Parthenolide Inhibits STAT3 Signaling by Covalently Targeting Janus Kinases


Abstract
1. Introduction
2. Result
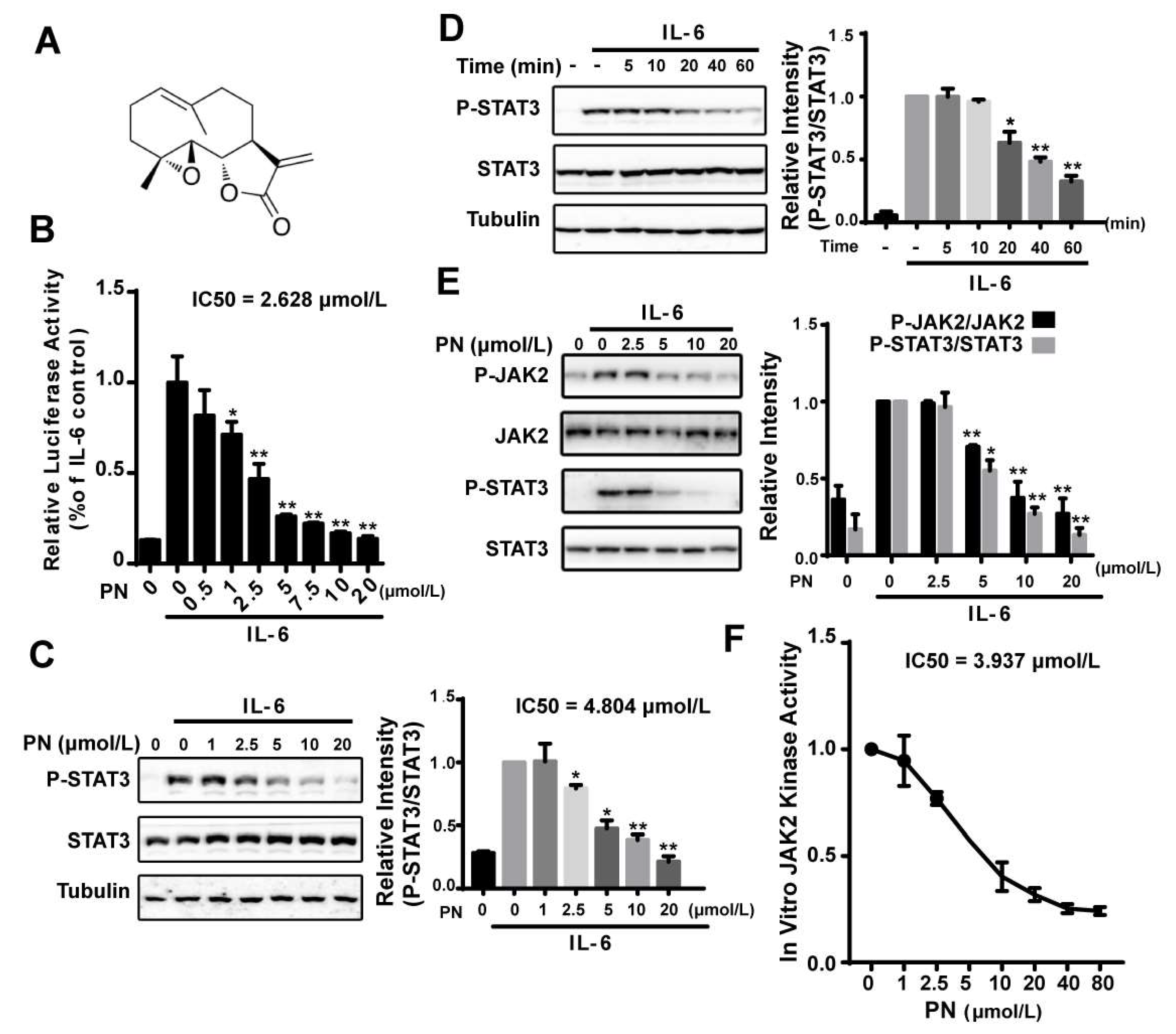
2.1. Parthenolide Inhibited the IL-6-Induced STAT3 Phosphorylation by Inhibiting JAK2 Kinase Activity
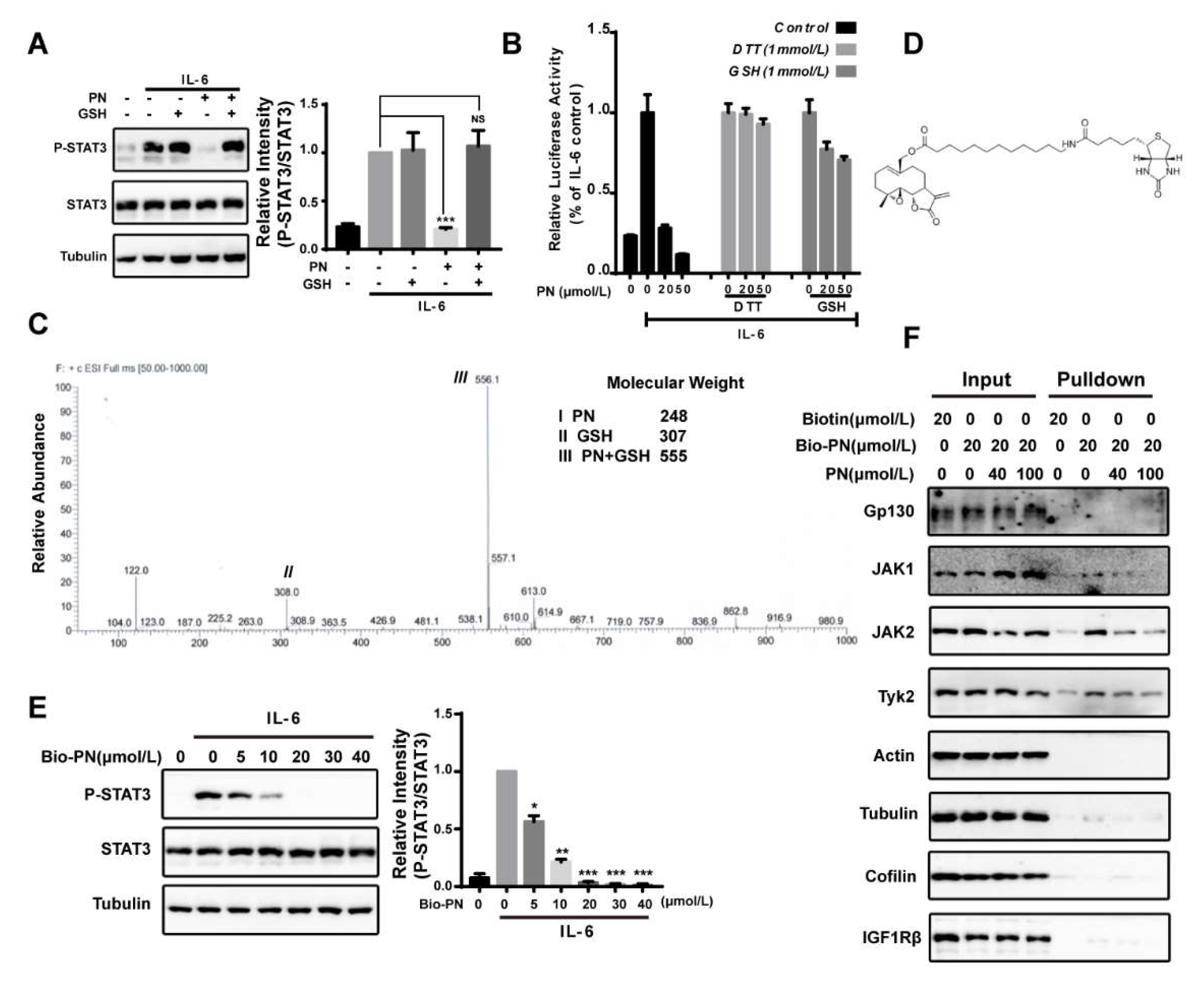
2.2. Parthenolide Was a Covalent Pan-JAK Inhibitor
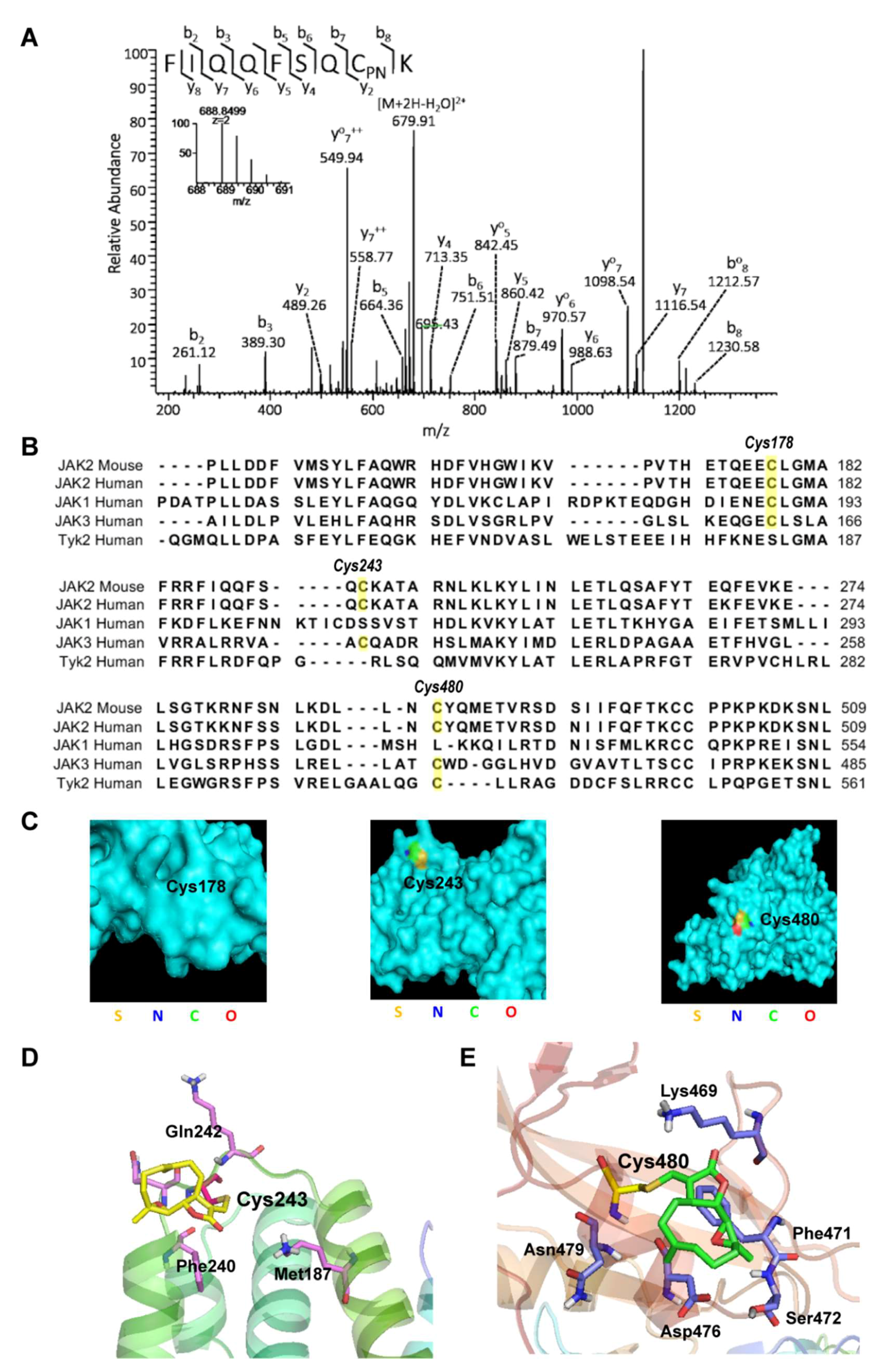
2.3. Parthenolide Covalently Modified Cys178, Cys243, Cys335, and Cys480 of Mouse JAK2
2.4. Virtual Docking Demonstrated the Preference of Parthenolide to Cys243 and Cys480 of JAK2
2.5. The Effects of Parthenolide on JAKs Were Selective
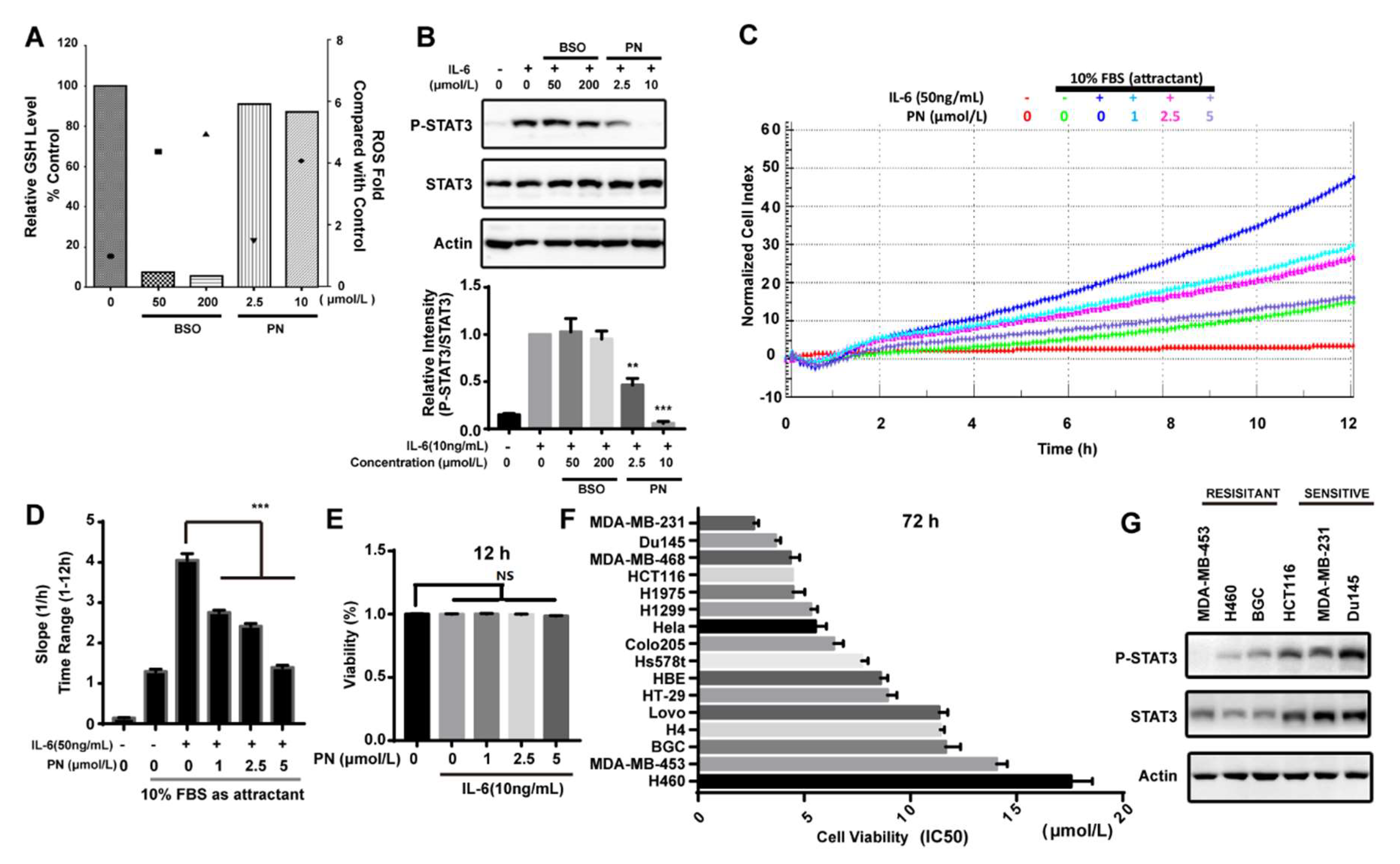
2.6. Reactive Oxygen Species Produced by Parthenolide Did Not Participate in the Inhibition of JAK/STAT3 Signaling
2.7. Parthenolide Selectively Inhibited the IL-6-Induced MDA-MB-231 Migration
2.8. Parthenolide Selectively Inhibited the Growth of Cancer Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals and Reagents
4.3. Antibodies
4.4. Luciferase Assay
4.5. Western Blot Analysis
4.6. JAK2 In Vitro Kinase Assay
4.7. HPLC-MS Analysis of GSH-PN Adduct
4.8. Syntheses of Biotinylated Parthenolide
4.9. Pull-Down Assay
4.10. In Vitro Kinase Assay
4.11. HPLC–MS/MS Analysis of JAK2
4.12. Docking Study
4.13. Determination of Cellular ROS
4.14. Determination of Cellular GSH Level
4.15. MTT Assay
4.16. Fluorimetric Method to Determine the Viability of MDA-MB-231
4.17. Migration Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ward, A.C.; Touw, I.; Yoshimura, A. The jak-stat pathway in normal and perturbed hematopoiesis. Blood 2000, 95, 19–29. [Google Scholar] [PubMed]
- O’Shea, J.J.; Plenge, R. Jak and stat signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. Il-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of jak2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The jak2/stat3 signaling pathway is required for growth of cd44(+)cd24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The il-6/jak/stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Witthuhn, B.A.; Yoshida, K.; Silvennoinen, O.; Yasukawa, K.; Ihle, J.N.; Kishimoto, T.; Taga, T. Activation of jak2 kinase mediated by the interleukin 6 signal transducer gp130. Proc. Natl. Acad. Sci. USA 1994, 91, 2285–2289. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Gao, F.H.; Wang, J.Y.; Liu, F.; Yuan, H.H.; Zhang, W.Y.; Jiang, B. Jak2/stat3 signaling pathway activation mediates tumor angiogenesis by upregulation of vegf and bfgf in non-small-cell lung cancer. Lung Cancer 2011, 73, 366–374. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal jak2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase jak2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Seidel, H.M.; Lamb, P.; Rosen, J. Pharmaceutical intervention in the jak/stat signaling pathway. Oncogene 2000, 19, 2645–2656. [Google Scholar] [CrossRef] [PubMed]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting jak kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Laborde, R.R.; Lasho, T.L.; Finke, C.; Begna, K.; Al-Kali, A.; Hogan, W.J.; Litzow, M.R.; Leontovich, A.; Kowalski, M.; et al. Safety and efficacy of cyt387, a jak1 and jak2 inhibitor, in myelofibrosis. Leukemia 2013, 27, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Roberts, A.W.; Seymour, J.F.; Burbury, K.; Verstovsek, S.; Kantarjian, H.M.; Begna, K.; Yoshitsugu, H.; Gestone, T.A.; Phillips, P.; et al. Bms-911543, a selective jak2 inhibitor: A multicenter phase 1/2a study in myelofibrosis. Blood 2013, 122, 664. [Google Scholar]
- Pardanani, A.; Gotlib, J.R.; Jamieson, C.; Cortes, J.E.; Talpaz, M.; Stone, R.M.; Silverman, M.H.; Gilliland, D.G.; Shorr, J.; Tefferi, A. Safety and efficacy of tg101348, a selective jak2 inhibitor, in myelofibrosis. J. Clin. Oncol. 2011, 29, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 1670–1671. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Yasothan, U.; Kirkpatrick, P. Ruxolitinib. Nat. Rev. Drug Discov. 2012, 11, 103–104. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.; Mesa, R.; Ross, D.; Mead, A.; Keohane, C.; Gotlib, J.; Verstovsek, S. Practical management of patients with myelofibrosis receiving ruxolitinib. Expert Rev. Hematol. 2013, 6, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of incb018424, a jak1 and jak2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T.; Liu, F.; Saunders, L.M.; Mullally, A.; Abdel-Wahab, O.; et al. Heterodimeric jak-stat activation as a mechanism of persistence to jak2 inhibitor therapy. Nature 2012, 489. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Sobota, R.; Szwed, M.; Kasza, A.; Bugno, M.; Kordula, T. Parthenolide inhibits activation of signal transducers and activators of transcription (stats) induced by cytokines of the il-6 family. Biochem. Biophys. Res. Commun. 2000, 267, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The jak2 inhibitor azd1480 potently blocks stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Li, C.; Wang, X.; Dian, L.; Zhang, X.; Li, L.; Chen, S.; Cao, R.; Li, L.; Huang, N.; et al. Ainsliadimer a selectively inhibits ikkalpha/beta by covalently binding a conserved cysteine. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.F.; Ye, B.Q.; Li, Y.D.; Wang, J.G.; He, X.J.; Lin, X.; Yao, X.; Ma, D.; Slungaard, A.; Hebbel, R.P.; et al. Andrographolide attenuates inflammation by inhibition of nf-kappa b activation through covalent modification of reduced cysteine 62 of p50. J. Immunol. 2004, 173, 4207–4217. [Google Scholar] [CrossRef] [PubMed]
- Lyss, G.; Knorre, A.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor nf-kappab by directly targeting p65. J. Biol. Chem. 1998, 273, 33508–33516. [Google Scholar] [CrossRef] [PubMed]
- McNally, R.; Toms, A.V.; Eck, M.J. Crystal structure of the ferm-sh2 module of human jak2. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, M.; Booz, G.W. Evidence that il-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: Parthenolide targets jak1 activation by generating ros. J. Cell Physiol. 2007, 212, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Obata, N.H.; Tamakoshi, K.; Shibata, K.; Kikkawa, F.; Tomoda, Y. Effects of interleukin-6 on in vitro cell attachment, migration and invasion of human ovarian carcinoma. Anticancer Res. 1997, 17, 337–342. [Google Scholar] [PubMed]
- Walter, M.; Liang, S.; Ghosh, S.; Hornsby, P.J.; Li, R. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene 2009, 28, 2745–2755. [Google Scholar] [CrossRef] [PubMed]
- Di, G.H.; Liu, Y.; Lu, Y.; Liu, J.; Wu, C.T.; Duan, H.F. Il-6 secreted from senescent mesenchymal stem cells promotes proliferation and migration of breast cancer cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Skoumal, R.; Toth, M.; Serpi, R.; Rysa, J.; Leskinen, H.; Ulvila, J.; Saiho, T.; Aro, J.; Ruskoaho, H.; Szokodi, I.; et al. Parthenolide inhibits stat3 signaling and attenuates angiotensin ii-induced left ventricular hypertrophy via modulation of fibroblast activity. J. Mol. Cell. Cardiol. 2011, 50, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Hilkens, C.M.; Is’harc, H.; Lillemeier, B.F.; Strobl, B.; Bates, P.A.; Behrmann, I.; Kerr, I.M. A region encompassing the ferm domain of jak1 is necessary for binding to the cytokine receptor gp130. FEBS Lett. 2001, 505, 87–91. [Google Scholar] [CrossRef]
- Haan, S.; Margue, C.; Engrand, A.; Rolvering, C.; Schmitz-Van de Leur, H.; Heinrich, P.C.; Behrmann, I.; Haan, C. Dual role of the jak1 ferm and kinase domains in cytokine receptor binding and in stimulation-dependent jak activation. J. Immunol. 2008, 180, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.J.; Chen, M.; Cusack, N.A.; Kimmel, L.H.; Magnuson, K.S.; Boyd, J.G.; Lin, W.; Roberts, J.L.; Lengi, A.; Buckley, R.H.; et al. Unexpected effects of ferm domain mutations on catalytic activity of jak3: Structural implication for janus kinases. Mol. Cell 2001, 8, 959–969. [Google Scholar] [CrossRef]
- Radtke, S.; Haan, S.; Jorissen, A.; Hermanns, H.M.; Diefenbach, S.; Smyczek, T.; Schmitz-Vandeleur, H.; Heinrich, P.C.; Behrmann, I.; Haan, C. The jak1 sh2 domain does not fulfill a classical sh2 function in jak/stat signaling but plays a structural role for receptor interaction and up-regulation of receptor surface expression. J. Biol. Chem. 2005, 280, 25760–25768. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, R.; Wallweber, H.J.; Ho, H.; Tam, C.; Franke, Y.; Quinn, J.; Lupardus, P.J. The structural basis for class ii cytokine receptor recognition by jak1. Structure 2016, 24, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Wallweber, H.J.; Tam, C.; Franke, Y.; Starovasnik, M.A.; Lupardus, P.J. Structural basis of recognition of interferon-alpha receptor by tyrosine kinase 2. Nat. Struct. Mol. Biol. 2014, 21, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.; Dillon, M.B.; Bachovchin, D.A.; Mowen, K.; Baker, D.; Cravatt, B.F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Kwon, K.S.; Kim, S.R.; Rhee, S.G. Reversible inactivation of protein-tyrosine phosphatase 1b in a431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998, 273, 15366–15372. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cheung, S.H.; Evans, E.L.; Shaw, P.E. Modulation of gene expression and tumor cell growth by redox modification of stat3. Cancer Res. 2010, 70, 8222–8232. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Monti, M.; Pucci, P.; Dell’Orco, D.; Mariotto, S. S-glutathionylation at cys328 and cys542 impairs stat3 phosphorylation. ACS Chem. Biol. 2014, 9, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Lu, B.; Monroe, R.K.; Ward, S.M.; Halvorsen, S.W. Inducers of oxidative stress block ciliary neurotrophic factor activation of jak/stat signaling in neurons. J. Neurochem. 2005, 92, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.X.; Ai, M.D.; Wang, Y.; Shen, S.S.; Gu, Y.; Jin, Y.; Zhou, Z.Y.; Long, Y.Q.; Yu, Q. Selective induction of tumor cell apoptosis by a novel p450-mediated reactive oxygen species (ros) inducer methyl 3-(4-nitrophenyl) propiolate. J. Biol. Chem. 2013, 288, 8826–8837. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Qi, C.T.; Liu, J.W.; Sun, X.X.; Zhang, Q.; Sima, Z.H.; Liu, J.L.; Li, W.G.; Yu, Q. 2-methoxystypandrone inhibits signal transducer and activator of transcription 3 and nuclear factor-jb signaling by inhibiting janus kinase 2 and ijb kinase. Cancer Sci. 2014, 105, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Macias, F.A.; Galindo, J.C.G.; Massanet, G.M. Natural product models as allelochemicals. 1. Potential allelopathic activity of several sesquiterpene lactone models. Phytochemistry 1992, 31, 1969–1977. [Google Scholar] [CrossRef]
- Sun, M.W.; Xu, J.Y.; Wu, Z.X.; Zhai, L.H.; Liu, C.X.; Cheng, Z.Y.; Xu, G.F.; Tao, S.C.; Ye, B.C.; Zhao, Y.M.; et al. Characterization of protein lysine propionylation in Escherichia coli: Global profiling, dynamic change, and enzymatic regulation. J. Proteom. Res. 2016, 15, 4696–4708. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; You, K.R.; Lee, S.Y.; Song, C.H.; Kim, D.G. Oxidative stress-mediated apoptosis. The anticancer effect of the sesquiterpene lactone parthenolide. J. Biol. Chem. 2002, 277, 38954–38964. [Google Scholar] [CrossRef] [PubMed]
- Scrace, S.; O’Neill, E.; Hammond, E.M.; Pires, I.M. Use of the xcelligence system for real-time analysis of changes in cellular motility and adhesion in physiological conditions. Methods Mol. Biol. 2013, 1046, 295–306. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds parthenolide and Bio-PN are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | EGFR | PI3K | cSRC | MAPK1 |
|---|---|---|---|---|
| Activity (%) | 116 | 107 | 91 | 86 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Xiao, C.; Sun, M.; Tan, M.; Hu, L.; Yu, Q. Parthenolide Inhibits STAT3 Signaling by Covalently Targeting Janus Kinases. Molecules 2018, 23, 1478. https://doi.org/10.3390/molecules23061478
Liu M, Xiao C, Sun M, Tan M, Hu L, Yu Q. Parthenolide Inhibits STAT3 Signaling by Covalently Targeting Janus Kinases. Molecules. 2018; 23(6):1478. https://doi.org/10.3390/molecules23061478
Chicago/Turabian StyleLiu, Man, Chengqian Xiao, Mingwei Sun, Minjia Tan, Lihong Hu, and Qiang Yu. 2018. "Parthenolide Inhibits STAT3 Signaling by Covalently Targeting Janus Kinases" Molecules 23, no. 6: 1478. https://doi.org/10.3390/molecules23061478
APA StyleLiu, M., Xiao, C., Sun, M., Tan, M., Hu, L., & Yu, Q. (2018). Parthenolide Inhibits STAT3 Signaling by Covalently Targeting Janus Kinases. Molecules, 23(6), 1478. https://doi.org/10.3390/molecules23061478