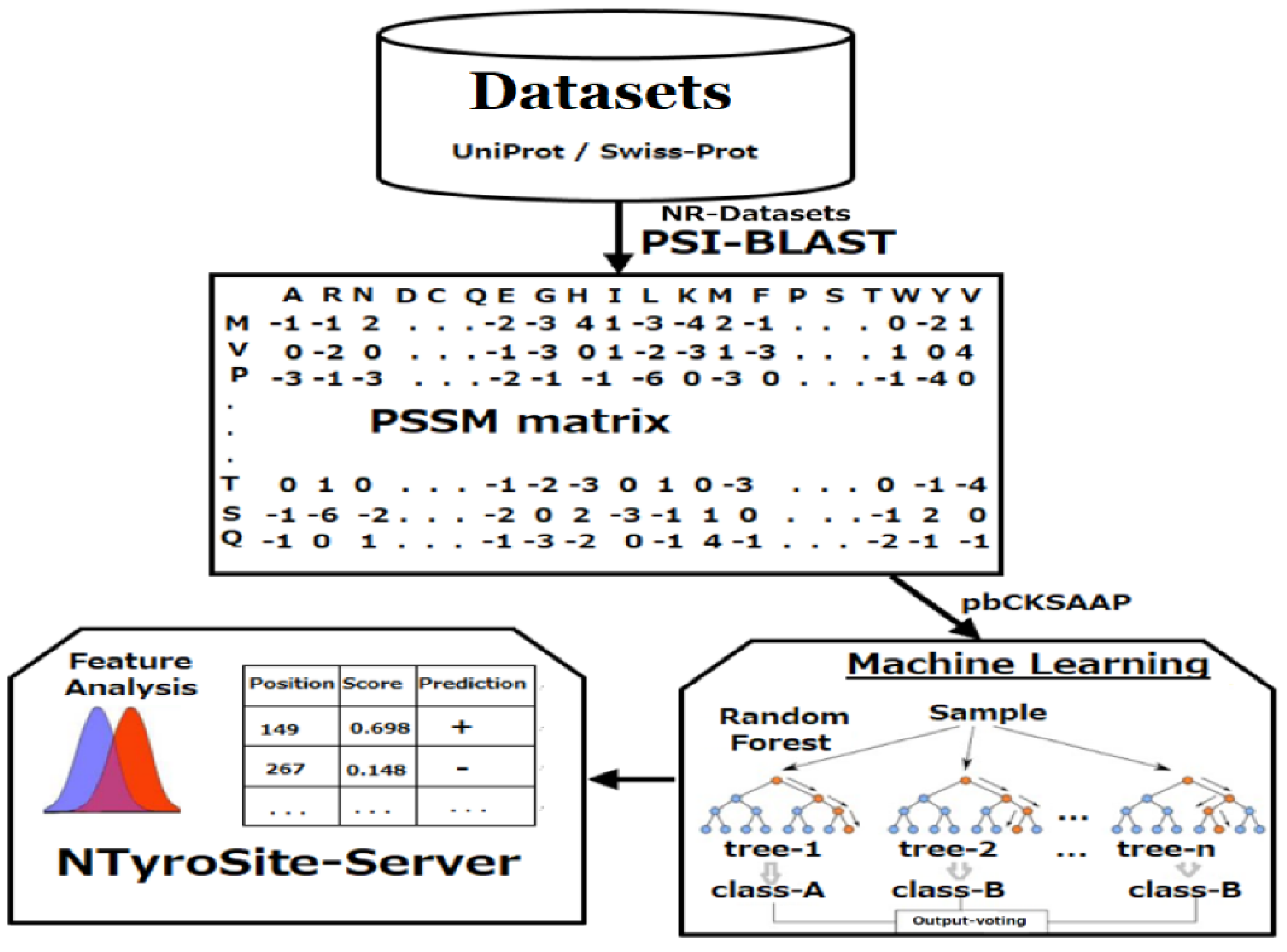
2. Materials and Methods
2.1. Data Preparation
2.2. Encoding Strategy of Sequence Evolutionary Information
2.3. KSAAP Encoding
2.4. AAindex Encoding
2.5. Binary Encoding
2.6. Random Forest
2.7. Rule Extraction
2.8. Feature Selection
2.9. Performance Evaluation
3. Results and Discussion
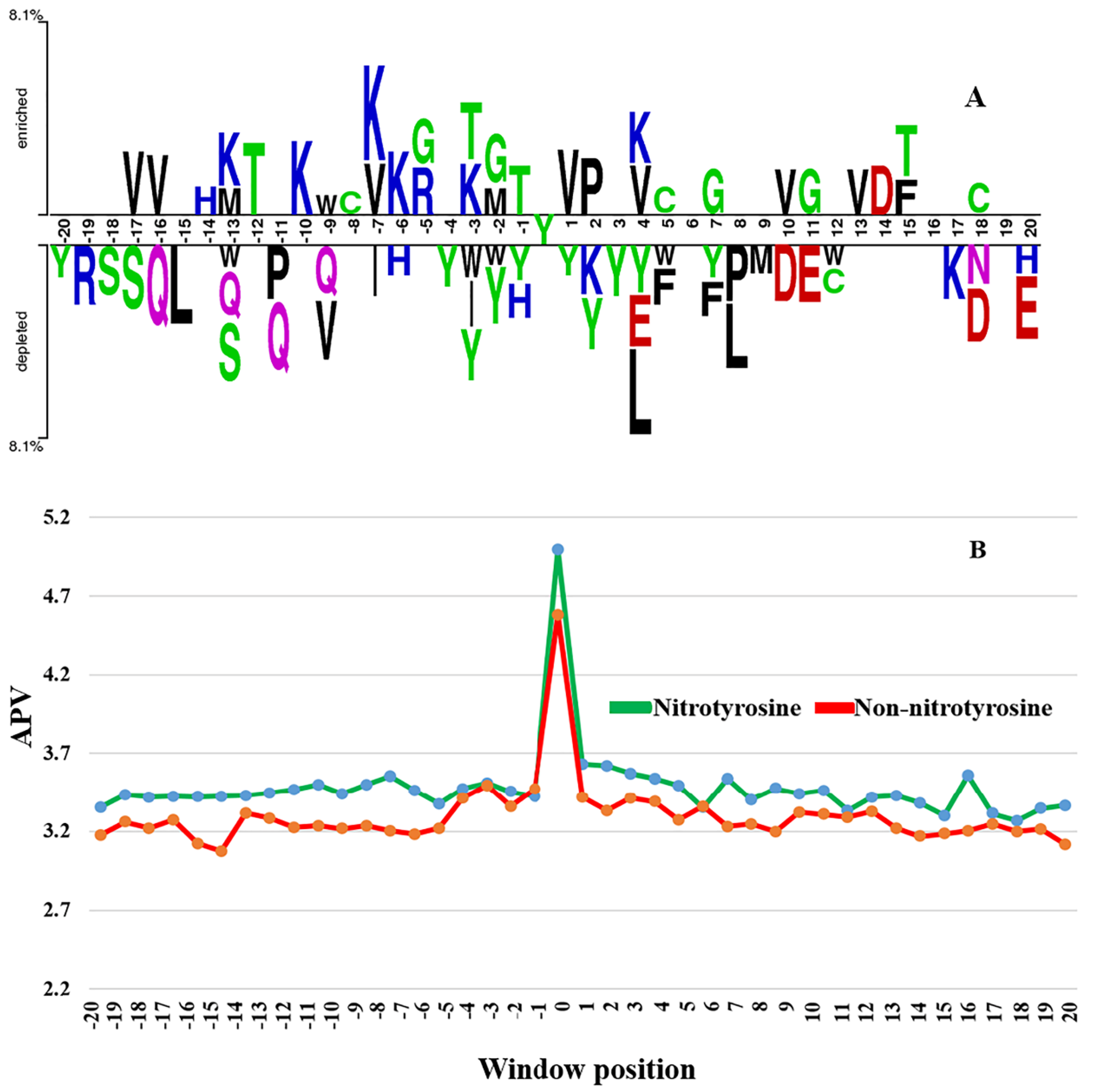
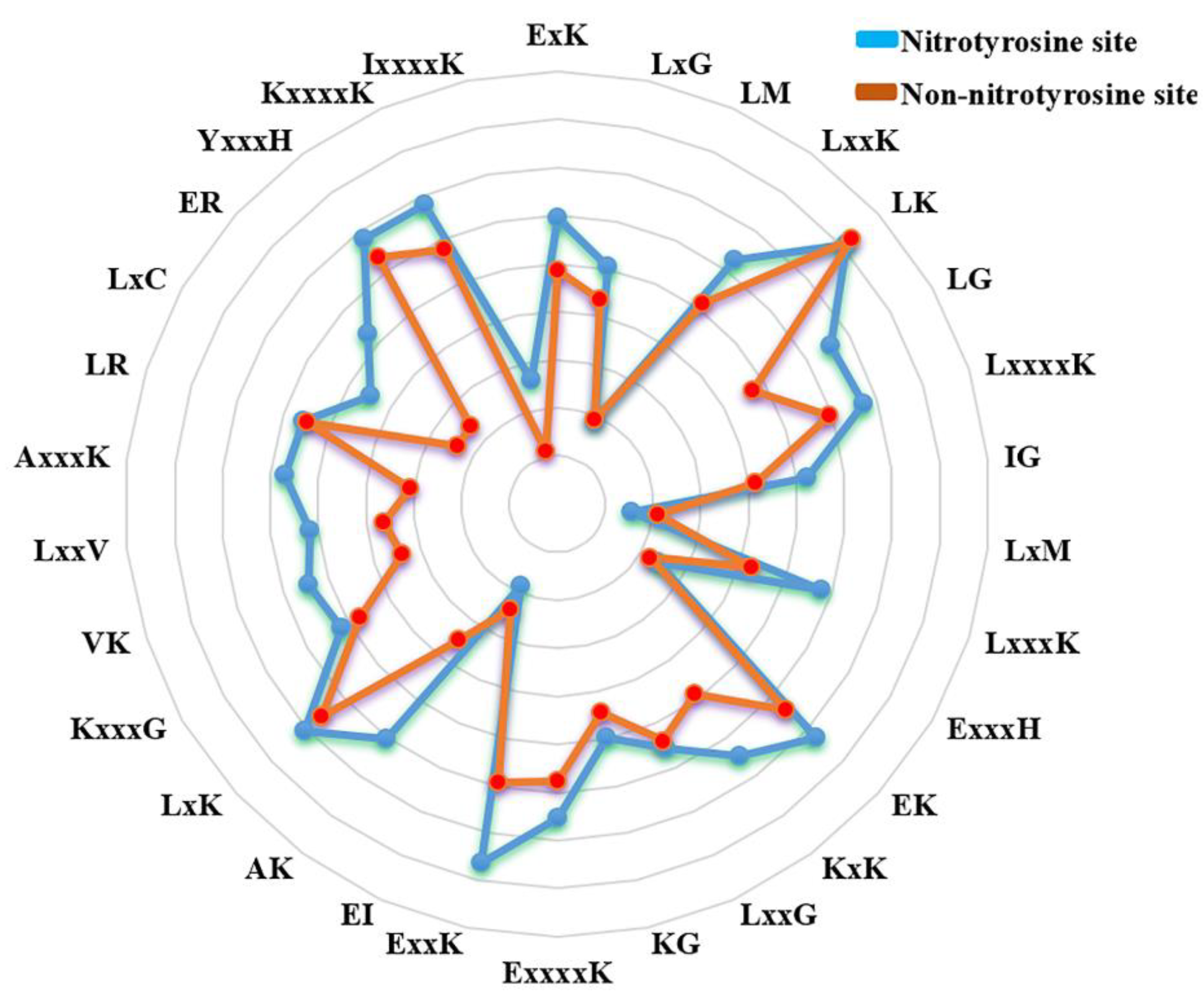
3.1. The Difference of Amino Acid Propensity in Nitrotyrosine Protein
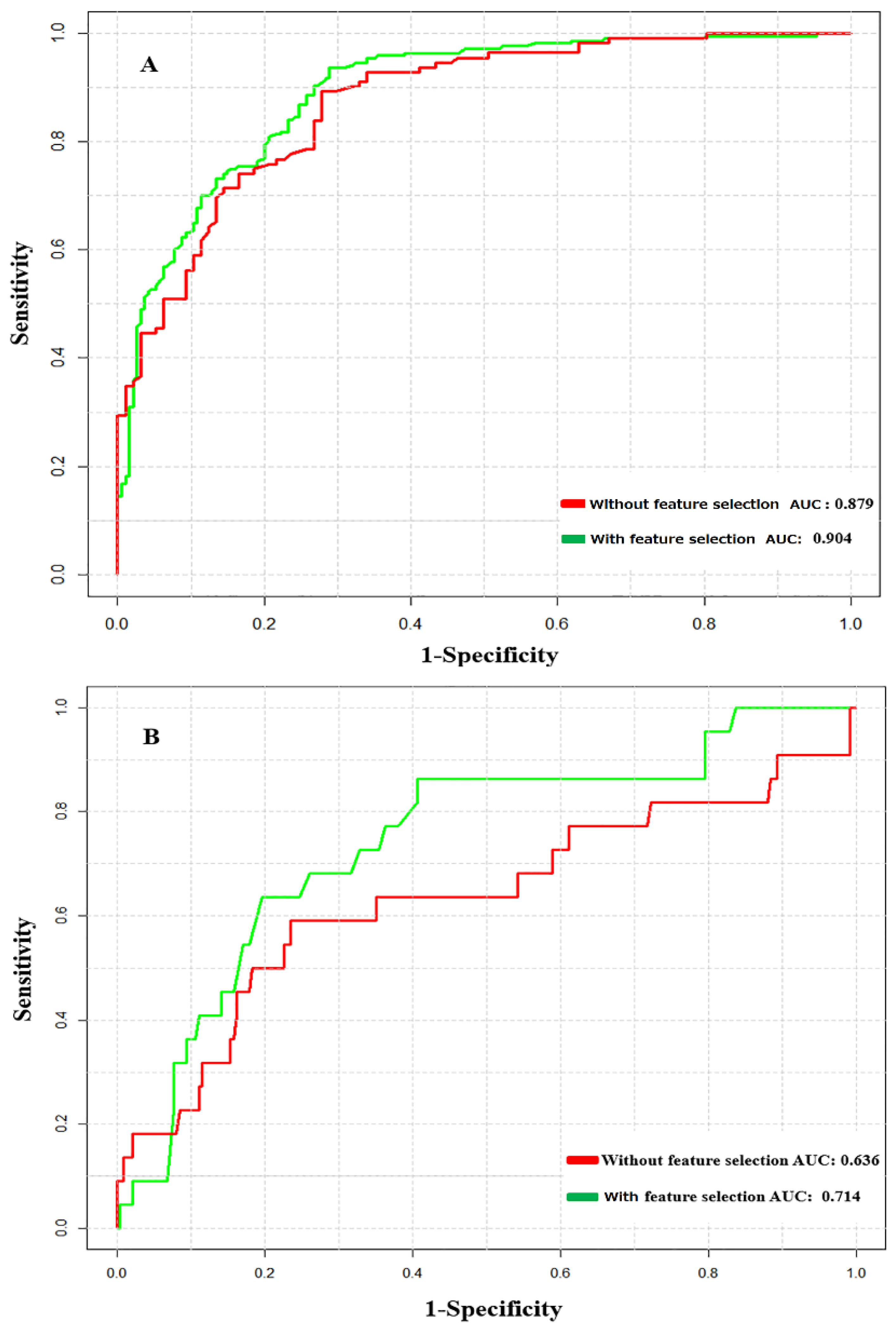
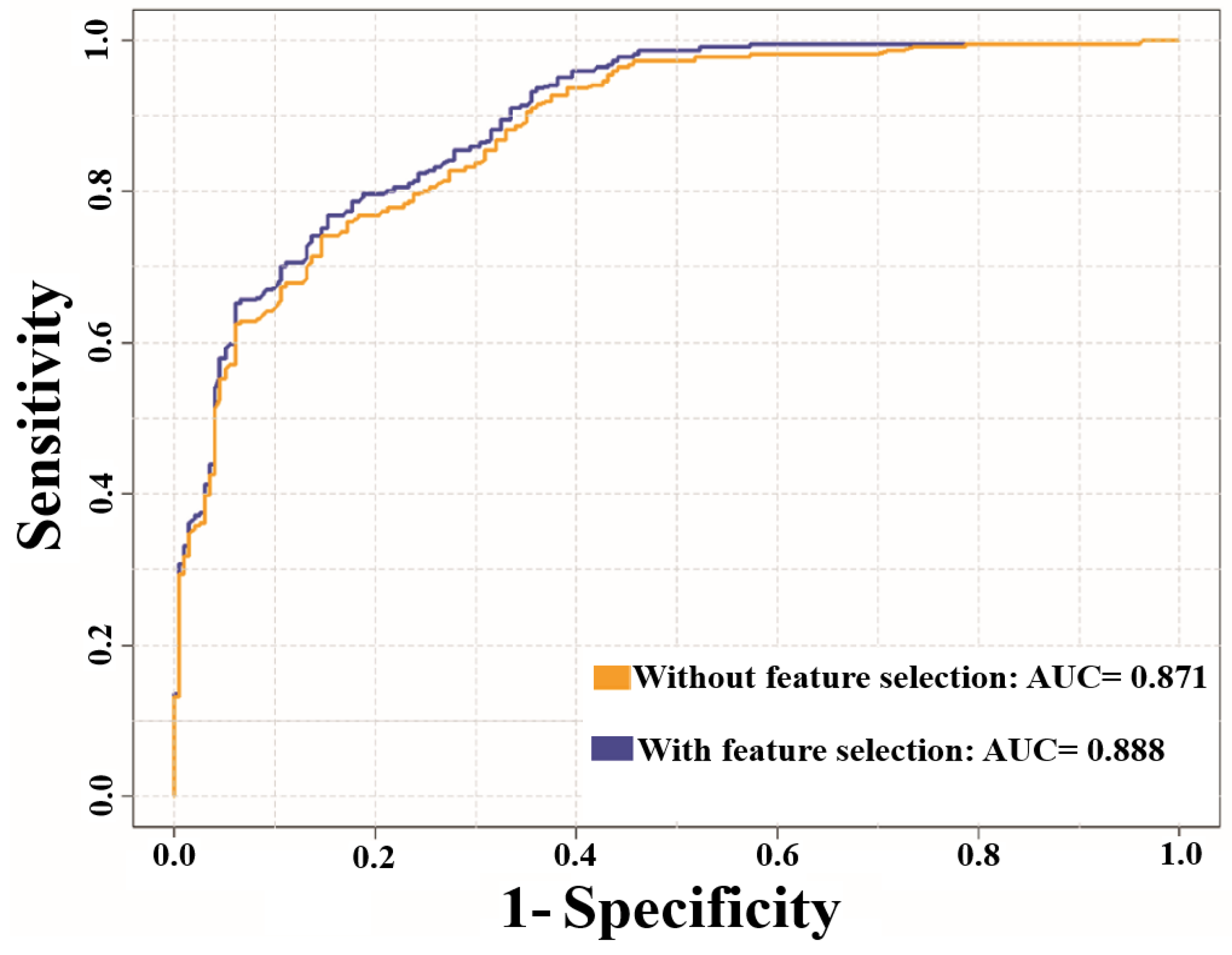
3.2. Prediction Performance of NTyroSite on the Cross-Validation Test
3.3. Analysis of Feature Optimization Results Using Wilcoxon Rank-Sum Test
3.4. Performance Comparison with Existing Tools on the Test Dataset
3.5. The Influence of Peptide Level Sequence Redundancy on the Predictive Model
3.6. Rules Extracted from NTyroSite Model
3.7. Case Studies
3.8. Comparison of WR with IG and mRMR Feature Selection Scheme
3.9. Comparison with Different Sequence-Based Features
3.10. Web Server
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Peng, F.; Li, J.; Guo, T.; Yang, H.; Li, M.; Sang, S.; Li, X.; Desiderio, D.M.; Zhan, X. Nitroproteins in Human Astrocytomas Discovered by Gel Electrophoresis and Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 2062–2076. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Wang, X.; Desiderio, D.M. Pituitary adenoma nitroproteomics: Current status and perspectives. Oxid. Med. Cell. Longev. 2013, 2013, 580710. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Wang, X.; Desiderio, D.M. Mass spectrometry analysis of nitrotyrosine-containing proteins. Mass Spectrom. Rev. 2015, 34, 423–448. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Scaloni, A.; Giustarini, D.; Cavarra, E.; Tell, G.; Lungarella, G.; Colombo, R.; Rossi, R.; Milzani, A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox proteomics. Mass Spectrom. Rev. 2005, 24, 55–99. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Kanski, J.; Hong, S.J.; Schoneich, C. Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J. Biol. Chem. 2005, 280, 24261–24266. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Desiderio, D.M. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal. Biochem. 2006, 354, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Desiderio, D.M. The human pituitary nitroproteome: Detection of nitrotyrosyl-proteins with two-dimensional Western blotting, and amino acid sequence determination with mass spectrometry. Biochem. Biophys. Res. Commun. 2004, 325, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Uhlin, U.; Stubbe, J. Site-specific incorporation of 3-nitrotyrosine as a probe of pKa perturbation of redox-active tyrosines in ribonucleotide reductase. J. Am. Chem. Soc. 2010, 132, 8385–8397. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Desiderio, D.M. Nitroproteins identified in human ex-smoker bronchoalveolar lavage fluid. Aging Dis. 2011, 2, 100–115. [Google Scholar] [PubMed]
- Song, Y.; Liao, J.; Zha, C.; Wang, B.; Liu, C.C. Simultaneous determination of 3-chlorotyrosine and 3-nitrotyrosine in human plasma by direct analysis in real time-tandem mass spectrometry. Acta Pharm. Sin. B 2015, 5, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Nuriel, T.; Whitehouse, J.; Ma, Y.; Mercer, E.J.; Brown, N.; Gross, S.S. ANSID: A solid-phase proteomic approach for identification and relative quantification of aromatic nitration sites. Front. Chem. 2015, 3, 70. [Google Scholar] [CrossRef] [PubMed]
- Seeley, K.W.; Fertig, A.R.; Dufresne, C.P.; Pinho, J.P.; Stevens, S.M., Jr. Evaluation of a method for nitrotyrosine site identification and relative quantitation using a stable isotope-labeled nitrated spike-in standard and high resolution fourier transform MS and MS/MS analysis. Int. J. Mol. Sci. 2014, 15, 6265–6285. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, J.R.; Kim, Y.H.; Park, Y.S.; Park, S.I.; Park, H.S.; Kim, K.P. Investigation of tyrosine nitration and nitrosylation of angiotensin II and bovine serum albumin with electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 2797–2804. [Google Scholar] [CrossRef] [PubMed]
- Sarver, A.; Scheffler, N.K.; Shetlar, M.D.; Gibson, B.W. Analysis of peptides and proteins containing nitrotyrosine by matrix-assisted laser desorption/ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2001, 12, 439–448. [Google Scholar] [CrossRef]
- Xu, Y.; Wen, X.; Wen, L.S.; Wu, L.Y.; Deng, N.Y.; Chou, K.C. iNitro-Tyr: Prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS ONE 2014, 9, e105018. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cao, J.; Ma, Q.; Gao, X.; Ren, J.; Xue, Y. GPS-YNO2: Computational prediction of tyrosine nitration sites in proteins. Mol. Biosyst. 2011, 7, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Zhou, Y.; Lu, X.; Li, J.; Song, J.; Zhang, Z. Computational identification of protein pupylation sites by using profile-based composition of k-spaced amino acid pairs. PLoS ONE 2015, 10, e0129635. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Han, Y.; Chen, T.; Tu, X.M. Causal inference for Mann-Whitney-Wilcoxon rank sum and other nonparametric statistics. Stat. Med. 2014, 33, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.Y.; Su, M.G.; Kao, H.J.; Hsieh, Y.C.; Jhong, J.H.; Cheng, K.H.; Huang, H.D.; Lee, T.Y. dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins. Nucleic Acids Res. 2016, 44, D435–D446. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, J.; Li, H.; Yu, J.; Sun, H.; He, Y.; Lv, D.; Yang, X.; Glocker, M.O.; Ma, L.; et al. SysPTM 2.0: An updated systematic resource for post-translational modification. Database J. Biol. Databases Curation 2014, 2014, bau025. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Provost, F. Machine Learning from Imbalanced Data Sets 101. In Proceedings of the AAAI Workshop on Learning from Imbalanced Data Set, Austin, TX, USA, 30 July 2000; pp. 1–3. [Google Scholar]
- Chen, Y.W.; Lin, C.J. Combining SVMs with Various Feature Selection Strategies. In Feature Extraction; Springer: Berlin, Germany, 2006; pp. 315–324. [Google Scholar]
- Radivojac, P.; Vacic, V.; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 2010, 78, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zhang, Y.J.; Zhang, Z. Using weakly conserved motifs hidden in secretion signals to identify type-III effectors from bacterial pathogen genomes. PLoS ONE 2013, 8, e56632. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Jiang, Y.; Du, L.; Kurgan, L. Prediction of integral membrane protein type by collocated hydrophobic amino acid pairs. J. Comput. Chem. 2009, 30, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Yang, S.; Zhou, Y.; Mollah, M.N. SuccinSite: A computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol. BioSyst. 2016, 12, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhou, Y.; Zhang, Z.; Song, J. Towards more accurate prediction of ubiquitination sites: A comprehensive review of current methods, tools and features. Brief. Bioinform. 2015, 16, 640–657. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef] [PubMed]
- Maclin, R.; Opitz, D. Popular ensemble methods: An empirical study. J. Artif. Intell. Res. 1999, 11, 169–198. [Google Scholar]
- Polikar, R. Ensemble based systems in decision making. Circuits Syst. Mag. IEEE 2006, 6, 21–45. [Google Scholar] [CrossRef]
- Rokach, L. Ensemble-based classifiers. Artif. Intell. Rev. 2010, 33, 1–39. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Yan, R. AAFreqCoil: A new classifier to distinguish parallel dimeric and trimeric coiled coils. Mol. BioSyst. 2015, 11, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Wang, H.; Tan, H.; Zhang, Z.; Webb, G.I.; Song, J. Accurate in silico identification of species-specific acetylation sites by integrating protein sequence-derived and functional features. Sci. Rep. 2014, 4, 5765. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Mollah, M.N.; Cao, Y.; Guo, D. A systematic identification of species-specific protein succinylation sites using joint element features information. Int. J. Nanomed. 2017, 12, 6303–6315. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S. Recent progress and challenges for protein pupylation sites prediction. EC Proteom. Bioinform. 2017, 2, 36–45. [Google Scholar]
- Hasan, M.M.; Guo, D.; Kurata, H. Computational identification of protein S-sulfenylation sites by incorporating the multiple sequence features information. Mol. BioSyst. 2017, 13, 2545–2550. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Kurata, H. Computational modeling of lysine post-translational modification: An overview. Curr. Synth. Syst. Biol. 2018, 6, 137. [Google Scholar] [CrossRef]
- Hasan, M.M.; Khatun, M.S. Prediction of protein post-translational modification sites: An overview. Ann. Proteom. Bioinform. 2018, 2, 049–057. [Google Scholar]
- Wei, L.; Tang, J.; Zhou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2016, 2017, 135–144. [Google Scholar] [CrossRef]
- Hasan, M.M.; Kurata, H. GPSuc: Global prediction of generic and species-specific succinylation sites by aggregating multiple sequence features. PLoS ONE 2018, in press. [Google Scholar]
- Jia, C.; Zuo, Y.; Zou, Q. O-GlcNAcPRED-II: An integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 2018, 34, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Tang, J.; Zou, Q. PhosPred-RF: A Novel Sequence-Based Predictor for Phosphorylation Sites Using Sequential Information Only. IEEE Trans. Nanobiosci. 2017, 16, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Shi, G.; Ji, Z.L.; Zou, Q. Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.P.; Shi, S.P.; Xu, H.D.; Wang, L.N.; Qiu, J.D. Accurate in silico prediction of species-specific methylation sites based on information gain feature optimization. Bioinformatics 2016, 32, 3107–3115. [Google Scholar] [CrossRef] [PubMed]
- Gribskov, M.; Robinson, N.L. Use of receiver operating characteristic (ROC) analysis to evaluate sequence matching. Comput. Chem. 1996, 20, 25–33. [Google Scholar] [CrossRef]
- Centor, R.M. Signal detectability—The use of roc curves and their analyses. Med. Decis. Mak. 1991, 11, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Iakoucheva, L.M.; Radivojac, P. Two Sample Logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.; Matsuyama, A.; Yoshida, M.; Kim, D. PostMod: Sequence based prediction of kinase-specific phosphorylation sites with indirect relationship. BMC Bioinform. 2010, 11 (Suppl. 1), S10. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Protein tyrosine nitration: Biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Bayden, A.S.; Yakovlev, V.A.; Graves, P.R.; Mikkelsen, R.B.; Kellogg, G.E. Factors influencing protein tyrosine nitration—Structure-based predictive models. Free Radic. Biol. Med. 2011, 50, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.M.; Daikhin, E.; Yudkoff, M.; Raman, C.S.; Ischiropoulos, H. Factors determining the selectivity of protein tyrosine nitration. Arch. Biochem. Biophys. 1999, 371, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Gondeau, C.; Chaloin, L.; Lallemand, P.; Roy, B.; Perigaud, C.; Barman, T.; Varga, A.; Vas, M.; Lionne, C.; Arold, S.T. Molecular basis for the lack of enantioselectivity of human 3-phosphoglycerate kinase. Nucleic Acids Res. 2008, 36, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Fiorillo, A.; Poser, E.; Lalioti, V.S.; Sundell, G.N.; Ivarsson, Y.; Genovese, I.; Colotti, G. Structural basis of Sorcin-mediated calcium-dependent signal transduction. Sci. Rep. 2015, 5, 16828. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Ratio (P/N) | Sp | Sn | Pr | Ac | MCC |
|---|---|---|---|---|---|
| 1:1 | 0.901 | 0.616 | 0.865 | 0.759 | 0.537 |
| 1:2 | 0.900 | 0.567 | 0.757 | 0.789 | 0.511 |
| 1:3 | 0.902 | 0.548 | 0.612 | 0.813 | 0.474 |
| 1:total | 0.904 | 0.523 | 0.438 | 0.856 | 0.416 |
| Measurement | Training Test | Independent Test | ||
|---|---|---|---|---|
| without Feature Selection | with Feature Selection | without Feature Selection | with Feature Selection | |
| Sp | 0.901 | 0.900 | 0.806 | 0.801 |
| Sn | 0.616 | 0.675 | 0.479 | 0.609 |
| Pr | 0.865 | 0.884 | 0.197 | 0.231 |
| Ac | 0.759 | 0.787 | 0.778 | 0.782 |
| MCC | 0.537 | 0.601 | 0.196 | 0.272 |
| Measurement | GPS-YNO2 | iNitro-Tyr | NTyroSite |
|---|---|---|---|
| Sp | 0.791 | 0.796 | 0.801 |
| Sn | 0.211 | 0.211 | 0.609 |
| Pr | 0.087 | 0.089 | 0.231 |
| Ac | 0.741 | 0.745 | 0.782 |
| MCC | 0.002 | 0.004 | 0.274 |
| No. | Individual Reports of Rule Extraction | No. of Samples Covered by Rule |
|---|---|---|
| 1 | I(K × L) > 0.197 | 172 |
| 2 | I(GK) > 0.113 & I(K××××L) ≤ 0.097 & I(ST) ≤ 0.038 | 112 |
| 3 | I(A××××K) > 0.118 & I(H××××A) ≤ 0.013 | 90 |
| 4 | I(YG) ≤ 0.068 & I(K×××A) ≤ 0.014 & I(S××××G) ≤ 0.039 & I(R××K) ≤ 0.089 & I(V×××M) ≤ 0.014 & I(E××C) ≤ 0.038 & I(EV) > 0.163 | 78 |
| 5 | I(R××K) ≤ 0.089 & I(L××××H) ≤ 0.118 & I(S×K) ≤ 0.092 & I(SR) > 0.229 | 67 |
| 6 | I(A××R) ≤ 0.068 & I(K×××A) ≤ 0.014 & I(S××××G) ≤ 0.039 & I(R××K) > 0.089 & I(V×R) ≤ 0.026 | 49 |
| 7 | I(A××××K) ≤ 0.118 & I(R××K) ≤ 0.089 & I(LG) ≤ 0.113 & I(G××H) ≤ 0.095 & I(EM) ≤ 0.013 & I(K××××G) ≤ 0.042 & I(G×A) > 0.141 | 47 |
| 8 | I(G××H) > 0.149 & I(L×××M) ≤ 0.069 & I(DG) ≤ 0.038 & I(Y×××K) ≤ 0.0139 & I(A×K) ≤ 0.034 | 44 |
| 9 | I(Y×H) > 0.163 & I(V×R) ≤ 0.013 & I(LG) ≤ 0.113 & I(K×××I) ≤ 0.128 | 39 |
| 10 | I(DK) ≤ 0.092 & I(G×××G) > 0.122 & I(S×K) ≤ 0.013 | 35 |
| Methods | IG | mRMR | WR |
|---|---|---|---|
| Sp | 0.897 | 0.899 | 0.900 |
| Sn | 0.601 | 0.596 | 0.675 |
| Pr | 0.861 | 0.855 | 0.884 |
| Ac | 0.749 | 0.748 | 0.787 |
| MCC | 0.511 | 0.507 | 0.601 |
| Methods | Sp | Sn | Pr | Ac | MCC |
|---|---|---|---|---|---|
| AAindex | 0.896 | 0.442 | 0.802 | 0.669 | 0.424 |
| BE | 0.899 | 0.435 | 0.799 | 0.667 | 0.402 |
| KSAAP | 0.900 | 0.587 | 0.857 | 0.744 | 0.501 |
| pbCKSAAP | 0.901 | 0.617 | 0.865 | 0.759 | 0.538 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, M.M.; Khatun, M.S.; Mollah, M.N.H.; Yong, C.; Dianjing, G. NTyroSite: Computational Identification of Protein Nitrotyrosine Sites Using Sequence Evolutionary Features. Molecules 2018, 23, 1667. https://doi.org/10.3390/molecules23071667
Hasan MM, Khatun MS, Mollah MNH, Yong C, Dianjing G. NTyroSite: Computational Identification of Protein Nitrotyrosine Sites Using Sequence Evolutionary Features. Molecules. 2018; 23(7):1667. https://doi.org/10.3390/molecules23071667
Chicago/Turabian StyleHasan, Md. Mehedi, Mst. Shamima Khatun, Md. Nurul Haque Mollah, Cao Yong, and Guo Dianjing. 2018. "NTyroSite: Computational Identification of Protein Nitrotyrosine Sites Using Sequence Evolutionary Features" Molecules 23, no. 7: 1667. https://doi.org/10.3390/molecules23071667
APA StyleHasan, M. M., Khatun, M. S., Mollah, M. N. H., Yong, C., & Dianjing, G. (2018). NTyroSite: Computational Identification of Protein Nitrotyrosine Sites Using Sequence Evolutionary Features. Molecules, 23(7), 1667. https://doi.org/10.3390/molecules23071667