
Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity


Abstract
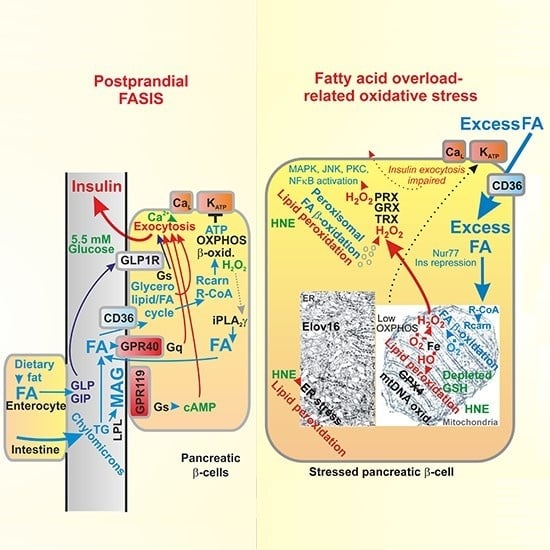
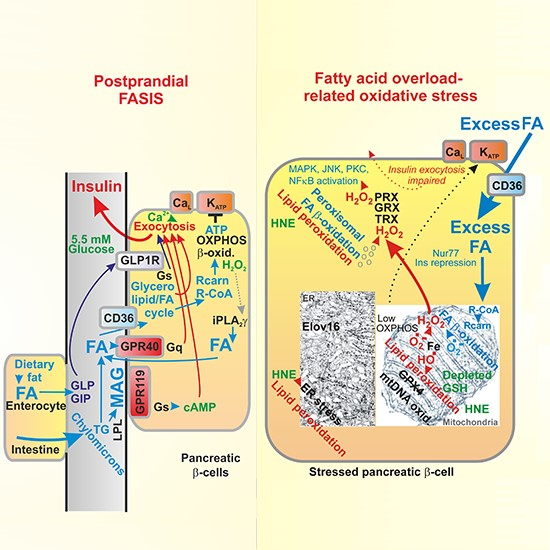{kind=link}
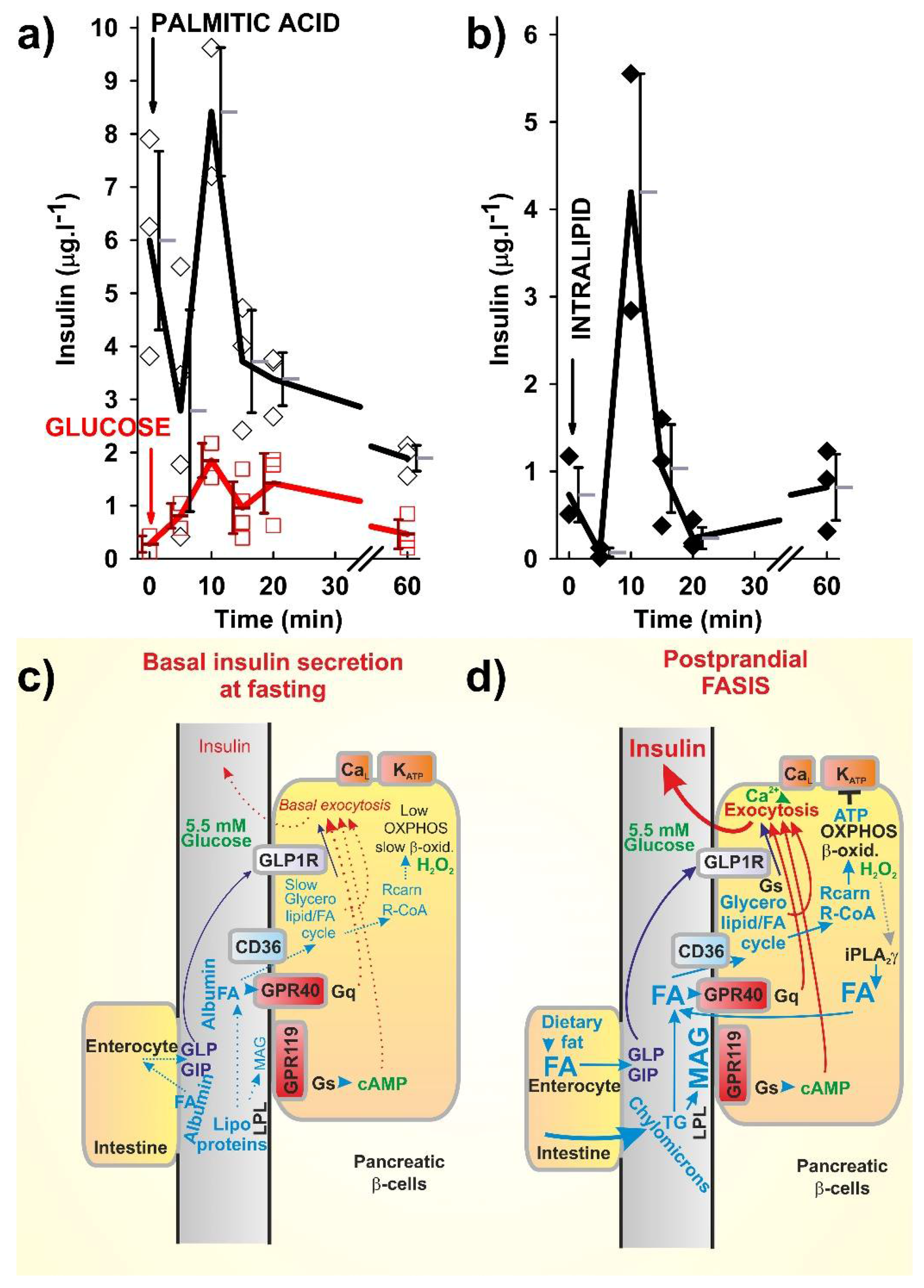{kind=link}
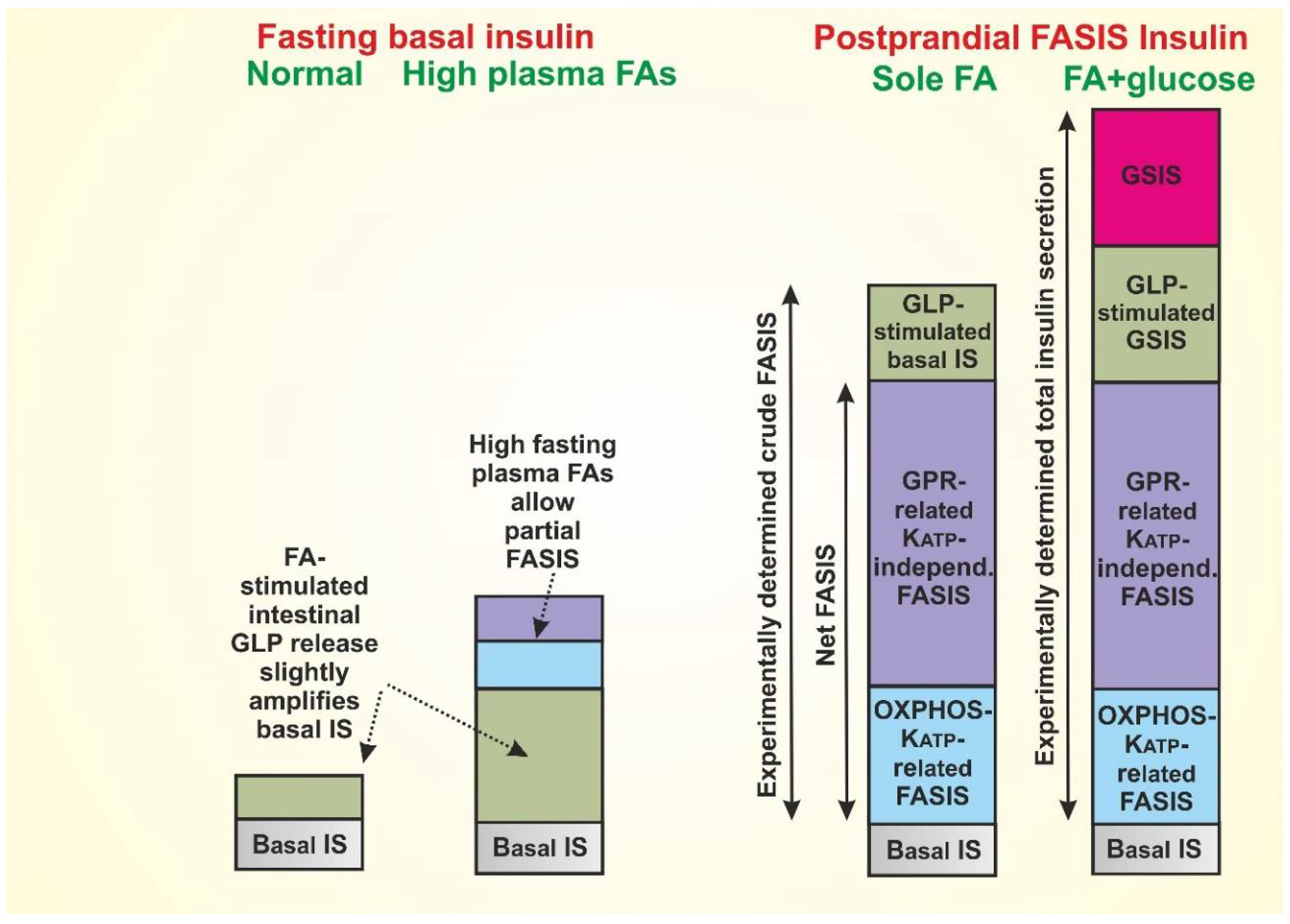{kind=link}
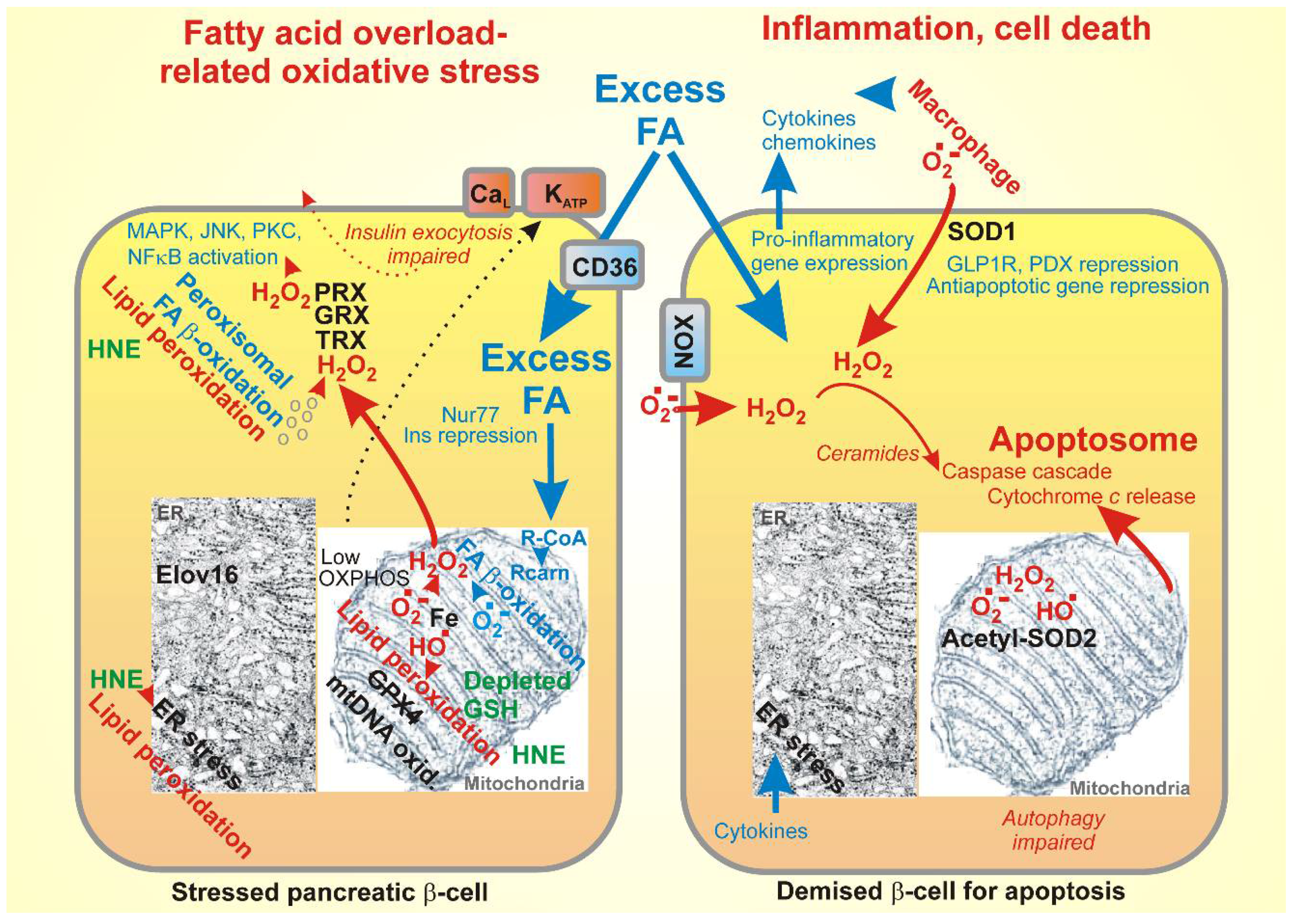{kind=link}
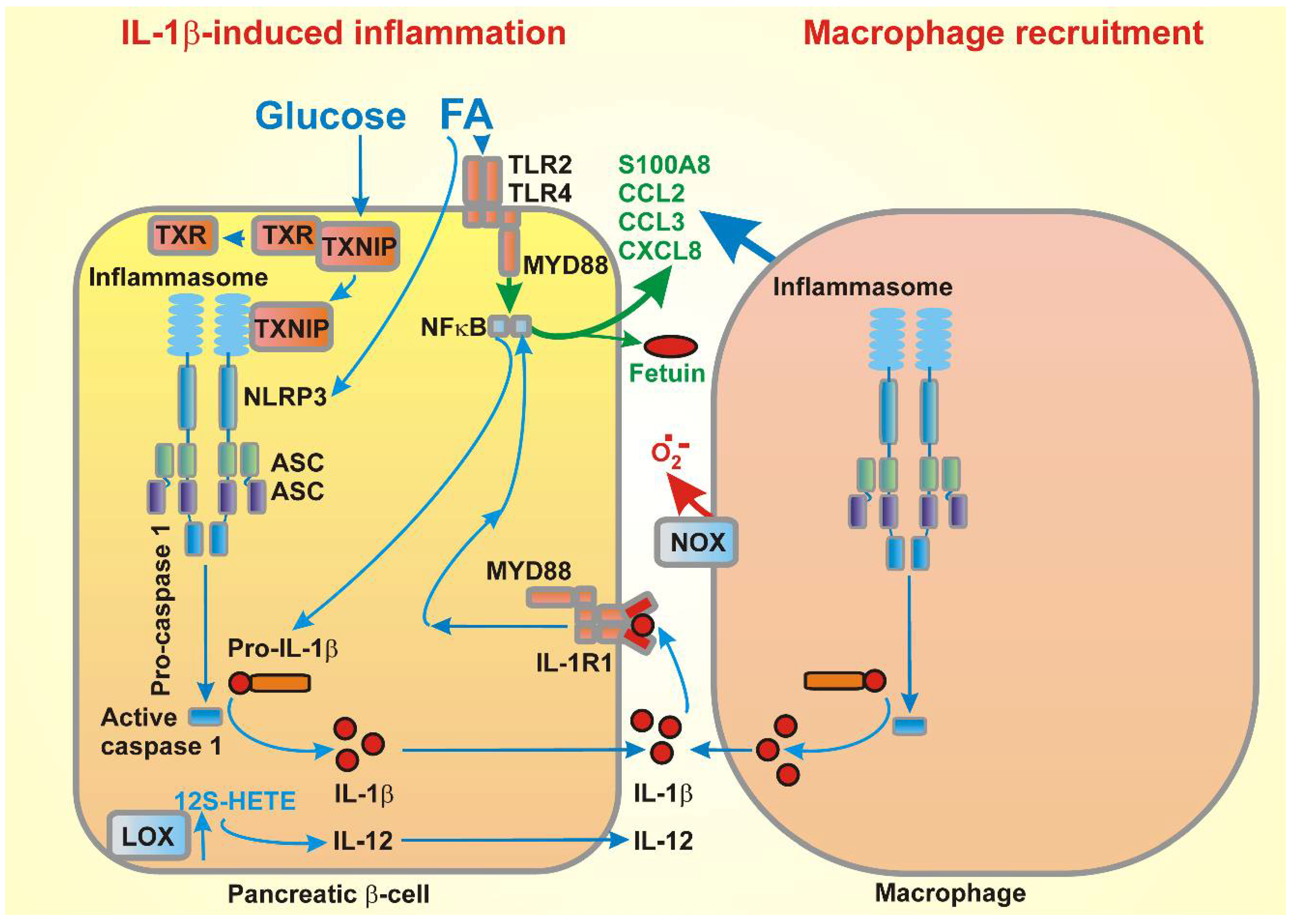{kind=link}
1. Introduction
1.1. Canonical Mechanism of Insulin Secretion
1.1.1. Glucose-Stimulated Insulin Secretion (GSIS)
1.1.2. Impaired GSIS in Type 2 Diabetes
2. Physiological Involvement of LCFAs in Insulin Secretion
2.1. FA-Stimulated Insulin Secretion (FASIS)
2.1.1. Dietary vs. Cleaved FAs as Relevant Secretagogues for Insulin Secretion
2.1.2. Experimental Determination of FASIS
2.2. Specificity of Distinct Classes of Fatty Acids
2.2.1. Polyunsaturated FAs (PUFAs)
2.2.2. ω-3 Polyunsaturated FAs
2.2.3. Oxidized FAs—Specific Messengers vs. Pathology Markers
3. Pathology Related to LCFAs
3.1. Oxidative Stress Related to LCFA Metabolism
3.1.1. Pro-Oxidant Role of Fatty Acids
3.1.2. Experimental Models of LCFA-Induced Lipotoxicity
3.1.3. LCFA Metabolism in β-Cells May Cause Lipotoxicity
3.1.4. Lipotoxicity Due to a Type of FA Species
3.1.5. Lipotoxicity Due to Lower Antioxidant Enzyme Expression and Function
3.1.6. Chronic LCFA Lipotoxicity
3.2. Chronic Low-Grade Inflammation Related to LCFA Metabolism in β-Cells
3.2.1. Systemic Pro-Inflammatory Roles of Fatty Acids
3.2.2. Fatty Acids and Pancreatic Islet Inflammation
3.2.3. Consequences of Exaggerated Pro-Inflammatory Milieu
3.3. Native Antilipotoxic Factors
3.3.1. Incretins
3.3.2. Irisin
3.3.3. Neutral Ceramidase
3.3.4. Other Native Antilipotoxic Factors
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R.M. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef] [PubMed]
- Ježek, J.; Dlasková, A.; Zelenka, J.; Jabůrek, M.; Ježek, P. H2O2-Activated Mitochondrial Phospholipase iPLA2γ Prevents Lipotoxic Oxidative Stress in Synergy with UCP2, Amplifies Signaling via G-Protein–Coupled Receptor GPR40, and Regulates Insulin Secretion in Pancreat. Antioxid. Redox Signal. 2015, 23, 958–972. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Montaño, P.; García-González, V. Effects of Dietary Fatty Acids in Pancreatic Beta Cell Metabolism, Implications in Homeostasis. Nutrients 2018, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, B.; Ortega-Gomez, A.; Varela, L.M.; Villar, J.; Abia, R.; Muriana, F.J.G.; Lopez, S. Clustering effects on postprandial insulin secretion and sensitivity in response to meals with different fatty acid compositions. Food Funct. 2014, 5, 1374. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kadowaki, T. Imbalanced Insulin Actions in Obesity and Type 2 Diabetes: Key Mouse Models of Insulin Signaling Pathway. Cell Metab. 2017, 25, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Akash, M.S.H. Mechanisms of inflammatory responses and development of insulin resistance: How are they interlinked? J. Biomed. Sci. 2016, 23, 87. [Google Scholar] [CrossRef] [PubMed]
- Van Greevenbroek, M.M.J.; Schalkwijk, C.G.; Stehouwer, C.D.A. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: causes and consequences. Neth. J. Med. 2013, 71, 174–187. [Google Scholar] [PubMed]
- Boulinguiez, A.; Staels, B.; Duez, H.; Lancel, S. Mitochondria and endoplasmic reticulum: Targets for a better insulin sensitivity in skeletal muscle? Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2017, 1862, 901–916. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.M.; Rorsman, P. Diabetes mellitus and the β cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Pullen, T.J.; Hodson, D.J.; Martinez-Sanchez, A. Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem. J. 2015, 466, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Electrophysiology of islet cells. Adv. Exp. Med. Biol. 2010, 654, 115–163. [Google Scholar] [CrossRef] [PubMed]
- Graaf, C.D.; Donnelly, D.; Wootten, D.; Lau, J.; Sexton, P.M.; Miller, L.J.; Ahn, J.-M.; Liao, J.; Fletcher, M.M.; Yang, D.; et al. Glucagon-Like Peptide-1 and Its Class B G Protein-Coupled Receptors: A Long March to Therapeutic Successes. Pharmacol. Rev. 2016, 68, 954–1013. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, C.H.S.; Widenmaier, S.; Kim, S.J. Glucose-dependent insulinotropic polypeptide signaling in pancreatic β-cells and adipocytes. J. Diabetes Investig. 2012, 3, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. A fresh view of glycolysis and glucokinase regulation: history and current status. J. Biol. Chem. 2014, 289, 12189–12194. [Google Scholar] [CrossRef] [PubMed]
- Kahancová, A.; Sklenář, F.; Ježek, P.; Dlasková, A. Regulation of glucose-stimulated insulin secretion by ATPase Inhibitory Factor 1 (IF1). FEBS Lett. 2018, 592, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Špaček, T.; Šantorová, J.; Zacharovová, K.; Berková, Z.; Hlavatá, L.; Saudek, F.; Ježek, P. Glucose-stimulated insulin secretion of insulinoma INS-1E cells is associated with elevation of both respiration and mitochondrial membrane potential. Int. J. Biochem. Cell Biol. 2008, 40, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.J.; Ade, L.; Ntambi, J.M.; Ansari, I.-U.H.; Stoker, S.W. Characterization of phospholipids in insulin secretory granules and mitochondria in pancreatic beta cells and their changes with glucose stimulation. J. Biol. Chem. 2015, 290, 11075–11092. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef]
- Hoehn, K.L.; Salmon, A.B.; Hohnen-Behrens, C.; Turner, N.; Hoy, A.J.; Maghzal, G.J.; Stocker, R.; Van Remmen, H.; Kraegen, E.W.; Cooney, G.J.; et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 17787–17792. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Araki, E. Impact of Mitochondrial ROS Production in the Pathogenesis of Diabetes Mellitus and Its Complications. Antioxid. Redox Signal. 2007, 9, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Koshkin, V.; Allister, E.M.; Gyulkhandanyan, A.V.; Wheeler, M.B. Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes. Diabetes 2010, 59, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Jonas, J.-C.; Laybutt, D.R. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef] [PubMed]
- Nyrén, R.; Chang, C.L.; Lindström, P.; Barmina, A.; Vorrsjö, E.; Ali, Y.; Juntti-Berggren, L.; Bensadoun, A.; Young, S.G.; Olivecrona, T.; et al. Localization of lipoprotein lipase and GPIHBP1 in mouse pancreas: Effects of diet and leptin deficiency. BMC Physiol. 2012, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Ström, K.; Holm, C.; Ahrén, B. Glucose-stimulated insulin secretion correlates with β-cell lipolysis. Nutr. Metab. Cardiovasc. Dis. 2006, 16, S11–S16. [Google Scholar] [CrossRef] [PubMed]
- Cruz, W.S.; Kwon, G.; Marshall, C.A.; McDaniel, M.L.; Semenkovich, C.F. Glucose and insulin stimulate heparin-releasable lipoprotein lipase activity in mouse islets and INS-1 cells. A potential link between insulin resistance and β-cell dysfunction. J. Biol. Chem. 2001, 276, 12162–12168. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.A.; Tordjman, K.; Host, H.H.; Ensor, N.J.; Kwon, G.; Marshall, C.A.; Coleman, T.; McDaniel, M.L.; Semenkovich, C.F. Relative hypoglycemia and hyperinsulinemia in mice with heterozygous lipoprotein lipase (LPL) deficiency. Islet LPL regulates insulin secretion. J. Biol. Chem. 1999, 274, 27426–27432. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.M.; Abdel-Wahab, Y.H.A.; Flatt, P.R.; McKillop, A.M. Activation of GPR119 by fatty acid agonists augments insulin release from clonal β-cells and isolated pancreatic islets and improves glucose tolerance in mice. Biol. Chem. 2014, 395, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Hauge, M.; Vestmar, M.A.; Husted, A.S.; Ekberg, J.P.; Wright, M.J.; Di Salvo, J.; Weinglass, A.B.; Engelstoft, M.S.; Madsen, A.N.; Lückmann, M.; et al. GPR40 (FFAR1)—Combined Gs and Gq signaling in vitro is associated with robust incretin secretagogue action ex vivo and in vivo. Mol. Metab. 2015, 4, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.D.; Bertrand, G.; Vivot, K.; Carpentier, É.; Tremblay, C.; Ghislain, J.; Bouvier, M.; Poitout, V. Β-Arrestin Recruitment and Biased Agonism At Free Fatty Acid Receptor 1. J. Biol. Chem. 2015, 290, 21131–21140. [Google Scholar] [CrossRef] [PubMed]
- Graciano, M.F.; Valle, M.M.; Curi, R.; Carpinelli, A.R. Evidence for the involvement of GPR40 and NADPH oxidase in palmitic acid-induced superoxide production and insulin secretion. Islets 2013, 5, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Gu, Y.; Wu, C.; Yu, F.; Chen, Y.; Zhu, J.; Yao, X.; Bei, C.; Zhu, Q. Agonist-induced activation of human FFA1 receptor signals to extracellular signal-regulated kinase 1 and 2 through Gq- and Gi-coupled signaling cascades. Cell. Mol. Biol. Lett. 2017, 22, 13. [Google Scholar] [CrossRef] [PubMed]
- Sabrautzki, S.; Kaiser, G.; Przemeck, G.K.H.; Gerst, F.; Lorza-Gil, E.; Panse, M.; Sartorius, T.; Hoene, M.; Marschall, S.; Häring, H.-U.; et al. Point mutation of Ffar1 abrogates fatty acid-dependent insulin secretion, but protects against HFD-induced glucose intolerance. Mol. Metab. 2017, 6, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, H.; Bergsten, P.; Sargsyan, E. Free fatty acid receptor 1 (FFAR1/GPR40) signaling affects insulin secretion by enhancing mitochondrial respiration during palmitate exposure. Biochim. Biophys. Acta. 2015, 1853, 3248–3257. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Maekawa, F.; Yada, T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: Mediation by PLC and L-type Ca2+ channel and link to insulin release. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E670–E677. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Kowluru, A. CD36 mediates lipid accumulation in pancreatic beta cells under the duress of glucolipotoxic conditions: Novel roles of lysine deacetylases. Biochem. Biophys. Res. Commun. 2018, 495, 2221–2226. [Google Scholar] [CrossRef] [PubMed]
- Leguina-Ruzzi, A.; Průchová, P.; Holendová, B.; Ježek, P.; Jabůrek, M. iPLA2γ Ablation Alters Glucose Homeostasis and Insulin Secretion in Response to Fatty Acids. Free Radic. Biol. Med. 2017, 112, 152–153. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Kamp, F. How are free fatty acids transported in membranes?—Is it by proteins or by free diffusion through the lipids? Diabetes 1999, 48, 2255–2269. [Google Scholar] [CrossRef] [PubMed]
- Cen, J.; Sargsyan, E.; Bergsten, P. Fatty acids stimulate insulin secretion from human pancreatic islets at fasting glucose concentrations via mitochondria-dependent and -independent mechanisms. Nutr. Metab. 2016, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Mugabo, Y.; Zhao, S.; Seifried, A.; Gezzar, S.; Al-Mass, A.; Zhang, D.; Lamontagne, J.; Attane, C.; Poursharifi, P.; Iglesias, J.; et al. Identification of a mammalian glycerol-3-phosphate phosphatase: Role in metabolism and signaling in pancreatic β-cells and hepatocytes. Proc. Natl. Acad. Sci. USA 2016, 113, E430–E439. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, M.; Flachs, P.; Brauner, P.; Sponarova, J.; Matejkova, O.; Prazak, T.; Ruzickova, J.; Bardova, K.; Kuda, O.; Kopecky, J. Role of energy charge and amp-activated protein kinase in adipocytes in the control of body fat stores. Int. J. Obes. 2004, 28, S38–S44. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.M.; Abdel-Wahab, Y.H.A.; Flatt, P.R.; Mckillop, A.M. Evaluation of the insulin-releasing and glucose-lowering effects of GPR120 activation in pancreatic β-cells. Diabetes Obes. Metab. 2014, 16, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- El-Azzouny, M.; Evans, C.R.; Treutelaar, M.K.; Kennedy, R.T.; Burant, C.F. Increased glucose metabolism and glycerolipid formation by fatty acids and GPR40 receptor signaling underlies the fatty acid potentiation of insulin secretion. J. Biol. Chem. 2014, 289, 13575–13588. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic β-cells. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 1929–1942. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, W.; Elsner, M.; Lenzen, S. Role of metabolically generated reactive oxygen species for lipotoxicity in pancreatic β-cells. Diabetes Obes. Metab. 2010, 12, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, H.; Sargsyan, E.; Manell, H.; Smith, D.M.; Göpel, S.O.; Bergsten, P. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci. Rep. 2017, 7, 4657. [Google Scholar] [CrossRef] [PubMed]
- Baynes, H.W.; Mideksa, S.; Ambachew, S. The role of polyunsaturated fatty acids (n-3 PUFAs) on the pancreatic β-cells and insulin action. Adipocyte 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Neuman, J.C.; Schaid, M.D.; Brill, A.L.; Fenske, R.J.; Kibbe, C.R.; Fontaine, D.A.; Sdao, S.M.; Brar, H.K.; Connors, K.M.; Wienkes, H.N.; et al. Enriching Islet Phospholipids With Eicosapentaenoic Acid Reduces Prostaglandin E2 Signaling and Enhances Diabetic β-Cell Function. Diabetes 2017, 66, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Badolato, M.; Carullo, G.; Perri, M.; Cione, E.; Manetti, F.; Di Gioia, M.L.; Brizzi, A.; Caroleo, M.C.; Aiello, F. Quercetin/oleic acid-based G-protein-coupled receptor 40 ligands as new insulin secretion modulators. Future Med. Chem. 2017, 9, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Bhaswant, M.; Poudyal, H.; Brown, L. Mechanisms of enhanced insulin secretion and sensitivity with n-3 unsaturated fatty acids. J. Nutr. Biochem. 2015, 26, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Layé, S.; Nadjar, A.; Joffre, C.; Bazinet, R.P. Anti-Inflammatory Effects of Omega-3 Fatty Acids in the Brain: Physiological Mechanisms and Relevance to Pharmacology. Pharmacol. Rev. 2018, 70, 12–38. [Google Scholar] [CrossRef] [PubMed]
- Kuda, O. Bioactive metabolites of docosahexaenoic acid. Biochimie 2017, 136, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Is there a role for bioactive lipids in the pathobiology of diabetes mellitus? Front. Endocrinol. 2017, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Minuz, P.; Jiang, H.; Fava, C.; Turolo, L.; Tacconelli, S.; Ricci, M.; Patrignani, P.; Morganti, A.; Lechi, A.; McGiff, J.C. Altered release of cytochrome p450 metabolites of arachidonic acid in renovascular disease. Hypertension 2008, 51, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Tunaru, S.; Bonnavion, R.; Brandenburger, I.; Preussner, J.; Thomas, D.; Scholich, K.; Offermanns, S. 20-HETE promotes glucose-stimulated insulin secretion in an autocrine manner through FFAR1. Nat. Commun. 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Janikiewicz, J.; Hanzelka, K.; Kozinski, K.; Kolczynska, K.; Dobrzyn, A. Islet β-cell failure in type 2 diabetes—Within the network of toxic lipids. Biochem. Biophys. Res. Commun. 2015, 460, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I.H. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef] [PubMed]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Johns, I.; Goff, L.; Bluck, L.J.; Griffin, B.A.; Jebb, S.A.; Lovegrove, J.A.; Sanders, T.A.B.; Frost, G.; Dornhorst, A. Plasma free fatty acids do not provide the link between obesity and insulin resistance or β-cell dysfunction: Results of the Reading, Imperial, Surrey, Cambridge, Kings (RISCK) study. Diabet. Med. 2014, 31, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Hlavatá, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef] [PubMed]
- Plecitá-Hlavatá, L.; Ježek, P. Integration of superoxide formation and cristae morphology for mitochondrial redox signaling. Int. J. Biochem. Cell Biol. 2016, 80, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Victor, V.M.; Rocha, M.; Herance, R.; Hernandez-Mijares, A. Oxidative stress and mitochondrial dysfunction in type 2 diabetes. Curr. Pharm. Des. 2011, 17, 3947–3958. [Google Scholar] [CrossRef] [PubMed]
- Supale, S.; Li, N.; Brun, T.; Maechler, P. Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol. Metab. 2012, 23, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Vatamaniuk, M.Z.; Roneker, C.A.; Pepper, M.P.; Hu, L.G.; Simmons, R.A.; Lei, X.G. Knockouts of SOD1 and GPX1 exert different impacts on murine islet function and pancreatic integrity. Antioxid. Redox Signal. 2011, 14, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Imai, H.; Koumura, T.; Kobayashi, T.; Nakagawa, Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem. J. 2000, 351, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.P.; Schafer, F.Q.; Goswami, P.C.; Oberley, L.W.; Buettner, G.R. Phospholipid hydroperoxide glutathione peroxidase induces a delay in G1 of the cell cycle. Free Radic. Res. 2003, 37, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Koulajian, K.; Ivovic, A.; Ye, K.; Desai, T.; Shah, A.; Fantus, I.G.; Ran, Q.; Giacca, A. Overexpression of glutathione peroxidase 4 prevents β-cell dysfunction induced by prolonged elevation of lipids in vivo. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E254–E262. [Google Scholar] [CrossRef] [PubMed]
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes 2011, 60, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Maulucci, G.; Daniel, B.; Cohen, O.; Avrahami, Y.; Sasson, S. Hormetic and regulatory effects of lipid peroxidation mediators in pancreatic beta cells. Mol. Asp. Med. 2016, 49, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wohltmann, M.; Tan, M.; Ladenson, J.H.; Turk, J. Group VIA phospholipase A2 mitigates palmitate-induced beta-cell mitochondrial injury and apoptosis. J. Biol. Chem. 2014, 289, 14194–14210. [Google Scholar] [CrossRef] [PubMed]
- Shida, T.; Kamei, N.; Takeda-Morishita, M.; Isowa, K.; Takayama, K. Colonic delivery of docosahexaenoic acid improves impaired glucose tolerance via GLP-1 secretion and suppresses pancreatic islet hyperplasia in diabetic KK-A(y) mice. Int. J. Pharm. 2013, 450, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Tersey, S.A.; Bolanis, E.; Holman, T.R.; Maloney, D.J.; Nadler, J.L.; Mirmira, R.G. Minireview: 12-Lipoxygenase and Islet β-Cell Dysfunction in Diabetes. Mol. Endocrinol. 2015, 29, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.R.; Holman, T.R.; Imai, Y.; Jadhav, A.; Kenyon, V.; Maloney, D.J.; Nadler, J.L.; Rai, G.; Simeonov, A.; Taylor-Fishwick, D.A. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol. Cell. Endocrinol. 2012, 358, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Fishwick, D.A.; Weaver, J.; Glenn, L.; Kuhn, N.; Rai, G.; Jadhav, A.; Simeonov, A.; Dudda, A.; Schmoll, D.; Holman, T.R.; et al. Selective inhibition of 12-lipoxygenase protects islets and beta cells from inflammatory cytokine-mediated beta cell dysfunction. Diabetologia 2015, 58, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Riahi, Y.; Sunda, V.; Deplano, S.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Signaling properties of 4-hydroxyalkenals formed by lipid peroxidation in diabetes. Free Radic. Biol. Med. 2013, 65, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Grankvist, K.; Marklund, S.L.; Täljedal, I.B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J. 1981, 199, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Bachnoff, N.; Trus, M.; Atlas, D. Alleviation of oxidative stress by potent and selective thioredoxin-mimetic peptides. Free Radic. Biol. Med. 2011, 50, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Reinbothe, T.M.; Ivarsson, R.; Li, D.-Q.; Niazi, O.; Jing, X.; Zhang, E.; Stenson, L.; Bryborn, U.; Renström, E. Glutaredoxin-1 Mediates NADPH-Dependent Stimulation of Calcium-Dependent Insulin Secretion. Mol. Endocrinol. 2009, 23, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wang, Q. The protective effect of peroxiredoxin II on oxidative stress induced apoptosis in pancreatic β-cells. Cell Biosci. 2012, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.-S.; Kang, S.W.; Woo, H.A.; Hwang, S.C.; Chae, H.Z.; Kim, K.; Rhee, S.G. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J. Biol. Chem. 2002, 277, 38029–38036. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, G.; Horta, B.B.; Pimenta, D.C.; Augusto, O.; Netto, L.E.S. Reduction of 1-Cys peroxiredoxins by ascorbate changes the thiol-specific antioxidant paradigm, revealing another function of vitamin C. Proc. Natl. Acad. Sci. USA 2007, 104, 4886–4891. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondrial complex III: An essential component of universal oxygen sensing machinery? Respir. Physiol. Neurobiol. 2010, 174, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Edalat, A.; Schulte-Mecklenbeck, P.; Bauer, C.; Undank, S.; Krippeit-Drews, P.; Drews, G.; Düfer, M. Mitochondrial succinate dehydrogenase is involved in stimulus-secretion coupling and endogenous ROS formation in murine beta cells. Diabetologia 2015, 58, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Martino, L.; Masini, M.; Novelli, M.; Beffy, P.; Bugliani, M.; Marselli, L.; Masiello, P.; Marchetti, P.; De Tata, V. Palmitate activates autophagy in INS-1E β-cells and in isolated rat and human pancreatic islets. PLoS ONE 2012, 7, e36188. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.-H.; Kim, J.-W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Biden, T.J.; Boslem, E.; Chu, K.Y.; Sue, N. Lipotoxic endoplasmic reticulum stress, β cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 2014, 25, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Ma, J.; Wang, X.; Yang, W.; Zhang, J.; Ji, Q. Free fatty acid induces endoplasmic reticulum stress and apoptosis of β-cells by Ca2+/calpain-2 pathways. PLoS ONE 2013, 8, e59921. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, P.; Zhang, X.; Liu, H.; Jiang, H.; Zhu, W.; Wang, H. Inhibition of GPR40 protects MIN6 β cells from palmitate-induced ER stress and apoptosis. J. Cell. Biochem. 2012, 113, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Mehmeti, I.; Gurgul-Convey, E.; Lenzen, S.; Lortz, S. Induction of the intrinsic apoptosis pathway in insulin-secreting cells is dependent on oxidative damage of mitochondria but independent of caspase-12 activation. Biochim. Biophys. Acta 2011, 1813, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.; Torii, S.; Saito, N.; Hosaka, M.; Takeuchi, T. Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology 2008, 149, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Chen, H.; Zhang, H.; Wan, X.; Su, Q. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 beta cell death. Endocrine 2012, 42, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Shrivastava, I.; Tyurin, V.A.; Mao, G.; Dar, H.H.; Watkins, S.; Epperly, M.; Bahar, I.; Shvedova, A.A.; Pitt, B.; et al. “Only a Life Lived for Others Is Worth Living”: Redox Signaling by Oxygenated Phospholipids in Cell Fate Decisions. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.R.; Choi, S.E.; Jung, J.G.; Lee, S.A.; Han, S.J.; Kim, H.J.; Kim, D.J.; Lee, K.W.; Kang, Y. Involvement of iron depletion in palmitate-induced lipotoxicity of beta cells. Mol. Cell. Endocrinol. 2015, 407, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Purves, T.; Middlemas, A.; Agthong, S.; Jude, E.B.; Boulton, A.J.; Fernyhough, P.; Tomlinson, D.R. A role for mitogen-activated protein kinases in the etiology of diabetic neuropathy. FASEB J. 2001, 15, 2508–2514. [Google Scholar] [CrossRef] [PubMed]
- Koya, D.; King, G.L. Protein kinase C activation and the development of diabetic complications. Diabetes 1998, 47, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic β-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Harmon, J.S.; Stein, R.; Robertson, R.P. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J. Biol. Chem. 2005, 280, 11107–11113. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Santos da Rocha, M.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Cacicedo, J.M.; Benjachareowong, S.; Chou, E.; Ruderman, N.B.; Ido, Y. Palmitate-induced apoptosis in cultured bovine retinal pericytes: Roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes 2005, 54, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patané, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that β-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Beeharry, N.; Chambers, J.A.; Green, I.C. Fatty acid protection from palmitic acid-induced apoptosis is lost following PI3-kinase inhibition. Apoptosis 2004, 9, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic β-cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef] [PubMed]
- Engin, A.B. What Is Lipotoxicity? Adv. Exp. Med. Biol. 2017, 960, 197–220. [Google Scholar] [CrossRef] [PubMed]
- Giacca, A.; Xiao, C.; Oprescu, A.I.; Carpentier, A.C.; Lewis, G.F. Lipid-induced pancreatic β-cell dysfunction: focus on in vivo studies. AJP Endocrinol. Metab. 2011, 300, E255–E262. [Google Scholar] [CrossRef] [PubMed]
- Graciano, M.F.; Valle, M.M.; Kowluru, A.; Curi, R.; Carpinelli, A.R. Regulation of insulin secretion and reactive oxygen species production by free fatty acids in pancreatic islets. Islets 2011, 3, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A. Structure and lipid binding properties of serum albumin. Methods Enzymol. 1986, 128, 320–339. [Google Scholar] [CrossRef] [PubMed]
- Richieri, G.V.; Kleinfeld, A.M. Unbound free fatty acid levels in human serum. J. Lipid Res. 1995, 36, 229–240. [Google Scholar] [PubMed]
- Richieri, G.V.; Anel, A.; Kleinfeld, A.M. Interactions of Long-Chain Fatty Acids and Albumin: Determination of Free Fatty Acid Levels Using the Fluorescent Probe ADIFAB. Biochemistry 1993, 32, 7574–7580. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Nielsen, S. Insulin dose response analysis of free fatty acid kinetics. Metabolism 2007, 56, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Van der Vusse, G.J. Albumin as Fatty Acid Transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.H.; Kleinfeld, A.M. Unbound free fatty acid profiles in human plasma and the unexpected absence of unbound palmitoleate. J. Lipid Res. 2017, 58, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Alsabeeh, N.; Chausse, B.; Kakimoto, P.A.; Kowaltowski, A.J.; Shirihai, O. Cell culture models of fatty acid overload: Problems and solutions. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2018, 1863, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Plötz, T.; Krümmel, B.; Laporte, A.; Pingitore, A.; Persaud, S.J.; Jörns, A.; Elsner, M.; Mehmeti, I.; Lenzen, S. The monounsaturated fatty acid oleate is the major physiological toxic free fatty acid for human beta cells. Nutr. Diabetes 2017, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, W.; Würdemann, W.; Plötz, T.; Jörns, A.; Lenzen, S.; Elsner, M. Antagonism between Saturated and Unsaturated Fatty Acids in ROS Mediated Lipotoxicity in Rat Insulin-Producing Cells. Cell. Physiol. Biochem. 2015, 36, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.B.; Tan, K.C.B.; Hales, C.N.; Betteridge, D.J. Postprandial lipid metabolism and β-cell function in non-insulin-dependent (Type 2) diabetes mellitus after a mixed meal with a high fat content. Diabet. Med. 1996, 13, 816–827. [Google Scholar] [CrossRef]
- Malin, S.K.; Kashyap, S.R.; Hammel, J.; Miyazaki, Y.; DeFronzo, R.A.; Kirwan, J.P. Adjusting glucose-stimulated insulin secretion for adipose insulin resistance: An index of β-cell function in obese adults. Diabetes Care 2014, 37, 2940–2946. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Giacca, A.; Carpentier, A.; Lewis, G.F. Differential effects of monounsaturated, polyunsaturated and saturated fat ingestion on glucose-stimulated insulin secretion, sensitivity and clearance in overweight and obese, non-diabetic humans. Diabetologia 2006, 49, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. Oxidative stress: The vulnerable β-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Szkudlinska, M.A.; von Frankenberg, A.D.; Utzschneider, K.M. The antioxidant N-Acetylcysteine does not improve glucose tolerance or β-cell function in type 2 diabetes. J. Diabetes Complicat. 2016, 30, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Ansari, I.H.; Longacre, M.J.; Stoker, S.W.; Kendrick, M.A.; O’Neill, L.M.; Zitur, L.J.; Fernandez, L.A.; Ntambi, J.M.; MacDonald, M.J. Characterization of Acyl-CoA synthetase isoforms in pancreatic beta cells: Gene silencing shows participation of ACSL3 and ACSL4 in insulin secretion. Arch. Biochem. Biophys. 2017, 618, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Kim-Muller, J.Y.; Kim, Y.J.R.; Fan, J.; Zhao, S.; Banks, A.S.; Prentki, M.; Accili, D. FoxO1 Deacetylation Decreases Fatty Acid Oxidation in β-Cells and Sustains Insulin Secretion in Diabetes. J. Biol. Chem. 2016, 291, 10162–10172. [Google Scholar] [CrossRef] [PubMed]
- Ježek, J.; Jabůrek, M.; Zelenka, J.; Ježek, P. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol. Res. 2010, 59, 737–747. [Google Scholar] [PubMed]
- Ježek, P.; Olejár, T.; Smolková, K.; Ježek, J.; Dlasková, A.; Plecitá-Hlavatá, L.; Zelenka, J.; Špaček, T.; Engstová, H.; Pajuelo Reguera, D.; et al. Antioxidant and regulatory role of mitochondrial uncoupling protein UCP2 in pancreatic beta-cells. Physiol. Res. 2014, 63 (Suppl. 1), S73–S91. [Google Scholar] [PubMed]
- Ježek, P.; Holendová, B.; Garlid, K.D.; Jabůrek, M. Mitochondrial uncoupling proteins: Subtle regulators of cellular redox signaling. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, C.; Vasquez, J.S.; Balcazar, N. In vitro effect of fatty acids identified in the plasma of obese adolescents on the function of pancreatic β-cells. Diabetes Metab. J. 2017, 41, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Shamni, O.; Avrahami, Y.; Cohen, O.; Broner, E.C.; Filippov-Levy, N.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Beta cell response to nutrient overload involves phospholipid remodelling and lipid peroxidation. Diabetologia 2015, 58, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The peroxisome-mitochondria connection: How and why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.L.; Pomatto, L.C.D.; Tripathi, D.N.; Davies, K.J.A. Redox Regulation of Homeostasis and Proteostasis in Peroxisomes. Physiol. Rev. 2018, 98, 89–115. [Google Scholar] [CrossRef] [PubMed]
- Kunau, W.H.; Dommes, V.; Schulz, H. beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: A century of continued progress. Prog. Lipid Res. 1995, 34, 267–342. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front. Cell Dev. Biol. 2016, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [PubMed]
- Azevedo-Martins, A.K.; Monteirom, A.P.; Lima, C.L.; Lenzen, S.; Curi, R. Fatty acid-induced toxicity and neutral lipid accumulation in insulin-producing RINm5F cells. Toxicol. In Vitro 2006, 20, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, W.; Elsner, M.A. specific fluorescence probe for hydrogen peroxide detection in peroxisomes. Free Radic. Res. 2011, 45, 501–506. [Google Scholar] [CrossRef]
- Bao, S.; Song, H.; Tan, M.; Wohltmann, M.; Ladenson, J.H.; Turk, J. Group VIB Phospholipase A(2) promotes proliferation of INS-1 insulinoma cells and attenuates lipid peroxidation and apoptosis induced by inflammatory cytokines and oxidant agents. Oxid. Med. Cell Longev. 2012. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Matsuzaka, T.; Nakano, Y.; Motomura, K.; Tang, N.; Yokoo, T.; Okajima, Y.; Han, S.; Takeuchi, Y.; Aita, Y.; et al. Elovl6 Deficiency Improves Glycemic Control in Diabetic db/db Mice by Expanding β-Cell Mass and Increasing Insulin Secretory Capacity. Diabetes 2017, 66, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Cruciani-Guglielmacci, C.; Bellini, L.; Denom, J.; Oshima, M.; Fernandez, N.; Normandie-Levi, P.; Berney, X.P.; Kassis, N.; Rouch, C.; Dairou, J.; et al. Molecular phenotyping of multiple mouse strains under metabolic challenge uncovers a role for Elovl2 in glucose-induced insulin secretion. Mol. Metab. 2017, 6, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Graciano, M.F.; Leonelli, M.; Curi, R.; R Carpinelli, A. Omega-3 fatty acids control productions of superoxide and nitrogen oxide and insulin content in INS-1E cells. J. Physiol. Biochem. 2016, 72, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Modak, M.A.; Datar, S.P.; Bhonde, R.R.; Ghaskadbi, S.S. Differential susceptibility of chick and mouse islets to streptozotocin and its co-relation with islet antioxidant status. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 2007, 177, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Modak, M.A.; Parab, P.B.; Ghaskadbi, S.S. Pancreatic islets are very poor in rectifying oxidative DNA damage. Pancreas 2009, 38, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, R.; Quintens, R.; Dejonghe, S.; Tsukamoto, K.; In’t Veld, P.; Renström, E.; Schuit, F.C. Redox control of exocytosis: Regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 2005, 54, 2132–2142. [Google Scholar] [CrossRef] [PubMed]
- Welsh, N.; Margulis, B.; Borg, L.A.; Wiklund, H.J.; Saldeen, J.; Flodström, M.; Mello, M.A.; Andersson, A.; Pipeleers, D.G.; Hellerström, C. Differences in the expression of heat-shock proteins and antioxidant enzymes between human and rodent pancreatic islets: Implications for the pathogenesis of insulin-dependent diabetes mellitus. Mol. Med. 1995, 1, 806–820. [Google Scholar] [PubMed]
- Tonooka, N.; Oseid, E.; Zhou, H.; Harmon, J.S.; Robertson, R.P. Glutathione peroxidase protein expression and activity in human islets isolated for transplantation. Clin. Transplant. 2007, 21, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Homem De Bittencourt, P.I.; O’ Hagan, C.; De Vito, G.; Murphy, C.; Krause, M.S. Exercise and possible molecular mechanisms of protection from vascular disease and diabetes: The central role of ROS and nitric oxide. Clin. Sci. 2010, 118, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Traba, J.; Geiger, S.S.; Kwarteng-Siaw, M.; Han, K.; Ra, O.H.; Siegel, R.M.; Gius, D.; Sack, M.N. Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3-mediated activation of superoxide dismutase 2. J. Biol. Chem. 2017, 292, 12153–12164. [Google Scholar] [CrossRef] [PubMed]
- Ciregia, F.; Bugliani, M.; Ronci, M.; Giusti, L.; Boldrini, C.; Mazzoni, M.R.; Mossuto, S.; Grano, F.; Cnop, M.; Marselli, L.; et al. Palmitate-induced lipotoxicity alters acetylation of multiple proteins in clonal β cells and human pancreatic islets. Sci. Rep. 2017, 7, 13445. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chung, A.C.K.; Fan, R.; Lee, H.M.; Xu, G.; Tomlinson, B.; Chan, J.C.N.; Kong, A.P.S. Sirt3 Deficiency Increased the Vulnerability of Pancreatic Beta Cells to Oxidative Stress-Induced Dysfunction. Antioxid. Redox Signal. 2017, 27, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, J.S.; Oh, J.E.; Nan, J.; Lee, H.; Jung, H.S.; Chung, S.S.; Park, K.S. SIRT3 overexpression attenuates palmitate-induced pancreatic β-cell dysfunction. PLoS ONE 2015, 10, e0124744. [Google Scholar] [CrossRef] [PubMed]
- Mehmeti, I.; Lortz, S.; Avezov, E.; Jörns, A.; Lenzen, S. ER-resident antioxidative GPx7 and GPx8 enzyme isoforms protect insulin-secreting INS-1E β-cells against lipotoxicity by improving the ER antioxidative capacity. Free Radic. Biol. Med. 2017, 112, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Volkov, P.; Dayeh, T.; Bacos, K.; Rönn, T.; Nitert, M.D.; Ling, C. Effects of palmitate on genome-wide mRNA expression and DNA methylation patterns in human pancreatic islets. BMC Med. 2014, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Briand, O.; Helleboid-Chapman, A.; Ploton, M.; Hennuyer, N.; Carpentier, R.; Pattou, F.; Vandewalle, B.; Moerman, E.; Gmyr, V.; Kerr-Conte, J.; et al. The Nuclear Orphan Receptor Nur77 Is a Lipotoxicity Sensor Regulating Glucose-Induced Insulin Secretion in Pancreatic β-Cells. Mol. Endocrinol. 2012, 26, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Hwang, S.; Lee, S.H.; Lee, Y.R.; Shin, J.; Park, K.S.; Cho, Y.M. Genome-wide identification of palmitate-regulated immediate early genes and target genes in pancreatic beta-cells reveals a central role of NF-κB. Mol. Biol. Rep. 2012, 39, 6781–6789. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Y.; Feng, D.D.; Wang, D.F.; Sun, J.; Li, S.B.; Chen, C. Long-term in vitro treatment of INS-1 rat pancreatic β-cells by unsaturated free fatty acids protects cells against gluco- and lipotoxicities via activation of GPR40 receptors. Clin. Exp. Pharmacol. Physiol. 2012, 39, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Keane, D.C.; Takahashi, H.K.; Dhayal, S.; Morgan, N.G.; Curi, R.; Newsholme, P. Arachidonic acid actions on functional integrity and attenuation of the negative effects of palmitic acid in a clonal pancreatic β-cell line. Clin. Sci. 2011, 120, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Hagman, D.K.; Hays, L.B.; Parazzoli, S.D.; Poitout, V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J. Biol. Chem. 2005, 280, 32413–32418. [Google Scholar] [CrossRef] [PubMed]
- Roomp, K.; Kristinsson, H.; Schvartz, D.; Ubhayasekera, K.; Sargsyan, E.; Manukyan, L.; Chowdhury, A.; Manell, H.; Satagopam, V.; Groebe, K.; et al. Combined lipidomic and proteomic analysis of isolated human islets exposed to palmitate reveals time-dependent changes in insulin secretion and lipid metabolism. PLoS ONE 2017, 12, e0176391. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Kawai, T.; Hirose, H.; Tanaka, K.; Kurosawa, H.; Fujii, C.; Fujita, H.; Seto, Y.; Matsumoto, H.; Itoh, H. Palmitic acid-rich diet suppresses glucose-stimulated insulin secretion (GSIS) and induces endoplasmic reticulum (ER) stress in pancreatic islets in mice. Endocr. Res. 2016, 41, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Hoppa, M.B.; Collins, S.; Ramracheya, R.; Hodson, L.; Amisten, S.; Zhang, Q.; Johnson, P.; Ashcroft, F.M.; Rorsman, P. Chronic Palmitate Exposure Inhibits Insulin Secretion by Dissociation of Ca2+ Channels from Secretory Granules. Cell Metab. 2009, 10, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; Biondi, G.; Marrano, N.; Labarbuta, R.; Tortosa, F.; Spagnuolo, R.; D’Oria, R.; Carchia, E.; Leonardini, A.; Cignarelli, A.; et al. Long-term exposure of pancreatic β-cells to palmitate results in SREBP-1C-dependent decreases in GLP-1 receptor signaling via CREB and AKT and insulin secretory response. Endocrinology 2016, 157, 2243–2258. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tong, Y.; Gong, M.; Lu, Y.; Wang, C.; Zhou, M.; Yang, Q.; Mao, T.; Tong, N. Activation of PPARβ/δ protects pancreatic β cells from palmitate-induced apoptosis by upregulating the expression of GLP-1 receptor. Cell. Signal. 2014, 26, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Sommerweiss, D.; Gorski, T.; Richter, S.; Garten, A.; Kiess, W. Oleate rescues INS-1E β-cells from palmitate-induced apoptosis by preventing activation of the unfolded protein response. Biochem. Biophys. Res. Commun. 2013, 441, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Maria Sol, E.R.; Xu, Y.; Shuai, H.; Tengholm, A. Impaired cAMP generation contributes to defective glucose-stimulated insulin secretion after long-term exposure to palmitate. Diabetes 2015, 64, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Hao, L.; Li, S.; Lin, S.; Lv, L.; Chen, Y.; Cui, H.; Zi, T.; Chu, X.; Na, L.; et al. Elevated circulating stearic acid leads to a major lipotoxic effect on mouse pancreatic beta cells in hyperlipidaemia via a miR-34a-5p-mediated PERK/p53-dependent pathway. Diabetologia 2016, 59, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lai, G.; Wu, J.; Sun, R.; Xu, R.; Yang, X.; Qi, Y.; Zhao, Y. 20-HETE attenuates the response of glucose-stimulated insulin secretion through the AKT/GSK-3β/Glut2 pathway. Endocrine 2016, 54, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, J.; Hollern, D.; Stamateris, R.; Abbasi, M.; Romano, L.C.; Zou, B.; O’Donnell, C.P.; Garcia-Ocana, A.; Alonso, L.C. Free fatty acids block glucose-induced β-cell proliferation in mice by inducing cell cycle inhibitors p16 and p18. Diabetes 2012, 61, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Hodson, D.J.; Mitchell, R.K.; Bellomo, E.A.; Sun, G.; Vinet, L.; Meda, P.; Li, D.; Li, W.H.; Bugliani, M.; Marchetti, P.; et al. Lipotoxicity disrupts incretin-regulated human β cell connectivity. J. Clin. Investig. 2013, 123, 4182–4194. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Shimajiri, Y.; Sakagashira, S.; Furuta, M.; Sanke, T. Effect of exposure to non-esterified fatty acid on progressive deterioration of insulin secretion in patients with Type2 diabetes: A long-term follow-up study. Diabet. Med. 2012, 29, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P. Islet inflammation in type 2 diabetes. Diabetologia 2016, 59, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Innes, J.K.; Calder, P.C. Omega-6 fatty acids and inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2018. [Google Scholar] [CrossRef] [PubMed]
- Flachs, P.; Horakova, O.; Brauner, P.; Rossmeisl, M.; Pecina, P.; Franssen-Van Hal, N.; Ruzickova, J.; Sponarova, J.; Drahota, Z.; Vlcek, C.; et al. Polyunsaturated fatty acids of marine origin upregulate mitochondrial biogenesis and induce β-oxidation in white fat. Diabetologia 2005, 48, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, C.; Yang, L.; Yoshizaki, T.; Nakagawa, F.; Ishikado, A.; Kondo, M.; Morino, K.; Sekine, O.; Ugi, S.; Nishio, Y.; et al. Omega-3 polyunsaturated fatty acid has an anti-oxidant effect via the Nrf-2/HO-1 pathway in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2013, 430, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Shin, Y.; Moon, S.; Kim, S.; Kim, Y. Effects of Eicosapentaenoic Acid and Docosahexaenoic Acid on Mitochondrial DNA Replication and PGC-1α Gene Expression in C2C12 Muscle Cells. Prev. Nutr. Food Sci. 2016, 21, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Lepretti, M.; Martucciello, S.; Burgos Aceves, M.; Putti, R.; Lionetti, L. Omega-3 Fatty Acids and Insulin Resistance: Focus on the Regulation of Mitochondria and Endoplasmic Reticulum Stress. Nutrients 2018, 10, 350. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chen, X.; Chen, M.; Li, Y.; Li, Q.; Jiang, X.; Yang, Y.; Ling, W. Fish oil supplementation inhibits endoplasmic reticulum stress and improves insulin resistance: Involvement of AMP-activated protein kinase. Food Funct. 2017, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Mena, S.J.; Manosalva, C.; Carretta, M.D.; Teuber, S.; Olmo, I.; Burgos, R.A.; Hidalgo, M.A. Differential free fatty acid receptor-1 (FFAR1/GPR40) signalling is associated with gene expression or gelatinase granule release in bovine neutrophils. Innate Immun. 2016, 22, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Roelofsen, H.; Priebe, M.G.; Vonk, R.J. The interaction of short-chain fatty acids with adipose tissue: Relevance for prevention of type 2 diabetes. Benef. Microbes 2010, 1, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Ren, X.; Han, T.; Chen, Y.; Qiu, H.; Liu, W.; Hu, Y. Fenofibrate attenuates fatty acid-induced islet β-cell dysfunction and apoptosis via inhibiting the NF-κB/MIF dependent inflammatory pathway. Metabolism 2017, 77, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Mukhuty, A.; Fouzder, C.; Mukherjee, S.; Malick, C.; Mukhopadhyay, S.; Bhattacharya, S.; Kundu, R. Palmitate induced Fetuin-A secretion from pancreatic β-cells adversely affects its function and elicits inflammation. Biochem. Biophys. Res. Commun. 2017, 491, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Dobrian, A.D.; Morris, M.A.; Taylor-Fishwick, D.A.; Nadler, J.L. Lipids and immunoinflammatory pathways of beta cell destruction. Diabetologia 2016, 59, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.J.; Sparer, T.E.; Karlstad, M.D.; Burke, S.J. Pancreatic islet inflammation: An emerging role for chemokines. J. Mol. Endocrinol. 2017, 59, R33–R46. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Marselli, L.; Cunha, D.A.; Ladrière, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.V.; Griebel, T.; Villate, O.; Santin, I.; et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014, 63, 1978–1993. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Shirakawa, J.; Togashi, Y.; Tajima, K.; Okuyama, T.; Kyohara, M.; Tanaka, Y.; Orime, K.; Saisho, Y.; Yamada, T.; et al. Signaling between pancreatic β-cells and macrophages via S100 calcium-binding protein A8 exacerbates β-cell apoptosis and islet inflammation. J. Biol. Chem. 2018, 293, 5934–5946. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, T.; Hänzelmann, S.; Salehi, A.; Muhammed, S.J.; Reinbothe, T.M.; Tang, Y.; Axelsson, A.S.; Zhou, Y.; Jing, X.; Almgren, P.; et al. Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2 diabetes. Cell Metab. 2012, 16, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Taneera, J.; Lang, S.; Sharma, A.; Fadista, J.; Zhou, Y.; Ahlqvist, E.; Jonsson, A.; Lyssenko, V.; Vikman, P.; Hansson, O.; et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012, 16, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Marselli, L.; Thorne, J.; Dahiya, S.; Sgroi, D.C.; Sharma, A.; Bonner-Weir, S.; Marchetti, P.; Weir, G.C. Gene expression profiles of beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS ONE 2010, 5, e11499. [Google Scholar] [CrossRef] [PubMed]
- Böni-Schnetzler, M.; Thorne, J.; Parnaud, G.; Marselli, L.; Ehses, J.A.; Kerr-Conte, J.; Pattou, F.; Halban, P.A.; Weir, G.C.; Donath, M.Y. Increased interleukin (IL)-1β messenger ribonucleic acid expression in β-cells of individuals with type 2 diabetes and regulation of IL-1β in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 2008, 93, 4065–4074. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Manabe, I. Macrophages and islet inflammation in type 2 diabetes. Diabetes Obes. Metab. 2013, 15, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Kugelberg, E. Diabetes: Macrophages mediate β-cell loss in T2DM. Nat. Rev. Endocrinol. 2013, 9, 626. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C.; et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 2013, 19, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Azevedo-Martins, A.K.; Lortz, S.; Lenzen, S.; Curi, R.; Eizirik, D.L.; Tiedge, M. Improvement of the mitochondrial antioxidant defense status prevents cytokine-induced nuclear factor-κB activation in insulin-producing cells. Diabetes 2003, 52, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M.; et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Zummo, F.P.; Cullen, K.S.; Honkanen-Scott, M.; Shaw, J.A.M.; Lovat, P.E.; Arden, C. Glucagon-Like Peptide 1 Protects Pancreatic β-Cells From Death by Increasing Autophagic Flux and Restoring Lysosomal Function. Diabetes 2017, 66, 1272–1285. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; Marrano, N.; Biondi, G.; Spagnuolo, R.; Labarbuta, R.; Porreca, I.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; Perrini, S.; et al. The Myokine Irisin Is Released in Response to Saturated Fatty Acids and Promotes Pancreatic β-Cell Survival and Insulin Secretion. Diabetes 2017, 66, 2849–2856. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Feng, Y.; Ma, H.; Liu, C.; Chen, G.; Wei, X.; Mao, X.; Li, X.; Xu, Y.; Tang, S.; et al. Neutral ceramidase activity inhibition is involved in palmitate-induced apoptosis in INS-1 cells. Endocr. J. 2017, 64, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Cito, M.; Grieco, F.A.; Cosentino, C.; Danilova, T.; Ladrière, L.; Lindahl, M.; Domanskyi, A.; Bugliani, M.; Marchetti, P.; et al. Pancreatic β-cell protection from inflammatory stress by the endoplasmic reticulum proteins thrombospondin 1 and mesencephalic astrocyte-derived neutrotrophic factor (MANF). J. Biol. Chem. 2017, 292, 14977–14988. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ježek, P.; Jabůrek, M.; Holendová, B.; Plecitá-Hlavatá, L. Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules 2018, 23, 1483. https://doi.org/10.3390/molecules23061483
Ježek P, Jabůrek M, Holendová B, Plecitá-Hlavatá L. Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules. 2018; 23(6):1483. https://doi.org/10.3390/molecules23061483
Chicago/Turabian StyleJežek, Petr, Martin Jabůrek, Blanka Holendová, and Lydie Plecitá-Hlavatá. 2018. "Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity" Molecules 23, no. 6: 1483. https://doi.org/10.3390/molecules23061483
APA StyleJežek, P., Jabůrek, M., Holendová, B., & Plecitá-Hlavatá, L. (2018). Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules, 23(6), 1483. https://doi.org/10.3390/molecules23061483