
Structure-Activity Relationship of Cannabis Derived Compounds for the Treatment of Neuronal Activity-Related Diseases




Abstract
1. Introduction
1.1. Cannabinoids Receptors and Endocannabinoids
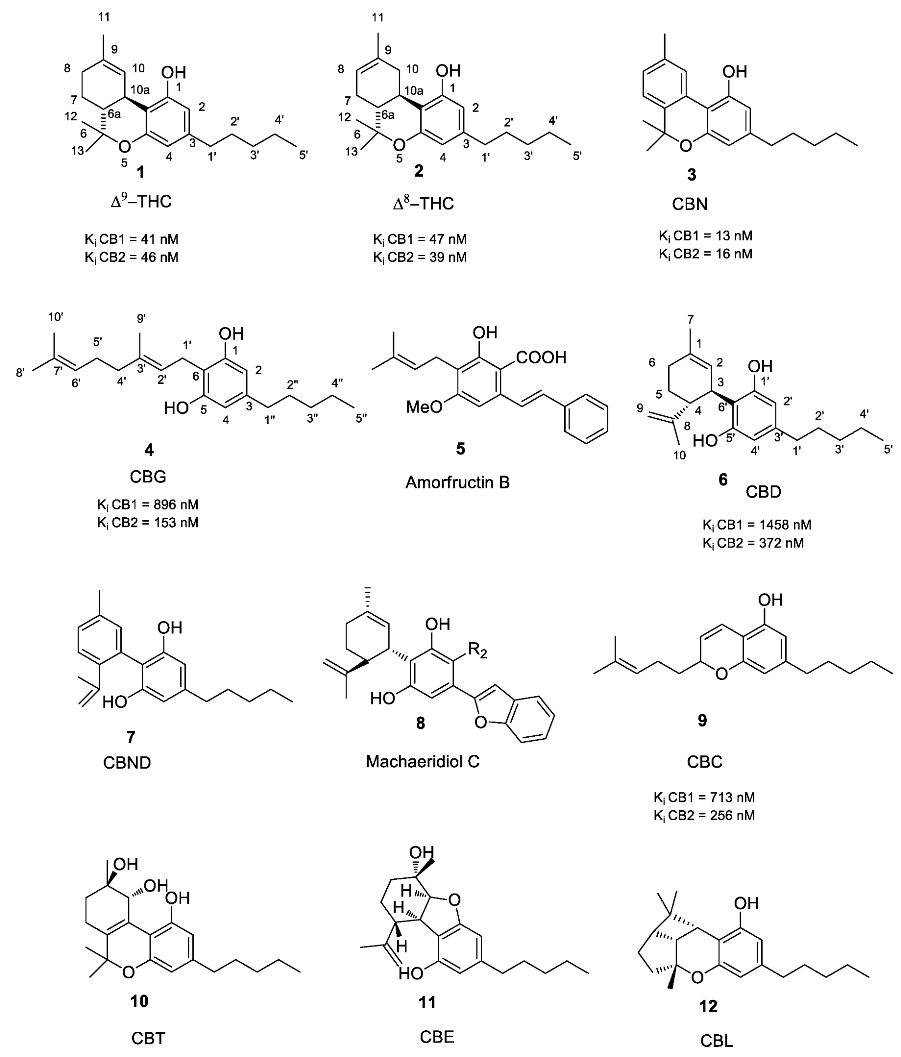
1.2. Phytocannabinoids
1.2.1. Cannabigerol (CBG) Compounds
1.2.2. Cannabichromene (CBC) Compounds
1.2.3. Cannabidiol (CBD) Compounds
1.2.4. Tetrahydrocannabinol (THC) Compounds
1.2.5. Cannabitriol (CBT) and Cannabelsoin (CBE) Compounds
1.2.6. Cannabinoids Degradant: Cannabinol (CBN) and Cannabicyclol (CBL) Compounds
1.2.7. Miscellaneous Cannabinoids
2. Discussion
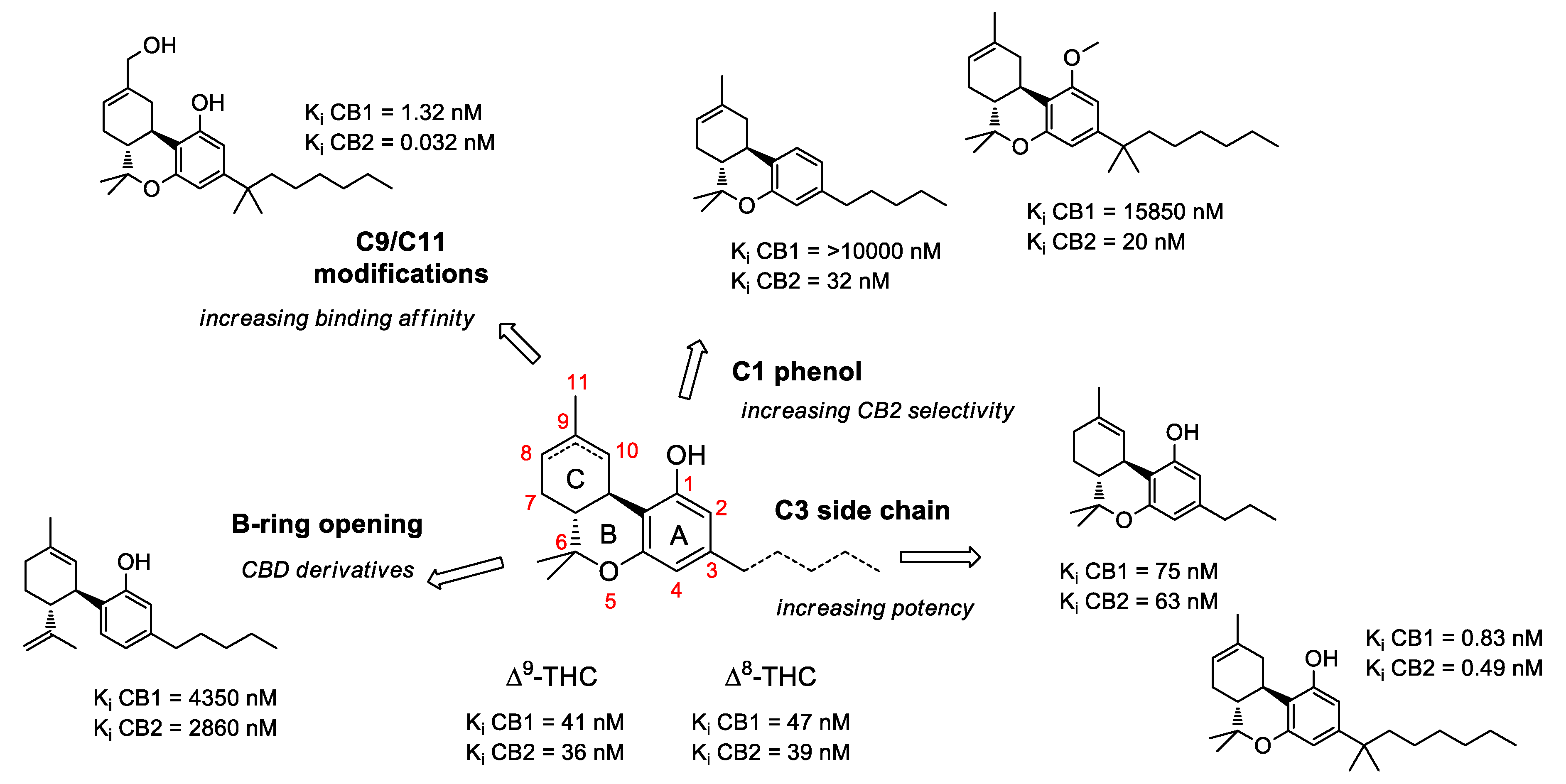
2.1. Structure-Activity Relationship (SAR) of Cannabis-Derived Compounds for the Cannabinoid Receptors
2.2. SAR of Cannabinoids and Their Receptors in Treating Neuronal Diseases
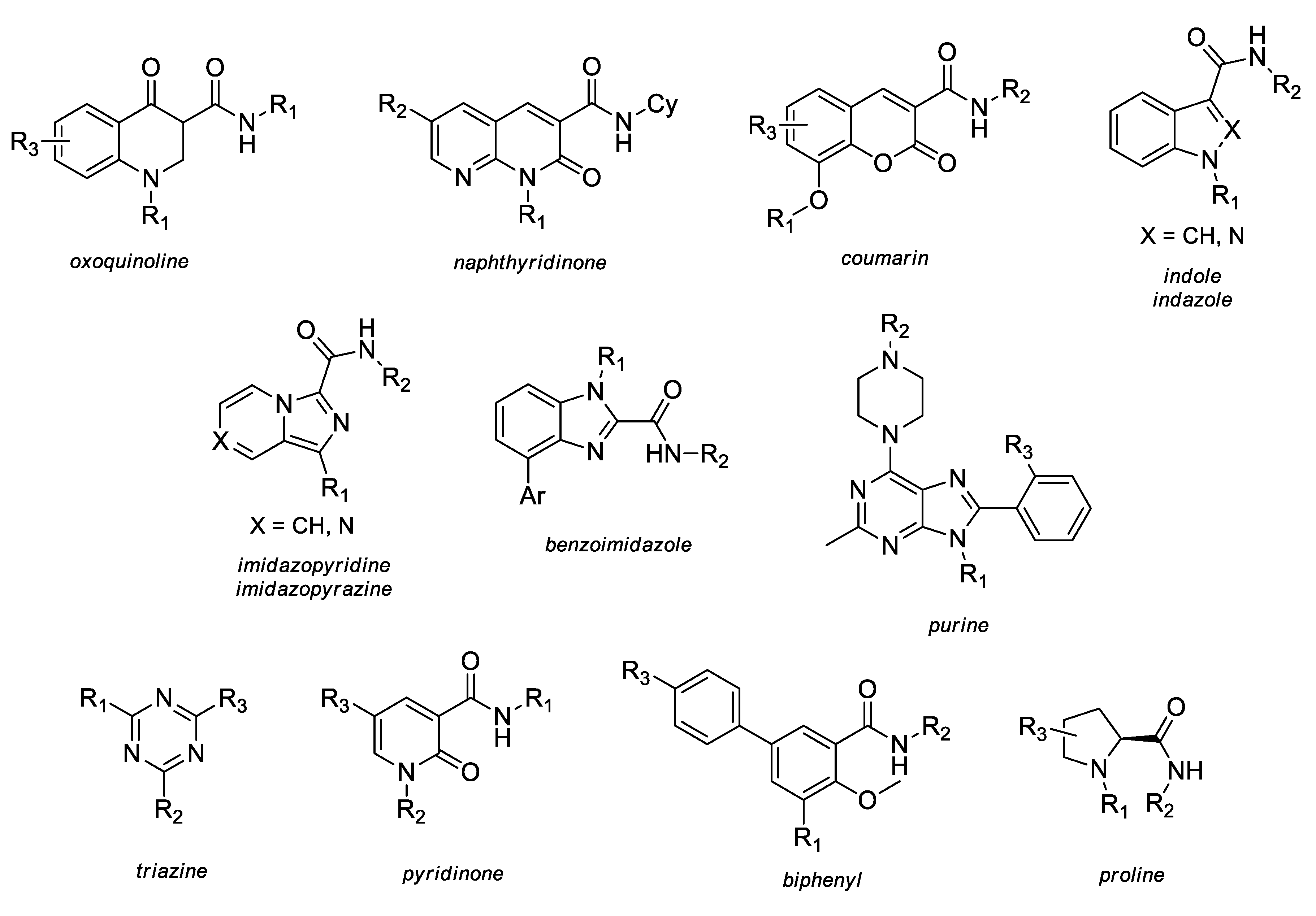
2.3. SAR and Activity Profiles of Several CB2 Selective Derivatives
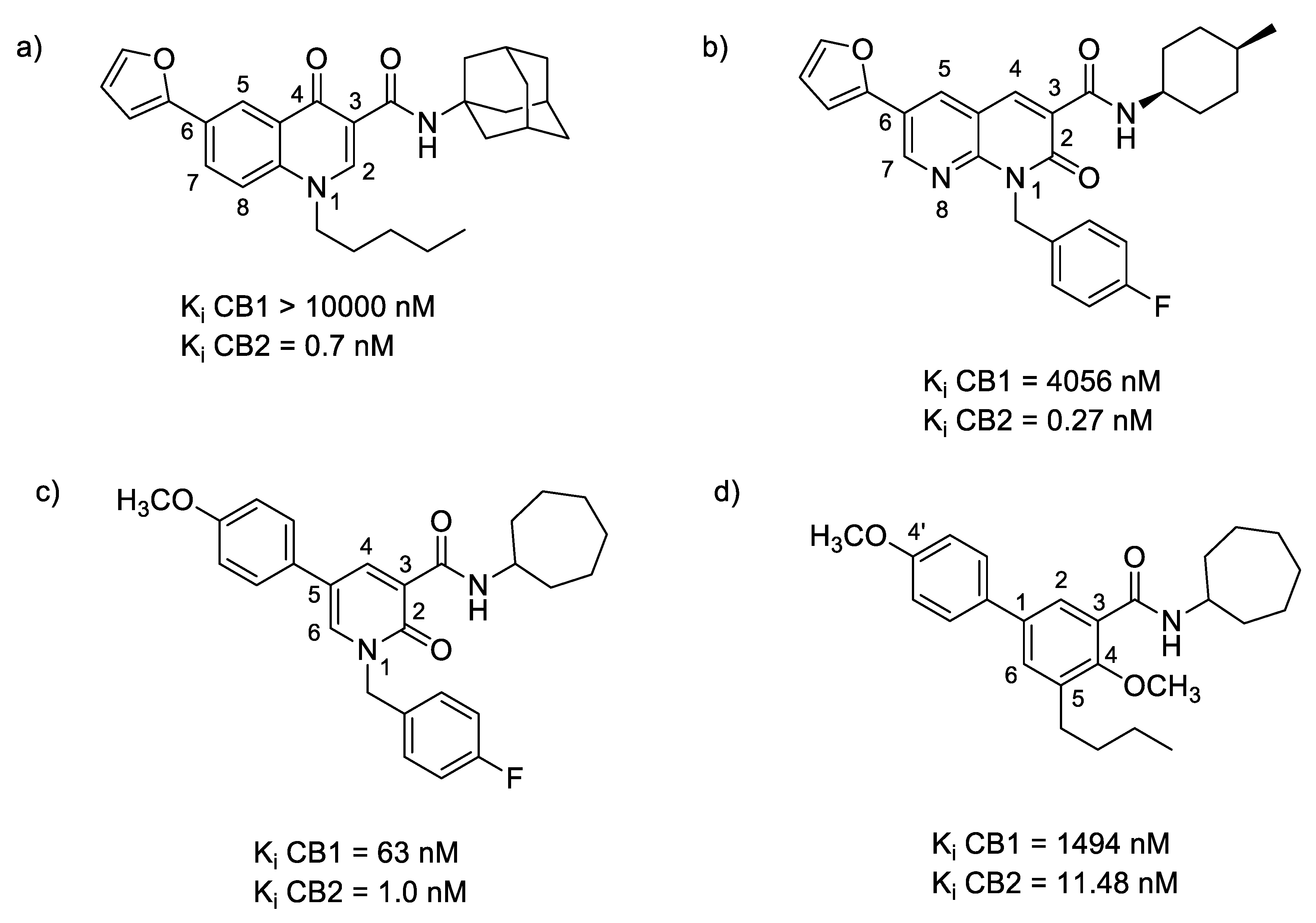
2.3.1. Oxoquinoline
2.3.2. Naphthyridinone
2.3.3. Coumarin
2.3.4. Indole and Indazole
2.3.5. Imidazopyridine and Imidazopyrazine
2.3.6. Benzimidazole
2.3.7. Purine
2.3.8. Triazine
2.3.9. Proline
2.3.10. Pyridinone
2.3.11. Biphenyl
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanûs, L.O.; Meyer, S.M.; Munoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Gaoni, Y. Hashish—IV: The isolation and structure of cannabinolic cannabidiolic and cannabigerolic acids. Tetrahedron 1965, 21, 1223–1229. [Google Scholar] [CrossRef]
- Aizpurua-Olaizola, O.; Soydaner, U.; Öztürk, E.; Schibano, D.; Simsir, Y.; Navarro, P.; Etxebarria, N.; Usobiaga, A. Evolution of the cannabinoid and terpene content during the growth of cannabis sativa plants from different chemotypes. J. Nat. Prod. 2016, 79, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Grotenhermen, F. Review of therapeutic effects. In Cannabis and Cannabinoids. Pharmacology, Toxicology, and Therapeutic Potential; Haworth Press: Binghamton, NY, USA, 2002; pp. 123–142. [Google Scholar]
- Carlini, E. The good and the bad effects of (−) trans-delta-9-tetrahydrocannabinol (δ9-THC) on humans. Toxicon 2004, 44, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Zlas, J.; Stark, H.; Seligman, J.; Levy, R.; Werker, E.; Breuer, A.; Mechoulam, R. Early medical use of cannabis. Nature 1993, 363, 215. [Google Scholar] [CrossRef] [PubMed]
- Merrillees, R.S. Opium trade in the bronze age levant. Antiquity 1962, 36, 287–292. [Google Scholar] [CrossRef]
- Merlin, M.D. Cover article: Archaeological evidence for the tradition of psychoactive plant use in the old world. Econ. Bot. 2003, 57, 295–323. [Google Scholar] [CrossRef]
- Namdar, D.; Amrani, A.; Kletter, R. Chapter 17: Scopolin in iron age juglets from the philistine repository pit of yavneh. In Orbis Biblicus et Orientalis; Kletter, A.R., Ziffer, I., Yavneh, W.Z., II, Eds.; Academic Press: Fribourg, Switzerland, 2015; pp. 214–223. [Google Scholar]
- Gadot, Y.; Finkelstein, I.; Iserlis, M.; Maeir, A.M.; Nahshoni, P.; Namdar, D. Tracking down cult: Production, function and content of chalices in iron age philistia. Tel Aviv 2014, 41, 55–76. [Google Scholar] [CrossRef]
- Pennacchio, M.; Jefferson, L.; Havens, K. Uses and Abuses of Plant-Derived Smoke: Its Ethnobotany as Hallucinogen, Perfume, Incense, and Medicine; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Lynch, M.E.; Campbell, F. Cannabinoids for treatment of chronic non-cancer pain; a systematic review of randomized trials. Br. J. Clin. Pharmacol. 2011, 72, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Wilsey, B.; Marcotte, T.; Deutsch, R.; Gouaux, B.; Sakai, S.; Donaghe, H. Low-dose vaporized cannabis significantly improves neuropathic pain. J. Pain 2013, 14, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Wilsey, B.; Marcotte, T.; Tsodikov, A.; Millman, J.; Bentley, H.; Gouaux, B.; Fishman, S. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J. Pain 2008, 9, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Porter, B.E.; Jacobson, C. Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy Behav. 2013, 29, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Slatkin, N.E. Cannabinoids in the treatment of chemotherapy-induced nausea and vomiting: Beyond prevention of acute emesis. J. Support. Oncol. 2007, 5, 1–9. [Google Scholar] [PubMed]
- Kogan, N.M.; Mechoulam, R. Cannabinoids in health and disease. Dialogues Clin. Neurosci. 2007, 9, 413–430. [Google Scholar] [PubMed]
- Corey-Bloom, J.; Wolfson, T.; Gamst, A.; Jin, S.; Marcotte, T.D.; Bentley, H.; Gouaux, B. Smoked cannabis for spasticity in multiple sclerosis: A randomized, placebo-controlled trial. Can. Med. Assoc. J. 2012, 184, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Mestre, L.; Correa, F.; Docagne, F.; Clemente, D.; Ortega-Gutierrez, S.; Arevalo-Martin, A.; Molina-Holgado, E.; Borrell, J.; Guaza, C. Cannabinoid system and neuroinflammation: Therapeutic perspectives in multiple sclerosis. Rev. Neurol. 2006, 43, 541–548. [Google Scholar] [PubMed]
- Rog, D.J.; Nurmikko, T.J.; Friede, T.; Young, C.A. Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology 2005, 65, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.T.; Makela, P.; Robson, P.; House, H.; Bateman, C. Do cannabis-based medicinal extracts have general or specific effects on symptoms in multiple sclerosis? A double-blind, randomized, placebo-controlled study on 160 patients. Mult. Scler. J. 2004, 10, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Tomida, I.; Pertwee, R.G.; Azuara-Blanco, A. Cannabinoids and glaucoma. Br. J. Ophthalmol. 2004, 88, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Tashkin, D.P.; Shapiro, B.J.; Frank, I.M. Acute effects of smoked marijuana and oral δ9-tetrahydrocannabinol on specific airway conductance in asthmatic subjects. Am. Rev. Respir. Dis. 1974, 109, 420–428. [Google Scholar] [PubMed]
- Ahmed, S.A.; Ross, S.A.; Slade, D.; Radwan, M.M.; Khan, I.A.; ElSohly, M.A. Structure determination and absolute configuration of cannabichromanone derivatives from high potency cannabis sativa. Tetrahedron Lett. 2008, 49, 6050–6053. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cdna. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Abood, M.E. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol. Ther. 2010, 126, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.G.; Behm, D.J.; Walker, D.J.; Ao, Z.; Shapland, E.M.; Daniels, D.A.; Riddick, M.; Dowell, S.; Staton, P.C.; Green, P.; et al. The novel endocannabinoid receptor GPR55 is activated by atypical cannabinoids but does not mediate their vasodilator effects. Br. J. Pharmacol. 2007, 152, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, J.; Lehmann, C. GPR55—A putative “type 3” cannabinoid receptor in inflammation. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Marichal-Cancino, B.A.; Fajardo-Valdez, A.; Ruiz-Contreras, A.E.; Mendez-Díaz, M.; Prospero-García, O. Advances in the physiology of GPR55 in the central nervous system. Curr. Neuropharmacol. 2017, 15, 771–778. [Google Scholar] [CrossRef] [PubMed]
- O’sullivan, S. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Grotenhermen, F. Pharmacology of cannabinoids. Neuro Endocrinol. Lett. 2004, 25, 14–23. [Google Scholar] [PubMed]
- Breivogel, C.S.; Griffin, G.; Di Marzo, V.; Martin, B.R. Evidence for a new G protein-coupled cannabinoid receptor in mouse brain. Mol. Pharmacol. 2001, 60, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Fride, E.; Foox, A.; Rosenberg, E.; Faigenboim, M.; Cohen, V.; Barda, L.; Blau, H.; Mechoulam, R. Milk intake and survival in newborn cannabinoid CB1 receptor knockout mice: Evidence for a “CB3” receptor. Eur. J. Pharmacol. 2003, 461, 27–34. [Google Scholar] [CrossRef]
- Di Marzo, V.; Hill, M.P.; Bisogno, T.; Crossman, A.R.; Brotchie, J.M. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of parkinson’s disease. FASEB J. 2000, 14, 1432–1438. [Google Scholar] [PubMed]
- Wiley, J.L.; Martin, B.R. Cannabinoid pharmacology: Implications for additional cannabinoid receptor subtypes. Chem. Phys. Lipids 2002, 121, 57–63. [Google Scholar] [CrossRef]
- Scotter, E.L.; Abood, M.E.; Glass, M. The endocannabinoid system as a target for the treatment of neurodegenerative disease. Br. J. Pharmacol. 2010, 160, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Moreno-Martet, M.; Rodríguez-Cueto, C.; Palomo-Garo, C.; Gómez-Cañas, M.; Valdeolivas, S.; Guaza, C.; Romero, J.; Guzmán, M.; Mechoulam, R. Prospects for cannabinoid therapies in basal ganglia disorders. Br. J. Pharmacol. 2011, 163, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Bilkei-Gorzo, A. The endocannabinoid system in normal and pathological brain ageing. Philos. Trans. R. Soc. B 2012, 367, 3326–3341. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef] [PubMed]
- Kendall, D.A.; Yudowski, G.A. Cannabinoid receptors in the central nervous system: Their signaling and roles in disease. Front. Cell. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Heifets, B.D.; Castillo, P.E. Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 2009, 71, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Shani, A.; Edery, H.; Grunfeld, Y. Chemical basis of hashish activity. Science 1970, 169, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Endocannabinoids and their pharmacological actions. In Endocannabinoids; Springer: Berlin, Germany, 2015; pp. 1–37. [Google Scholar]
- Pertwee, R.G.; Howlett, A.; Abood, M.E.; Alexander, S.; Di Marzo, V.; Elphick, M.; Greasley, P.; Hansen, H.S.; Kunos, G.; Mackie, K. International union of basic and clinical pharmacology. Lxxix. Cannabinoid receptors and their ligands: Beyond cb1 and cb2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.; Costa, M.; Almada, M.; Correia-da-Silva, G.; Teixeira, N. Endogenous cannabinoids revisited: A biochemistry perspective. Prostaglandins Other Lipid Mediat. 2013, 102, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.L.; Khanolkar, A.D.; Makriyannis, A. Natural and synthetic endocannabinoids and their structure-activity relationships. Curr. Pharm. Des. 2000, 6, 1381–1397. [Google Scholar] [CrossRef] [PubMed]
- Thakur, G.A.; Duclos, R.I., Jr.; Makriyannis, A. Natural cannabinoids: Templates for drug discovery. Life Sci. 2005, 78, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Gomez-Canas, M.; Burgaz, S.; Palomares, B.; Gomez-Galvez, Y.; Palomo-Garo, C.; Campo, S.; Ferrer-Hernandez, J.; Pavicic, C.; Navarrete, C.; et al. Benefits of VCE-003.2, a cannabigerol quinone derivative, against inflammation-driven neuronal deterioration in experimental parkinson’s disease: Possible involvement of different binding sites at the ppar gamma receptor. J. Neuroinflamm. 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, L.; Rousseau, M.; Plauth, A.; Schroeder, F.C.; Sauer, S. Amorfrutins are natural pparγ agonists with potent anti-inflammatory properties. J. Nat. Prod. 2015, 78, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Sauer, S. Amorfrutins: A promising class of natural products that are beneficial to health. ChemBioChem 2014, 15, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Weidner, C.; de Groot, J.C.; Prasad, A.; Freiwald, A.; Quedenau, C.; Kliem, M.; Witzke, A.; Kodelja, V.; Han, C.-T.; Giegold, S. Amorfrutins are potent antidiabetic dietary natural products. Proc. Natl. Acad. Sci. USA 2012, 109, 7257–7262. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Onofri, C.; de Meijer, E.P.M.; Mandolino, G. Sequence heterogeneity of cannabidiolic- and tetrahydrocannabinolic acid-synthase in cannabis sativa l. And its relationship with chemical phenotype. Phytochemistry 2015, 116, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, I.; Li, X.C.; Jacob, M.R.; Tekwani, B.L.; Dunbar, D.C.; Ferreira, D. Antimicrobial and antiparasitic (+)-trans-hexahydrodibenzopyrans and analogues from machaerium multiflorum. J. Nat. Prod. 2003, 66, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Nadal, X.; Del Rio, C.; Casano, S.; Palomares, B.; Ferreiro-Vera, C.; Navarrete, C.; Sanchez-Carnerero, C.; Cantarero, I.; Bellido, M.L.; Meyer, S.; et al. Tetrahydrocannabinolic acid is a potent ppargamma agonist with neuroprotective activity. Br. J. Pharmacol. 2017, 174, 4263–4276. [Google Scholar] [CrossRef] [PubMed]
- Stern, E.; Lambert, D.M. Medicinal chemistry endeavors around the phytocannabinoids. Chem. Biodivers. 2007, 4, 1707–1728. [Google Scholar] [CrossRef] [PubMed]
- Obata, Y.; Ishikawa, Y. Studies on the constituents of hemp plant (Cannabis sativa L.). Agric. Biol. Chem. 1966, 30, 619–620. [Google Scholar] [CrossRef]
- Chan, W.; Magnus, K.; Watson, H. The structure of cannabitriol. Cell. Mol. Life Sci. 1976, 32, 283–284. [Google Scholar] [CrossRef]
- Kinghorn, A.D.; Falk, H.; Gibbons, S.; Kobayashi, J.I. Phytocannabinoids; Springer: Berlin, Germany, 2017. [Google Scholar]
- Brenneisen, R. Chemistry and analysis of phytocannabinoids and other cannabis constituents. In Marijuana and the Cannabinoids; Springer: Berlin, Germany, 2007; pp. 17–49. [Google Scholar]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal structure of the human cannabinoid receptor CB1. Cell 2016, 167, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Nadine, J.; Cristina, F.-F.; Pilar, G. CB1 cannabinoid antagonists: Structure-activity relationships and potential therapeutic applications. Curr. Top. Med. Chem. 2008, 8, 205–230. [Google Scholar]
- Russo, E.B. Clinical endocannabinoid deficiency (CECD): Can this concept explain therapeutic benefits of cannabis in migraine, fibromyalgia, irritable bowel syndrome and other treatment-resistant conditions? Neuro Endocrinol. Lett. 2008, 29, 192–200. [Google Scholar] [PubMed]
- Razdan, R.K. Structure-activity relationships in cannabinoids. Pharmacol. Rev. 1986, 38, 75. [Google Scholar] [PubMed]
- Gaoni, Y.; Mechoulam, R. Isolation, structure, and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Khanolkar, A.D.; Palmer, S.L.; Makriyannis, A. Molecular probes for the cannabinoid receptors. Chem. Phys. Lipids 2000, 108, 37–52. [Google Scholar] [CrossRef]
- Martin, B.R.; Jefferson, R.; Winckler, R.; Wiley, J.L.; Huffman, J.W.; Crocker, P.J.; Saha, B.; Razdan, R.K. Manipulation of the tetrahydrocannabinol side chain delineates agonists, partial agonists, and antagonists. J. Pharmacol. Exp. Ther. 1999, 290, 1065–1079. [Google Scholar] [PubMed]
- Andersson, D.A.; Gentry, C.; Alenmyr, L.; Killander, D.; Lewis, S.E.; Andersson, A.; Bucher, B.; Galzi, J.-L.; Sterner, O.; Bevan, S. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid δ 9-tetrahydrocannabiorcol. Nat. Commun. 2011, 2, 551. [Google Scholar] [CrossRef] [PubMed]
- Bow, E.W.; Rimoldi, J.M. The structure–function relationships of classical cannabinoids: CB1/CB2 modulation. Perspect. Med. Chem. 2016, 8, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Ross, R.A.; Saha, B.; Mahadevan, A.; Razdan, R.K.; Pertwee, R.G. 6″-azidohex-2″-yne-cannabidiol: A potential neutral, competitive cannabinoid cb1 receptor antagonist. Eur. J. Pharmacol. 2004, 487, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Rahn, E.J.; Hohmann, A.G. Cannabinoids as pharmacotherapies for neuropathic pain: From the bench to the bedside. Neurotherapeutics 2009, 6, 713–737. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and δ9-tetrahydrocannabivarin. Br. J. Pharmacol. 2009, 153, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol. Ther. 2012, 133, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Fezza, F.; Galati, S.; Battista, N.; Napolitano, S.; Finazzi-Agrò, A.; Bernardi, G.; Brusa, L.; Pierantozzi, M.; Stanzione, P. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated parkinson’s disease patients. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2005, 57, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Kreitzer, A.C.; Malenka, R.C. Endocannabinoid-mediated rescue of striatal ltd and motor deficits in parkinson’s disease models. Nature 2007, 445, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Cebeira, M.; Ceballos, M.D.; Zeng, B.Y.; Jenner, P.; Ramos, J.; Fernandez-Ruiz, J. Increased cannabinoid cb1 receptor binding and activation of gtp-binding proteins in the basal ganglia of patients with parkinson’s syndrome and of mptp-treated marmosets. Eur. J. Neurosci. 2001, 14, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Brotchie, J.M. CB1 cannabinoid receptor signalling in parkinson’s disease. Curr. Opin. Pharm. 2003, 3, 54–61. [Google Scholar] [CrossRef]
- Sieradzan, K.; Fox, S.; Hill, M.; Dick, J.; Crossman, A.; Brotchie, J. Cannabinoids reduce levodopa-induced dyskinesia in parkinson’s disease: A pilot study. Neurology 2001, 57, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gálvez, Y.; Palomo-Garo, C.; Fernández-Ruiz, J.; García, C. Potential of the cannabinoid CB2 receptor as a pharmacological target against inflammation in parkinson’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 64, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.; Bain, P.; Teare, L.; Liu, X.; Joint, C.; Wroath, C.; Parkin, S.; Fox, P.; Wright, D.; Hobart, J. Cannabis for dyskinesia in parkinson disease a randomized double-blind crossover study. Neurology 2004, 63, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. Taming THC: Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. [Google Scholar] [CrossRef] [PubMed]
- Marzo, V.D.; Petrocellis, L.D. Plant, synthetic, and endogenous cannabinoids in medicine. Annu. Rev. Med. 2006, 57, 553–574. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh Tabrizi, M.; Baraldi, P.G.; Borea, P.A.; Varani, K. Medicinal chemistry, pharmacology, and potential therapeutic benefits of cannabinoid CB2 receptor agonists. Chem. Rev. 2016, 116, 519–560. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, F.; Capparelli, E.; Abate, C.; Colabufo, N.A.; Contino, M. Perspectives of cannabinoid type 2 receptor (CB2R) ligands in neurodegenerative disorders: Structure–affinity relationship (SAfiR) and structure-activity relationship (SAR) studies. J. Med. Chem. 2017, 60, 9913–9931. [Google Scholar] [CrossRef] [PubMed]
- Mugnaini, C.; Nocerino, S.; Pedani, V.; Pasquini, S.; Tafi, A.; De Chiaro, M.; Bellucci, L.; Valoti, M.; Guida, F.; Luongo, L.; et al. Investigations on the 4-quinolone-3-carboxylic acid motif part 5: Modulation of the physicochemical profile of a set of potent and selective cannabinoid-2 receptor ligands through a bioisosteric approach. Chemmedchem 2012, 7, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, S.; Botta, L.; Scincraro, T.; Mugnaini, C.; Ligresti, A.; Palazzo, E.; Maione, S.; Di Marzo, V.; Corelli, F. Investigations on the 4-quinolone-3-carboxylic acid motif. 2. Synthesis and structure-activity relationship of potent and selective cannabinoid-2 receptor agonists endowed with analgesic activity in vivo. J. Med. Chem. 2008, 51, 5075–5084. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, S.; De Rosa, M.; Ligresti, A.; Mugnaini, C.; Brizzi, A.; Caradonna, N.P.; Cascio, M.G.; Bolognini, D.; Pertwee, R.G.; Di Marzo, V.; et al. Investigations on the 4-quinolone-3-carboxylic acid motif. 6. Synthesis and pharmacological evaluation of 7-substituted quinolone-3-carboxamide derivatives as high affinity ligands for cannabinoid receptors. Eur. J. Med. Chem. 2012, 58, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, S.; De Rosa, M.; Pedani, V.; Mugnaini, C.; Guida, F.; Luongo, L.; De Chiaro, M.; Maione, S.; Dragoni, S.; Frosini, M.; et al. Investigations on the 4-quinolone-3-carboxylic acid motif. 4. Identification of new potent and selective ligands for the cannabinoid type 2 receptor with diverse substitution patterns and antihyperalgesic effects in mice. J. Med. Chem. 2011, 54, 5444–5453. [Google Scholar] [CrossRef] [PubMed]
- Manera, C.; Saccomanni, G.; Malfitano, A.M.; Bertini, S.; Castelli, F.; Laezza, C.; Ligresti, A.; Lucchesi, V.; Tuccinardi, T.; Rizzolio, F.; et al. Rational design, synthesis and anti-proliferative properties of new CB2 selective cannabinoid receptor ligands: An investigation of the 1,8-naphthyridin-2(1H)-one scaffold. Eur. J. Med. Chem. 2012, 52, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, V.; Hurst, D.P.; Shore, D.M.; Bertini, S.; Ehrmann, B.M.; Allarà, M.; Lawrence, L.; Ligresti, A.; Minutolo, F.; Saccomanni, G.; et al. CB2-selective cannabinoid receptor ligands: Synthesis, pharmacological evaluation, and molecular modeling investigation of 1,8-naphthyridin-2(1H)-one-3-carboxamides. J. Med. Chem. 2014, 57, 8777–8791. [Google Scholar] [CrossRef] [PubMed]
- Manera, C.; Malfitano, A.M.; Parkkari, T.; Lucchesi, V.; Carpi, S.; Fogli, S.; Bertini, S.; Laezza, C.; Ligresti, A.; Saccomanni, G.; et al. New quinolone- and 1,8-naphthyridine-3-carboxamides as selective CB2 receptor agonists with anticancer and immuno–modulatory activity. Eur. J. Med. Chem. 2015, 97, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhang, F.-F.; Qian, H.-Y.; Chen, L.-L.; Pu, J.-B.; Xie, X.; Chen, J.-Z. Design, syntheses, structure-activity relationships and docking studies of coumarin derivatives as novel selective ligands for the CB2 receptor. Eur. J. Med. Chem. 2015, 93, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.; Banister, S.D.; Mack, J.B.C.; Glass, M.; Connor, M.; Kassiou, M. The 2-alkyl-2H-indazole regioisomers of synthetic cannabinoids AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA are possible manufacturing impurities with cannabimimetic activities. Forensic Toxicol. 2016, 34, 286–303. [Google Scholar] [CrossRef] [PubMed]
- Trotter, B.W.; Nanda, K.K.; Burgey, C.S.; Potteiger, C.M.; Deng, J.Z.; Green, A.I.; Hartnett, J.C.; Kett, N.R.; Wu, Z.; Henze, D.A.; et al. Imidazopyridine CB2 agonists: Optimization of CB2/CB1 selectivity and implications for in vivo analgesic efficacy. Bioorg. Med. Chem. Lett. 2011, 21, 2354–2358. [Google Scholar] [CrossRef] [PubMed]
- Nanda, K.K.; Henze, D.A.; Della Penna, K.; Desai, R.; Leitl, M.; Lemaire, W.; White, R.B.; Yeh, S.; Brouillette, J.N.; Hartman, G.D.; et al. Benzimidazole CB2 agonists: Design, synthesis and sar. Bioorg. Med. Chem. Lett. 2014, 24, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Hollinshead, S.P.; Tidwell, M.W.; Palmer, J.; Guidetti, R.; Sanderson, A.; Johnson, M.P.; Chambers, M.G.; Oskins, J.; Stratford, R.; Astles, P.C. Selective cannabinoid receptor type 2 (CB2) agonists: Optimization of a series of purines leading to the identification of a clinical candidate for the treatment of osteoarthritic pain. J. Med. Chem. 2013, 56, 5722–5733. [Google Scholar] [CrossRef] [PubMed]
- Yrjölä, S.; Sarparanta, M.; Airaksinen, A.J.; Hytti, M.; Kauppinen, A.; Pasonen-Seppänen, S.; Adinolfi, B.; Nieri, P.; Manera, C.; Keinänen, O.; et al. Synthesis, in vitro and in vivo evaluation of 1,3,5-triazines as cannabinoid CB2 receptor agonists. Eur. J. Pharm. Sci. 2015, 67, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Riether, D.; Zindell, R.; Wu, L.; Betageri, R.; Jenkins, J.E.; Khor, S.; Berry, A.K.; Hickey, E.R.; Ermann, M.; Albrecht, C.; et al. Selective CB2 receptor agonists. Part 2: Structure–activity relationship studies and optimization of proline-based compounds. Bioorg. Med. Chem. Lett. 2015, 25, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, V.; Parkkari, T.; Savinainen, J.R.; Malfitano, A.M.; Allarà, M.; Bertini, S.; Castelli, F.; Del Carlo, S.; Laezza, C.; Ligresti, A.; et al. 1,2-dihydro-2-oxopyridine-3-carboxamides: The C-5 substituent is responsible for functionality switch at CB2 cannabinoid receptor. Eur. J. Med. Chem. 2014, 74, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Bertini, S.; Chicca, A.; Arena, C.; Chicca, S.; Saccomanni, G.; Gertsch, J.; Manera, C.; Macchia, M. Synthesis and pharmacological evaluation of new biphenylic derivatives as CB2 receptor ligands. Eur. J. Med. Chem. 2016, 116, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Bertini, S.; Parkkari, T.; Savinainen, J.R.; Arena, C.; Saccomanni, G.; Saguto, S.; Ligresti, A.; Allarà, M.; Bruno, A.; Marinelli, L.; et al. Synthesis, biological activity and molecular modeling of new biphenylic carboxamides as potent and selective CB2 receptor ligands. Eur. J. Med. Chem. 2015, 90, 526–536. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkyl | βAralkyl | |
|---|---|---|
| Cannabigerol (CBG) |  |  |
| Cannabichromene (CBC) |  |  |
| Cannabidiol (CBD) |  |  |
| Tetrahydrocannabinol (THC) |  |  |
| Cannabinol (CBN) |  | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prandi, C.; Blangetti, M.; Namdar, D.; Koltai, H. Structure-Activity Relationship of Cannabis Derived Compounds for the Treatment of Neuronal Activity-Related Diseases. Molecules 2018, 23, 1526. https://doi.org/10.3390/molecules23071526
Prandi C, Blangetti M, Namdar D, Koltai H. Structure-Activity Relationship of Cannabis Derived Compounds for the Treatment of Neuronal Activity-Related Diseases. Molecules. 2018; 23(7):1526. https://doi.org/10.3390/molecules23071526
Chicago/Turabian StylePrandi, Cristina, Marco Blangetti, Dvora Namdar, and Hinanit Koltai. 2018. "Structure-Activity Relationship of Cannabis Derived Compounds for the Treatment of Neuronal Activity-Related Diseases" Molecules 23, no. 7: 1526. https://doi.org/10.3390/molecules23071526
APA StylePrandi, C., Blangetti, M., Namdar, D., & Koltai, H. (2018). Structure-Activity Relationship of Cannabis Derived Compounds for the Treatment of Neuronal Activity-Related Diseases. Molecules, 23(7), 1526. https://doi.org/10.3390/molecules23071526