Feature Selection via Swarm Intelligence for Determining Protein Essentiality


Abstract
1. Introduction
2. Preliminary
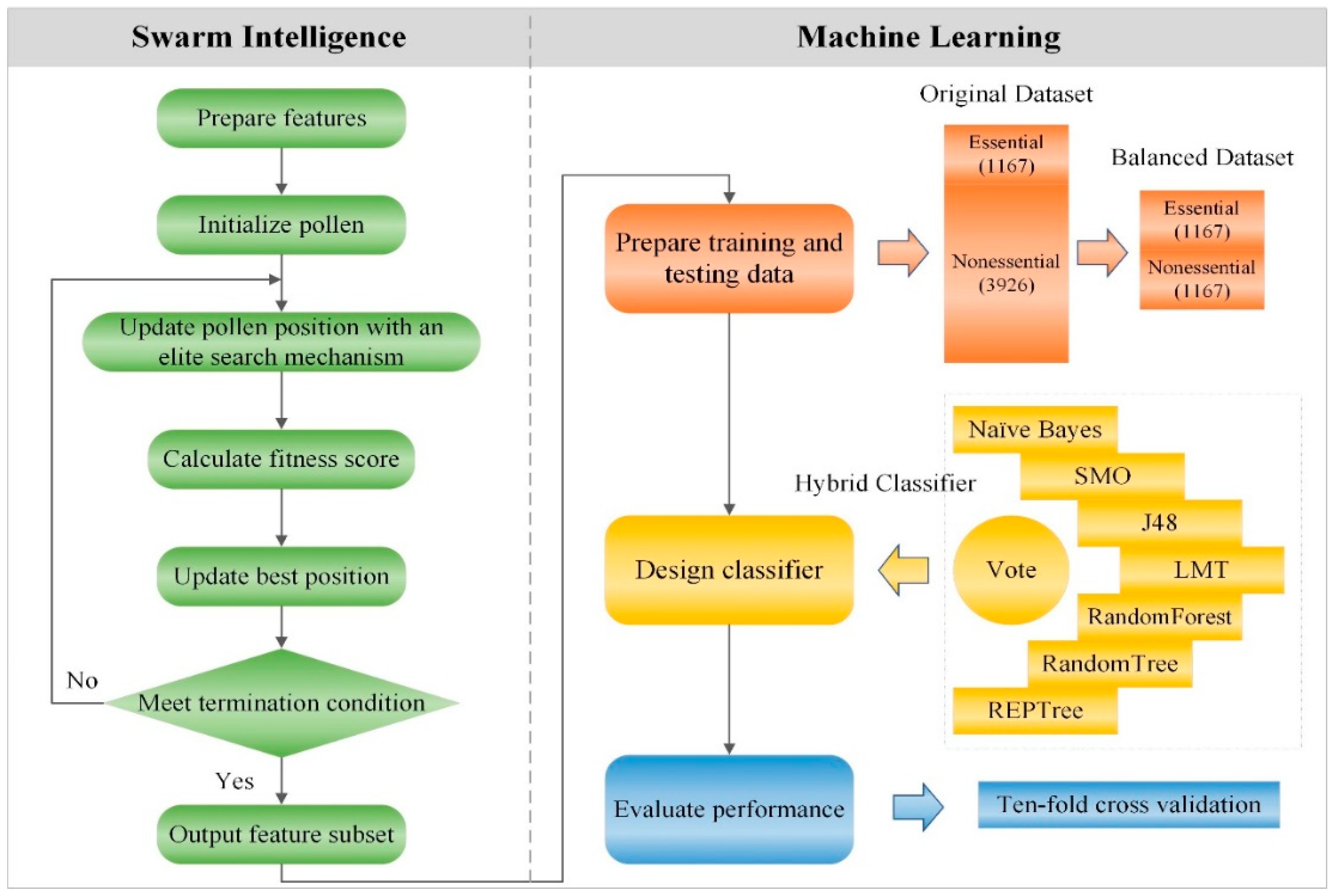
3. Materials and Methods
3.1. Data Source and Feature Preparation
3.1.1. Topological Features
3.1.2. Biological Features
3.2. Golden Standard of Features
3.3. Encoding and Initializing
3.4. Fitness Score
3.5. Update Rule with an Elite Search Mechanism
3.6. Outputting Feature Subset
3.7. Preparing Training and Testing Data
3.8. Classifier Design and Evaluation
4. Results and Discussion
4.1. Comparison of All the Balanced Datasets
4.2. Comparison of the Balanced Dataset with Imbalanced Dataset
4.3. Comparison of Different Feature Selection Methods
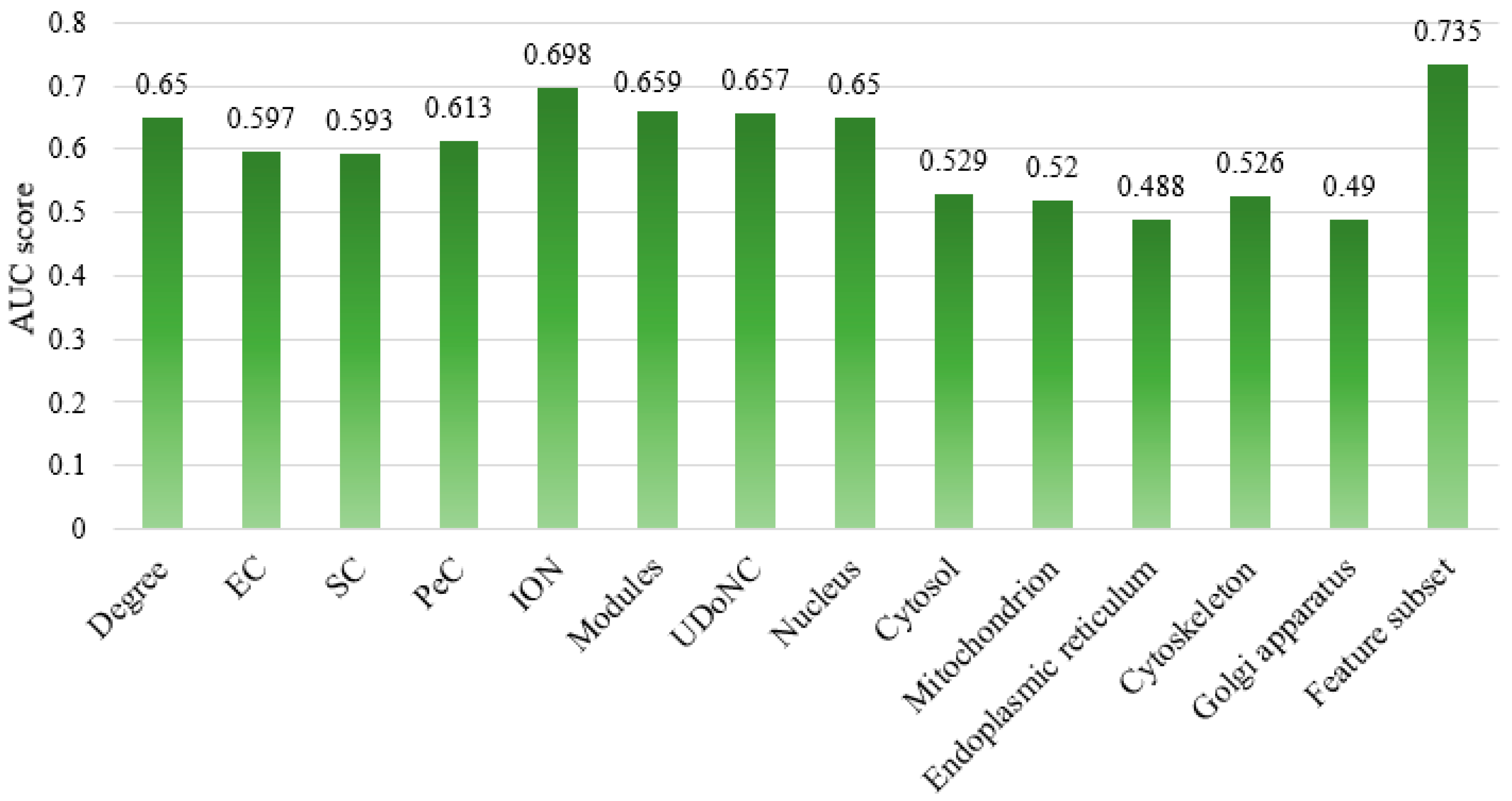
4.4. Comparison of Different Feature Subsets
4.5. Comparison of Diverse Classifiers Performance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lu, Y.; Deng, J.; Rhodes, J.C.; Lu, H.; Lu, L.J. Predicting essential genes for identifying potential drug targets in aspergillus fumigatus. Comput. Biol. Chem. 2014, 50, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, D. Understanding protein dispensability through machine-learning analysis of high-throughput data. Bioinformatics 2005, 21, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Veronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; Andre, B.; et al. Functional profiling of the saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Kamath, R.S.; Fraser, A.G.; Dong, Y.; Poulin, G.; Durbin, R.; Gotta, M.; Kanapin, A.; Le Bot, N.; Moreno, S.; Sohrmann, M.; et al. Systematic functional analysis of the caenorhabditis elegans genome using RNAi. Nature 2003, 421, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Roemer, T.; Jiang, B.; Davison, J.; Ketela, T.; Veillette, K.; Breton, A.; Tandia, F.; Linteau, A.; Sillaots, S.; Marta, C.; et al. Large-scale essential gene identification in candida albicans and applications to antifungal drug discovery. Mol. Microbiol. 2003, 50, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Mason, S.P.; Barabasi, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Joy, M.P.; Brock, A.; Ingber, D.E.; Huang, S. High-betweenness proteins in the yeast protein interaction network. J. Biomed. Biotechnol. 2005, 2005, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Wuchty, S.; Stadler, P.F. Centers of complex networks. J. Theor. Biol. 2003, 223, 45–53. [Google Scholar] [CrossRef]
- Bonacich, P. Power and centrality: A family of measures. Am. J. Sociol. 1987, 92, 1170–1182. [Google Scholar] [CrossRef]
- Stephenson, K.; Zelen, M. Rethinking centrality: Methods and examples. Soc. Netw. 1989, 11, 1–37. [Google Scholar] [CrossRef]
- Estrada, E.; Rodriguez-Velazquez, J.A. Subgraph centrality in complex networks. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2005, 71. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Wang, J.; Wang, W.; Liu, Q.; Wu, F.-X.; Pan, Y. Iteration method for predicting essential proteins based on orthology and protein-protein interaction networks. BMC Syst. Biol. 2012, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, J.; Xiao, W.-X. A new method for the discovery of essential proteins. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lu, Y.; Niu, Z.; Wu, F.-X. United complex centrality for identification of essential proteins from PPI networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, A.M.; Snitkin, E.S.; Parker, S.C.J.; DeLisi, C.; Kasif, S. Towards the identification of essential genes using targeted genome sequencing and comparative analysis. BMC Genom. 2006, 7, 265. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.-C.; Lin, C.-C.; Chang, J.-Y.; Mori, H.; Juan, H.-F.; Huang, H.-C. Predicting essential genes based on network and sequence analysis. Mol. Biosyst. 2009, 5, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Acencio, M.L.; Lemke, N. Towards the prediction of essential genes by integration of network topology, cellular localization and biological process information. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Wang, J.; Peng, W.; Zhang, Z.; Li, M. A feature selection method for prediction essential protein. Tsinghua Sci. Technol. 2015, 20, 491–499. [Google Scholar] [CrossRef]
- Moradi, P.; Gholampour, M. A hybrid particle swarm optimization for feature subset selection by integrating a novel local search strategy. Appl. Soft Comput. 2016, 43, 117–130. [Google Scholar] [CrossRef]
- Whitney, A.W. A direct method of nonparametric measurement selection. IEEE Trans. Comput. 1971, C-20, 1100–1103. [Google Scholar] [CrossRef]
- Marill, T.; Green, D. On the effectiveness of receptors in recognition systems. IEEE Trans. Inf. Theory 1963, 9, 11–17. [Google Scholar] [CrossRef]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Zeng, J.; Cao, L.; Ji, R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting tata binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Eberhart, R. Particle swarm optimization. In Proceedings of the IEEE International Conference on Neural Networks, Perth, Australia, 27 November–1 December 1995; pp. 1942–1948. [Google Scholar]
- Samanta, B.; Nataraj, C. Application of particle swarm optimization and proximal support vector machines for fault detection. Swarm Intell. 2009, 3, 303–325. [Google Scholar] [CrossRef]
- Unler, A.; Murat, A. A discrete particle swarm optimization method for feature selection in binary classification problems. Eur. J. Oper. Res. 2010, 206, 528–539. [Google Scholar] [CrossRef]
- Zhang, Y.; Gong, D.; Sun, X.; Guo, Y. A pso-based multi-objective multi-label feature selection method in classification. Sci. Rep. 2017, 7, 376. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Honavar, V. Feature subset selection using a genetic algorithm. IEEE Intell. Syst. Their Appl. 1998, 13, 44–49. [Google Scholar] [CrossRef]
- Oh, I.-S.; Lee, J.-S.; Moon, B.-R. Hybrid genetic algorithms for feature selection. IEEE Trans. Pattern Anal. Mach. Intell. 2004, 26, 1424–1437. [Google Scholar] [PubMed]
- Das, A.K.; Das, S.; Ghosh, A. Ensemble feature selection using bi-objective genetic algorithm. Knowl.-Based Syst. 2017, 123, 116–127. [Google Scholar] [CrossRef]
- Sivagaminathan, R.K.; Ramakrishnan, S. A hybrid approach for feature subset selection using neural networks and ant colony optimization. Expert Syst. Appl. 2007, 33, 49–60. [Google Scholar] [CrossRef]
- Moradi, P.; Rostami, M. Integration of graph clustering with ant colony optimization for feature selection. Knowl.-Based Syst. 2015, 84, 144–161. [Google Scholar] [CrossRef]
- Zhang, L.; Shan, L.; Wang, J. Optimal feature selection using distance-based discrete firefly algorithm with mutual information criterion. Neural Comput. Appl. 2017, 28, 2795–2808. [Google Scholar] [CrossRef]
- Rodrigues, D.; Yang, X.-S.; de Souza, A.N.; Papa, J.P. Binary flower pollination algorithm and its application to feature selection. In Recent Advances in Swarm Intelligence and Evolutionary Computation; Yang, X.-S., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 85–100. [Google Scholar]
- Lei, X.; Fang, M.; Wu, F.-X.; Chen, L. Improved flower pollination algorithm for identifying essential proteins. BMC Syst. Biol. 2018, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Landwehr, N.; Hall, M.; Frank, E. Logistic model trees. Mach. Learn. 2005, 59, 161–205. [Google Scholar] [CrossRef]
- Quinlan, J.R. C4.5: Programs for Machine Learning; Morgan Kaufmann Publishers Inc.: Burlington, MA, USA, 1993. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Wei, L.; Xing, P.; Shi, G.; Ji, Z.-L.; Zou, Q. Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- John, G.H.; Langley, P. Estimating continuous distributions in bayesian classifiers. In Proceedings of the Eleventh Conference on Uncertainty in Artificial Intelligence, Montréal, QC, Canada, 18–20 August 1995; pp. 338–345. [Google Scholar]
- Platt, J.C. Fast training of support vector machines using sequential minimal optimization. In Advances in Kernel Methods; Bernhard, S., Christopher, J.C.B., Alexander, J.S., Eds.; MIT Press: Cambridge, MA, USA, 1999; pp. 185–208. [Google Scholar]
- Yang, X.-S. Flower pollination algorithm for global optimization. In Proceedings of the Unconventional Computation and Natural Computation: 11th International Conference, Orléans, France, 3–7 September 2012; Durand-Lose, J., Jonoska, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 240–249. [Google Scholar]
- Hua, H.L.; Zhang, F.Z.; Alemayehu, L.A.; Dong, C.; Jin, Y.T.; Guo, F.B. An approach for predicting essential genes using multiple homology mapping and machine learning algorithms. Biomed. Res. Int. 2016, 2016, 7639397. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.; Chen, X.; Wang, H.; Pan, Y. A local average connectivity-based method for identifying essential proteins from the network level. Comput. Biol. Chem. 2011, 35, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, M.; Wang, H.; Pan, Y. Identification of essential proteins based on edge clustering coefficient. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Kittler, J.; Hatef, M.; Duin, R.P.W.; Matas, J. On combining classifiers. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 226–239. [Google Scholar] [CrossRef]
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. Dip: The database of interacting proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M.; Adler, C.; Ball, C.; Chervitz, S.A.; Dwight, S.S.; Hester, E.T.; Jia, Y.; Juvik, G.; Roe, T.; Schroeder, M.; et al. Sgd: Saccharomyces genome database. Nucleic Acids Res. 1998, 26, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ou, H.-Y.; Zhang, C.-T. Deg: A database of essential genes. Nucleic Acids Res. 2004, 32, D271–D272. [Google Scholar] [CrossRef] [PubMed]
- Mewes, H.W.; Frishman, D.; Mayer, K.F.; Munsterkotter, M.; Noubibou, O.; Pagel, P.; Rattei, T.; Oesterheld, M.; Ruepp, A.; Stumpflen, V. Mips: Analysis and annotation of proteins from whole genomes in 2005. Nucleic Acids Res. 2006, 34, D169–D172. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E. Virtual identification of essential proteins within the protein interaction network of yeast. Proteomics 2006, 6, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhang, N. Prediction of essential proteins based on edge clustering coefficient and gene ontology information. J. Biol. Syst. 2014, 22, 339–351. [Google Scholar] [CrossRef]
- Deng, J.; Deng, L.; Su, S.; Zhang, M.; Lin, X.; Wei, L.; Minai, A.A.; Hassett, D.J.; Lu, L.J. Investigating the predictability of essential genes across distantly related organisms using an integrative approach. Nucleic Acids Res. 2011, 39, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, M.; Wang, J.; Wu, J.; Wu, F.-X.; Pan, Y. Predicting essential proteins based on subcellular localization, orthology and PPI networks. BMC Bioinform. 2016, 17, 279. [Google Scholar] [CrossRef] [PubMed]
- Binder, J.X.; Pletscher-Frankild, S.; Tsafou, K.; Stolte, C.; O’Donoghue, S.I.; Schneider, R.; Jensen, L.J. Compartments: Unification and visualization of protein subcellular localization evidence. Database 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.T.; Lee, I.; Marcotte, E.R. A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinform. 2007, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Pu, S.; Wong, J.; Turner, B.; Cho, E.; Wodak, S.J. Up-to-date catalogues of yeast protein complexes. Nucleic Acids Res. 2009, 37, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, H.; Wang, J.-X.; Pan, Y. A new essential protein discovery method based on the integration of protein-protein interaction and gene expression data. BMC Syst. Biol. 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, J.; Zhong, J.; Pan, Y. Predicting essential proteins based on weighted degree centrality. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Wang, J.; Cheng, Y.; Lu, Y.; Wu, F.; Pan, Y. Udonc: An algorithm for identifying essential proteins based on protein domains and protein-protein interaction networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zheng, R.; Zhang, H.; Wang, J.; Pan, Y. Effective identification of essential proteins based on priori knowledge, network topology and gene expressions. Methods 2014, 67, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Visa, S.; Ralescu, A. Issues in mining imbalanced data sets—A review paper. In Proceedings of the Sixteen Midwest Artificial Intelligence and Cognitive Science Conference, Dayton, OH, USA, 16–17 April 2005; pp. 67–73. [Google Scholar]
- Wang, J.; Peng, W.; Wu, F.-X. Computational approaches to predicting essential proteins: A survey. Proteom. Clin. Appl. 2013, 7, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.; Hall, M.A.; Witten, I.H. The Weka Workbench, 4th ed.; Online Appendix for “Data Mining: Practical Machine Learning Tools and Techniques”; Morgan Kaufmann: Burlington, MA, USA, 2016. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature Selection Methods | Evaluation Criterion | ||
|---|---|---|---|
| Precision | Recall | F-Measure | |
| SFS | 0.689 | 0.571 | 0.624 |
| SBS | 0.708 | 0.530 | 0.607 |
| GA | 0.733 | 0.657 | 0.693 |
| ACO | 0.713 | 0.696 | 0.704 |
| PSO | 0.740 | 0.697 | 0.718 |
| FPA | 0.718 | 0.620 | 0.665 |
| ESFPA | 0.745 | 0.715 | 0.730 |
| Feature Selection Methods | Evaluation Criterion | ||
|---|---|---|---|
| Precision | Recall | F-Measure | |
| SFS | 0.554 | 0.158 | 0.245 |
| SBS | 0.544 | 0.134 | 0.215 |
| GA | 0.654 | 0.275 | 0.387 |
| ACO | 0.693 | 0.269 | 0.388 |
| PSO | 0.708 | 0.314 | 0.436 |
| FPA | 0.691 | 0.282 | 0.400 |
| ESFPA | 0.716 | 0.311 | 0.434 |
| Feature Subsets | Evaluation Criterion | ||
|---|---|---|---|
| Precision | Recall | F-Measure | |
| Degree | 0.667 | 0.598 | 0.631 |
| EC | 0.660 | 0.401 | 0.499 |
| SC | 0.665 | 0.376 | 0.481 |
| PeC | 0.747 | 0.341 | 0.468 |
| ION | 0.717 | 0.655 | 0.684 |
| Modules | 0.715 | 0.530 | 0.609 |
| UDoNC | 0.759 | 0.459 | 0.572 |
| Nucleus | 0.654 | 0.638 | 0.646 |
| Cytosol | 0.594 | 0.181 | 0.277 |
| Mitochondrion | 0.512 | 0.848 | 0.639 |
| Endoplasmic reticulum | 0.484 | 0.352 | 0.408 |
| Cytoskeleton | 0.736 | 0.081 | 0.147 |
| Golgi apparatus | 0.492 | 0.669 | 0.567 |
| ESFPA-feature subset | 0.745 | 0.715 | 0.730 |
| Feature | Classifier | Pre | Rec | F | Feature | Classifier | Pre | Rec | F |
|---|---|---|---|---|---|---|---|---|---|
| Degree | NB | 0.714 | 0.227 | 0.345 | EC | NB | 0.719 | 0.162 | 0.264 |
| SMO | 0.686 | 0.150 | 0.246 | SMO | 0.736 | 0.127 | 0.216 | ||
| J48 | 0.667 | 0.598 | 0.631 | J48 | 0.653 | 0.509 | 0.572 | ||
| LMT | 0.667 | 0.598 | 0.631 | LMT | 0.634 | 0.546 | 0.587 | ||
| RF | 0.661 | 0.576 | 0.615 | RF | 0.533 | 0.522 | 0.528 | ||
| RT | 0.664 | 0.586 | 0.623 | RT | 0.531 | 0.524 | 0.528 | ||
| REPT | 0.658 | 0.599 | 0.627 | REPT | 0.629 | 0.543 | 0.583 | ||
| NC | 0.667 | 0.598 | 0.631 | NC | 0.660 | 0.401 | 0.499 | ||
| SC | NB | 0.670 | 0.054 | 0.100 | PeC | NB | 0.810 | 0.244 | 0.375 |
| SMO | 0.714 | 0.017 | 0.033 | SMO | 0.846 | 0.175 | 0.290 | ||
| J48 | 0.655 | 0.513 | 0.575 | J48 | 0.759 | 0.295 | 0.425 | ||
| LMT | 0.638 | 0.542 | 0.586 | LMT | 0.682 | 0.402 | 0.506 | ||
| RF | 0.532 | 0.530 | 0.531 | RF | 0.542 | 0.538 | 0.540 | ||
| RT | 0.529 | 0.532 | 0.531 | RT | 0.541 | 0.538 | 0.540 | ||
| REPT | 0.618 | 0.559 | 0.587 | REPT | 0.660 | 0.402 | 0.499 | ||
| NC | 0.665 | 0.376 | 0.481 | NC | 0.747 | 0.341 | 0.468 | ||
| ION | NB | 0.722 | 0.638 | 0.678 | Modules | NB | 0.715 | 0.530 | 0.609 |
| SMO | 0.715 | 0.663 | 0.688 | SMO | 0.715 | 0.530 | 0.609 | ||
| J48 | 0.711 | 0.626 | 0.665 | J48 | 0.715 | 0.530 | 0.609 | ||
| LMT | 0.707 | 0.679 | 0.692 | LMT | 0.715 | 0.530 | 0.609 | ||
| RF | 0.662 | 0.568 | 0.611 | RF | 0.715 | 0.530 | 0.609 | ||
| RT | 0.659 | 0.584 | 0.619 | RT | 0.715 | 0.530 | 0.609 | ||
| REPT | 0.708 | 0.638 | 0.671 | REPT | 0.715 | 0.530 | 0.609 | ||
| NC | 0.717 | 0.655 | 0.684 | NC | 0.715 | 0.530 | 0.609 | ||
| Nucleus | NB | 0.654 | 0.638 | 0.646 | UDoNC | NB | 0.828 | 0.226 | 0.355 |
| SMO | 0.654 | 0.638 | 0.646 | SMO | 0.902 | 0.039 | 0.076 | ||
| J48 | 0.654 | 0.638 | 0.646 | J48 | 0.722 | 0.544 | 0.620 | ||
| LMT | 0.654 | 0.638 | 0.646 | LMT | 0.716 | 0.553 | 0.624 | ||
| RF | 0.654 | 0.638 | 0.646 | RF | 0.693 | 0.693 | 0.577 | ||
| RT | 0.654 | 0.638 | 0.646 | RT | 0.689 | 0.495 | 0.576 | ||
| REPT | 0.654 | 0.638 | 0.646 | REPT | 0.690 | 0.589 | 0.636 | ||
| NC | 0.654 | 0.638 | 0.646 | NC | 0.759 | 0.459 | 0.572 | ||
| Feature subset | NB | 0.807 | 0.380 | 0.516 | |||||
| SMO | 0.740 | 0.710 | 0.725 | ||||||
| J48 | 0.728 | 0.717 | 0.722 | ||||||
| LMT | 0.736 | 0.723 | 0.729 | ||||||
| RF | 0.710 | 0.729 | 0.720 | ||||||
| RT | 0.644 | 0.661 | 0.652 | ||||||
| REPT | 0.705 | 0.689 | 0.697 | ||||||
| NC | 0.745 | 0.715 | 0.730 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, M.; Lei, X.; Cheng, S.; Shi, Y.; Wu, F.-X. Feature Selection via Swarm Intelligence for Determining Protein Essentiality. Molecules 2018, 23, 1569. https://doi.org/10.3390/molecules23071569
Fang M, Lei X, Cheng S, Shi Y, Wu F-X. Feature Selection via Swarm Intelligence for Determining Protein Essentiality. Molecules. 2018; 23(7):1569. https://doi.org/10.3390/molecules23071569
Chicago/Turabian StyleFang, Ming, Xiujuan Lei, Shi Cheng, Yuhui Shi, and Fang-Xiang Wu. 2018. "Feature Selection via Swarm Intelligence for Determining Protein Essentiality" Molecules 23, no. 7: 1569. https://doi.org/10.3390/molecules23071569
APA StyleFang, M., Lei, X., Cheng, S., Shi, Y., & Wu, F.-X. (2018). Feature Selection via Swarm Intelligence for Determining Protein Essentiality. Molecules, 23(7), 1569. https://doi.org/10.3390/molecules23071569