3.2. Procedures
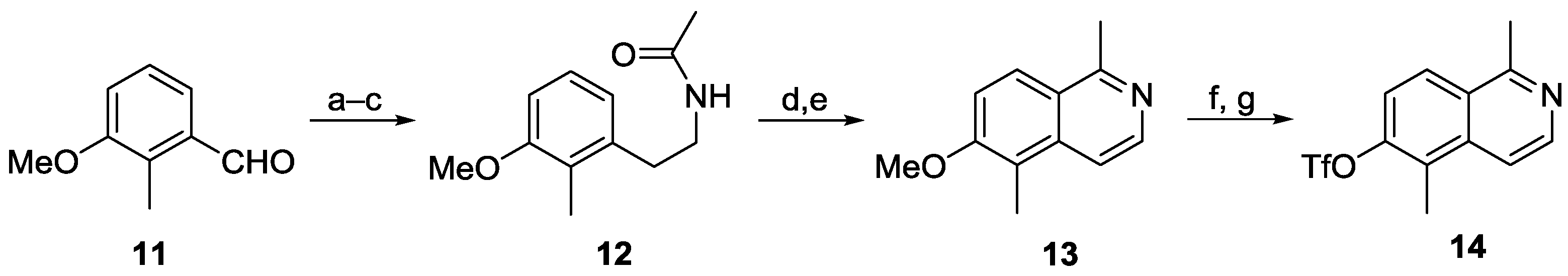
1-Methoxy-2-methyl-3-(2-nitrovinyl)benzene. Nitromethane (427 mg, 6.99 mmol) and freshly sublimated ammonium acetate (433 mg, 5.62 mmol) were added to a solution of 3-methoxy-2-methylbenzaldehyde (11, 800 mg, 5.33 mmol) in acetic acid (645 mg, 10.74 mmol), and the mixture was stirred at 80 °C for 1 h 50 min. After cooling to room temperature, the precipitate was dissolved by adding ethyl acetate. The mixture was transferred to a separatory funnel, and then washed twice with water and brine. The aqueous layer was extracted with ethyl acetate, the combined organic layers were dried (magnesium sulfate), and the solvent was evaporated. Purification of the residue by column chromatography (silica gel, petroleum ether/ethyl acetate, 1% to 15% ethyl acetate) provided 1-methoxy-2-methyl-3-(2-nitrovinyl)benzene (791 mg, 4.09 mmol, 77%) as yellow crystals. M.p. 97–98 °C; UV (MeOH): λ = 205, 228, 251, 317 nm; IR (ATR): ν = 3116, 2959, 2920, 2838, 1901, 1820, 1697, 1653, 1627, 1594, 1573, 1541, 1498, 1477, 1450, 1331, 1260, 1244, 1102, 1080, 1007, 968, 893, 873, 844, 806, 777, 725, 693 cm–1; 1H-NMR (500 MHz, CDCl3): δ = 2.33 (s, 3H), 3.86 (s, 3H), 6.96 (d, J = 8.2 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 7.22 (t, J = 8.0 Hz, 1H), 7.48 (d, J = 13.5 Hz, 1H), 8.33 (d, J = 13.5 Hz, 1H); 13C-NMR (125 MHz, CDCl3): δ = 11.96 (CH3), 55.84 (CH3), 113.12 (CH), 119.25 (CH), 127.11 (CH), 128.40 (C), 130.13 (C), 137.25 (CH), 138.20 (CH), 158.29 (C); MS (EI): m/z (%) = 193 (100, [M]+), 178 (6), 161 (7), 146 (70), 131 (52), 115 (54), 103 (67), 91 (37), 77 (47), 63 (18), 51 (18); HRMS: calcd. for C10H11NO3: 193.0738, found: 193.0733; elemental analysis: calcd. for C10H11NO3: C: 62.17, H: 5.74, N: 7.25; found: C: 62.16, H: 5.77, N: 7.50.
2-(3-Methoxy-2-methylphenyl)ethanamine. Over a period of 1 h, a solution of 1-methoxy-2-methyl-3(2-nitrovinyl)benzene (6.79 g, 35.2 mmol) in THF (95 mL) was added to a suspension of lithium aluminum hydride (6.83 g, 180 mmol) in THF (360 mL) at 0 °C. The cooling bath was removed, and the mixture was heated for 30 min at room temperature and 18 h at reflux. A second portion of lithium aluminum hydride (0.35 g, 9.1 mmol) was added to the slightly reddish-colored solution, and the mixture was heated at reflux for an additional hour. After cooling to room temperature, the reaction mixture was carefully quenched with saturated aqueous ammonium chloride, and the pH value was adjusted to nine. Diethyl ether was added, and the mixture was transferred into a separatory funnel. Still under argon, the layers were separated, and the aqueous layer was extracted three times with diethyl ether. The combined organic layers were washed with water and brine, dried (magnesium sulfate), and the solvent was evaporated to provide 2-(3-methoxy-2-methylphenyl)ethanamine (5.33 g, 32.3 mmol, 92%) as a yellow oil. UV (MeOH): λ = 204, 218, 273, 280 nm; IR (ATR): ν = 3402, 2989, 2923, 2848, 2659, 2480, 2065, 1658, 1604, 1581, 1510, 1463, 1395, 1293, 1256, 1194, 1171, 1149, 1122, 1096, 1006, 953, 875, 789, 776, 763, 719 cm–1; 1H-NMR (600 MHz, CDCl3): δ = 2.22 (s, 3H), 3.10–3.19 (m, 4H), 3.82 (s, 3H), 6.77 (d, J = 8.2 Hz, 1H), 6.83 (d, J = 7.6 Hz, 1H), 7.13 (t, J = 7.9 Hz, 1H); 13C-NMR (150 MHz, methanol-d4): δ = 11.47 (CH3), 32.26 (CH2), 40.35 (CH2), 55.49 (CH3), 109.10 (CH), 121.76 (CH), 125.07 (C), 126.59 (CH), 135.84 (C), 158.03 (C); MS (ESI, +10 V): m/z = 149.0 [M − NH3 + H]+, 166.0 [M + H]+, 331.2 [2M + H]+; HRMS: calcd. for C10H15NO: 165.1153, found: 165.1144.
2-(3-Methoxy-2-methylphenyl)ethyl acetamide (12). DMAP (14 mg, 0.11 mmol) was added to a solution of 2-(3-methoxy-2-methylphenyl)ethanamine (230 mg, 1.14 mmol) in pyridine (4.5 mL), and the mixture was cooled to 0 °C. Acetic anhydride (140 µL, 15 mmol) was added dropwise over a period of five minutes, and the reaction mixture was stirred for 4 h. The solvent was evaporated, and the raw material was purified by chromatography (Alox N, 5% H2O; ethyl acetate) to provide 2-(3-methoxy-2-methylphenyl)ethyl acetamide (12, 235 mg, 1.13 mmol, 99%) as a light yellow solid. M.p. 84–85 °C; UV (MeOH): λ = 205, 219, 229, 271, 279 nm; IR (ATR): ν = 3267, 3085, 2932, 2836, 2030, 2009, 1976, 1716, 1659, 1630, 1564, 1508, 1489, 1472, 1459, 1435, 1396, 1370, 1298, 1285, 1247, 1201, 1180, 1110, 1092, 1037, 1013, 896, 812, 776, 748, 723, 701, 651, 606 cm–1; 1H-NMR (500 MHz, CDCl3): δ = 1.95 (s, 3H), 2.19 (s, 3H), 2.84 (t, J = 6.9 Hz, 2H), 3.46 (q, J = 6.9 Hz, 2H), 3.82 (s, 3H), 5.46 (br s, 1H), 6.76 (d, J = 7.9 Hz, 2H), 7.12 (t, J = 7.9 Hz, 1H); 13C-NMR (125 MHz, CDCl3): δ = 11.53 (CH3), 23.51 (CH3), 33.38 (CH2), 39.87 (CH2), 55.64 (CH3), 108.61 (CH), 121.89 (CH), 125.24 (C), 126.36 (CH), 138.38 (C), 158.09 (C), 170.21 (C=O); MS (ESI, +10 V): m/z = 208.0 [M + H]+, 415.1 [2M + H]+, 437.1 [2M + Na]+; HRMS: calcd. for C12H17NO2: 207.1259, found: 207.1248; elemental analysis: calcd. for C12H17NO2: C: 69.54, H: 8.27, N: 6.76; found: C: 69.04, H: 8.73, N: 6.78.
6-Methoxy-1,5-dimethyl-3,4-dihydroisoquinoline. Phosphorus oxychloride (1.9 mL, 21 mmol) was added to a refluxing solution of acetamide 12 (433 mg, 2.09 mmol) in freshly distilled chloroform (23 mL), and the mixture was stirred for one hour. Subsequently, solvent and excess phosphorus oxychloride were removed under vigorous stirring by a nitrogen stream through a pair of soda lye-filled gas washing bottles. The remaining oily raw product was dissolved in ethyl acetate. Soda lye (10%) was added, and the pH value was adjusted to 8–9 using saturated aqueous ammonium chloride. The layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water and brine, and then dried (magnesium sulfate), and the solvent was evaporated. Purification of the crude product by chromatography (Alox N, 5% H2O; ethyl acetate + 3% triethylamine) afforded 6-methoxy-1,5-dimethyl-3,4-dihydroisoquinoline (393 mg, 2.08 mmol, 99%) as a yellow solid. M.p. 57–58 °C (subl.); UV (MeOH): λ = 229, 274, 319 nm; IR (ATR): ν = 3002, 2939, 2838, 1735, 1699, 1629, 1594, 1576, 1539, 1507, 1482, 1435, 1368, 1291, 1258, 1184, 1149, 1101, 1015, 922, 901, 873, 805, 751, 700, 666, 637 cm–1; 1H-NMR (500 MHz, CDCl3): δ = 2.15 (s, 3H), 2.35 (t, J = 1.4 Hz, 3H), 2.64 (t, J = 7.4 Hz, 2H), 3.63 (tq, J = 7.4, 1.4 Hz, 2H), 3.85 (s, 3H), 6.75 (d, J = 8.5 Hz, 1H), 7.37 (d, J = 8.5 Hz, 1H); 13C-NMR (125 MHz, CDCl3): δ = 11.08 (CH3), 23.32 (CH2), 23.62 (CH3), 46.91 (CH2), 55.61 (CH3), 107.50 (CH), 123.06 (C), 123.40 (C), 124.76 (CH), 137.70 (C), 159.47 (C), 164.80 (C); MS (EI): m/z (%) = 189 (95, [M]+), 174 (100), 158 (16), 144 (23), 131 (22), 115 (31), 105 (23), 91 (22), 77 (29), 63 (17), 51 (20); HRMS: calcd. for C12H15NO: 189.1154, found: 189.1147; elemental analysis: calcd. for C12H15NO: C: 76.16, H: 7.99, N: 7.40; found: C: 76.25, H: 7.98, N: 7.46.
6-Methoxy-1,5-dimethylisoquinoline (13). A flask filled with 6-methoxy-1,5-dimethyl-3,4-dihydroisoquinoline (90.3 mg, 0.48 mmol) and palladium on charcoal (10%, 93.6 mg) was evacuated under vigorous stirring for 15 min, and then filled with argon. Toluene (3.6 mL) and cyclohexene (1.3 mL, 13 mmol) were added, and the mixture was heated at reflux until full conversion was detected (TLC: Alox N; ethyl acetate/isohexane, 2:1 + 1 drop of ethanol). The catalyst was removed by filtration (ethyl acetate), and the crude product was purified by chromatography (Alox N, 5% H2O; petroleum ether/ethyl acetate, 5:1) to provide 6-methoxy-1,5-dimethylisoquinoline (13, 90 mg, 0.48 mmol, 100%) as a beige solid. M.p. 99–102 °C; UV (MeOH): λ = 203, 236, 301 nm; IR (ATR): ν = 3058, 3015, 2965, 2940, 2847, 1608, 1563, 1542, 1495, 1457, 1401, 1344, 1324, 1267, 1179, 1153, 1116, 1078, 1009, 984, 913, 848, 814, 774, 698, 673, 648, 581, 528 cm–1; 1H-NMR (600 MHz, CDCl3): δ = 2.50 (s, 3H), 2.97 (s, 3H), 4.00 (s, 3H), 7.35 (d, J = 9.2 Hz, 1H), 7.63 (d, J = 6.2 Hz, 1H), 8.05 (d, J = 9.2 Hz, 1H), 8.32 (d, J = 6.2 Hz, 1H); 13C-NMR (150 MHz, CDCl3): δ = 10.49 (CH3), 22.17 (CH3), 56.39 (CH3), 113.71 (CH), 115.83 (CH), 118.97 (C), 123.04 (C), 125.66 (CH), 127.47 (C), 136.96 (C), 140.72 (CH), 158.57 (C); MS (EI): m/z (%) = 187 (100, [M]+), 172 (26), 156 (16), 144 (80), 128 (19), 115 (43), 103 (21), 89 (11), 77 (30), 63 (24), 51 (22); HRMS: calcd. for C12H13NO: 187.0997, found: 187.0986; elemental analysis: calcd. for C12H13NO: C: 76.98, H: 7.00, N: 7.48; found: C: 76.43, H: 7.04, N: 7.53.
1,5-Dimethylisoquinolin-6-ol. For small amounts: In a microwave tube, a mixture of 6-methoxy-1,5-dimethylisoquinoline (13, 45 mg, 0.24 mmol) and pyridinium chloride (1 g, 8 mmol) was irradiated at 155 °C (300 W) for 30 min. After cooling to room temperature, the mixture was dissolved in water and ethyl acetate, and neutralized with a saturated aqueous solution of sodium bicarbonate. The layers were separated, and the aqueous layer was carefully extracted with ethyl acetate. The combined organic layers were dried (magnesium sulfate), and the solvent was evaporated to give 1,5-dimethylisoquinolin-6-ol (40 mg, 0.23 mmol, 96%) as a brownish solid.
For larger amounts: Freshly distilled hydrobromic acid (22 mL, 0.19 mol) was carefully added at 0 °C to 6-methoxy-1,5-dimethylisoquinoline (13, 3.01 g, 16.1 mmol). After the addition was completed, the cooling bath was removed, and the mixture was heated at reflux for five hours. Then, the excess of hydrobromic acid was removed under vacuo. The brownish raw material was completely dissolved in water (115 mL, ultrasound), filtered, and neutralized by the dropwise addition of a saturated aqueous solution of sodium bicarbonate. The resulting solid was carefully washed with water and dried in vacuo to provide 1,5-dimethylisoquinolin-6-ol (2.49 g, 14.4 mmol, 89%) as a brownish solid. M.p. 248–250 °C (sublimation); UV (MeOH): λ = 234, 279, 301, 328, 382 nm; IR (ATR): ν = 2920, 2850, 2475 (br), 1808 (br), 1617, 1599, 1564, 1479, 1423, 1385, 1356, 1337, 1279, 1202, 1057, 1006, 939, 813, 774, 718, 672, 660 cm–1; 1H-NMR (500 MHz, methanol-d4): δ = 2.43 (s, 3H), 2.84 (s, 3H), 7.22 (d, J = 9.1 Hz, 1H), 7.64 (d, J = 6.2 Hz, 1H), 7.95 (d, J = 9.1 Hz, 1H), 8.12 (d, J = 6.2 Hz, 1H); 13C-NMR (125 MHz, methanol-d4): δ = 10.13 (CH3), 21.33 (CH3), 116.26 (C), 116.72 (CH), 119.91 (CH), 123.50 (C), 126.19 (CH), 138.79 (C), 140.60 (CH), 157.91 (C), 158.92 (C); MS (ESI, +10 V): m/z = 174.0 [M + H]+; HRMS: calcd. for C11H11NO: 173.0841, found: 173.0851; elemental analysis: calcd. for C11H11NO: C: 76.28, H: 6.40, N: 8.09; found: C: 76.00, H: 6.47, N: 8.21.
1,5-Dimethylisoquinolin-6-yl trifluoromethanesulfonate (14). Pyridine (1.1 mL, 12 mmol) was added to a suspension of 1,5-dimethylisoquinolin-6-ol (0.60 g, 3.5 mmol) in acetonitrile (66 mL) at 0 °C. Subsequently, trifluoromethanesulfonic anhydride (0.87 mL, 5.2 mmol) was added dropwise, and the reaction mixture was stirred at this temperature for 20 h. Ethyl acetate and water were added, and the layers were separated. The aqueous layer was extracted three times with ethyl acetate. The combined organic layers were washed with water and brine, and then dried (sodium sulfate). The solvent was evaporated, and the residue was purified by column chromatography (silica gel, pentane/ethyl acetate, 1:1) to provide 1,5-dimethylisoquinolin-6-yl trifluoromethanesulfonate (14, 0.92 g, 3.0 mmol, 87%) as a beige solid. M.p. 67–67.5 °C; UV (MeOH): λ = 198, 219, 274, 308, 321 nm; IR (ATR): ν = 3088, 3031, 2995, 2927, 2856, 1612, 1564, 1522, 1473, 1459, 1414, 1375, 1350, 1245, 1207, 1170, 1132, 1038, 994, 933, 861, 826, 815, 767, 663, 621 cm–1; 1H-NMR (500 MHz, CDCl3): δ = 2.68 (s, 3H), 3.00 (s, 3H), 7.48 (d, J = 9.3 Hz, 1H), 7.71 (d, J = 6.1 Hz, 1H), 8.10 (d, J = 9.3 Hz, 1H), 8.52 (d, J = 6.1 Hz, 1H); 13C-NMR (125 MHz, CDCl3): δ = 12.24 (CH3), 22.88 (CH3), 116.23 (CH), 118.77 (q, JC,F = 321 Hz, CF3), 120.75 (CH), 126.42 (CH and C), 126.64 (C), 136.97 (C), 143.40 (CH), 147.91 (C), 159.50 (C); 19F-NMR (282 MHz, CDCl3): δ = −73.58 (s, 3F); MS (EI): m/z (%) = 305 (1, [M]+), 172 (8), 144 (48), 128 (7), 115 (19), 103 (13), 89 (5), 77 (18), 69 (100), 63 (10), 51 (12); MS (ESI, +10 V): m/z = 306.0 [M + H]+; elemental analysis: calcd. for C12H10F3NO3S: C: 47.21, H: 3.30, N: 4.59, S: 10.50; found: C: 47.09, H: 3.02, N: 4.58, S: 10.45.
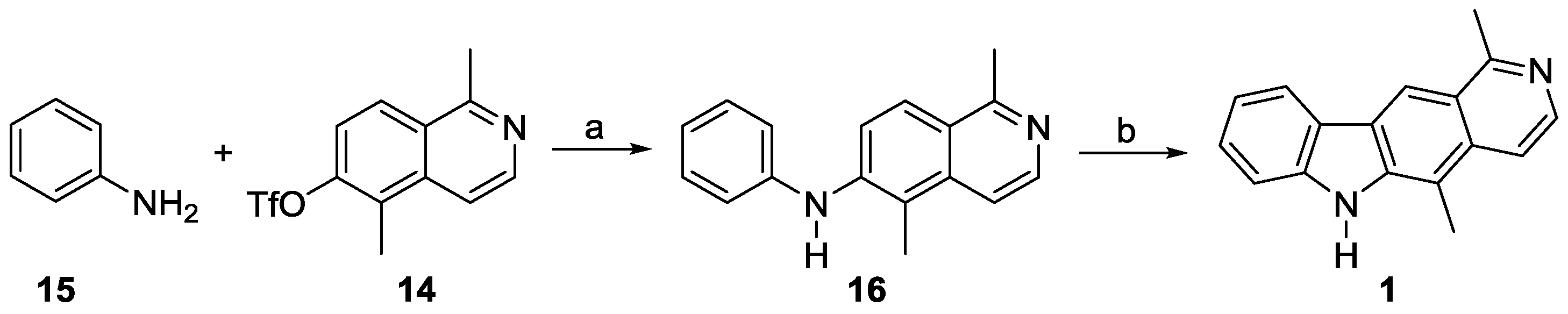
1,5-Dimethyl-N-phenylisoquinolin-6-amine (16). Aniline (15, 0.1 mL, 1.2 mmol) was added dropwise to a solution of 1,5-dimethylisoquinolin-6-yl trifluoromethanesulfonate (14, 0.235 g, 0.770 mmol), palladium(II) acetate (13 mg, 58 µmol), XPhos (55 mg, 0.12 mmol), and cesium carbonate (0.35 g, 1.1 mmol) in toluene (20 mL). The mixture was heated at reflux for 48 h. After cooling to room temperature, the reaction mixture was filtered over a short pad of Hyflo (ethyl acetate), and the solvent was evaporated. Purification of the residue by column chromatography (silica gel, dichloromethane/ethyl acetate 1:3 + 1% methanol) provided 1,5-dimethyl-N-phenylisoquinolin-6-amine (16, 0.19 g, 0.77 mmol, 100%) as a yellow solid. M.p. 175 °C (decomp.); UV (MeOH): λ = 223, 250, 280, 325, 358 (sh) nm; IR (ATR): ν = 3207, 3163, 3090, 3012, 2985, 2919, 2860, 1632, 1615, 1594, 1562, 1526, 1492, 1439, 1397, 1380, 1310, 1286, 1174, 1151, 1060, 990, 938, 864, 844, 819, 788, 748, 695, 678 cm–1; 1H-NMR (500 MHz, CDCl3): δ = 2.50 (s, 3H), 2.91 (s, 3H), 5.83 (br s, 1H), 7.01 (t, J = 7.4 Hz, 1H), 7.05 (d, J = 7.7 Hz, 2H), 7.29–7.33 (m, 2H), 7.53 (d, J = 9.1 Hz, 1H), 7.60 (d, J = 6.1 Hz, 1H), 7.92 (d, J = 9.1 Hz, 1H), 8.35 (d, J = 6.1 Hz, 1H); 13C-NMR (125 MHz, CDCl3): δ = 12.38 (CH3), 22.66 (CH3), 115.32 (CH), 118.78 (C), 118.88 (2 CH), 119.96 (CH), 121.99 (CH), 123.77 (C), 124.80 (CH), 129.63 (2 CH), 136.93 (C), 141.95 (C), 142.27 (CH), 142.91 (C), 158.59 (C); MS (EI): m/z (%) = 248 (100, [M]+), 233 (16), 171 (17); MS (ESI, +10 V): m/z = 249.1 [M + H]+; HRMS (ESI): calcd. for C17H16N2: 248.1313, found: 248.1310.
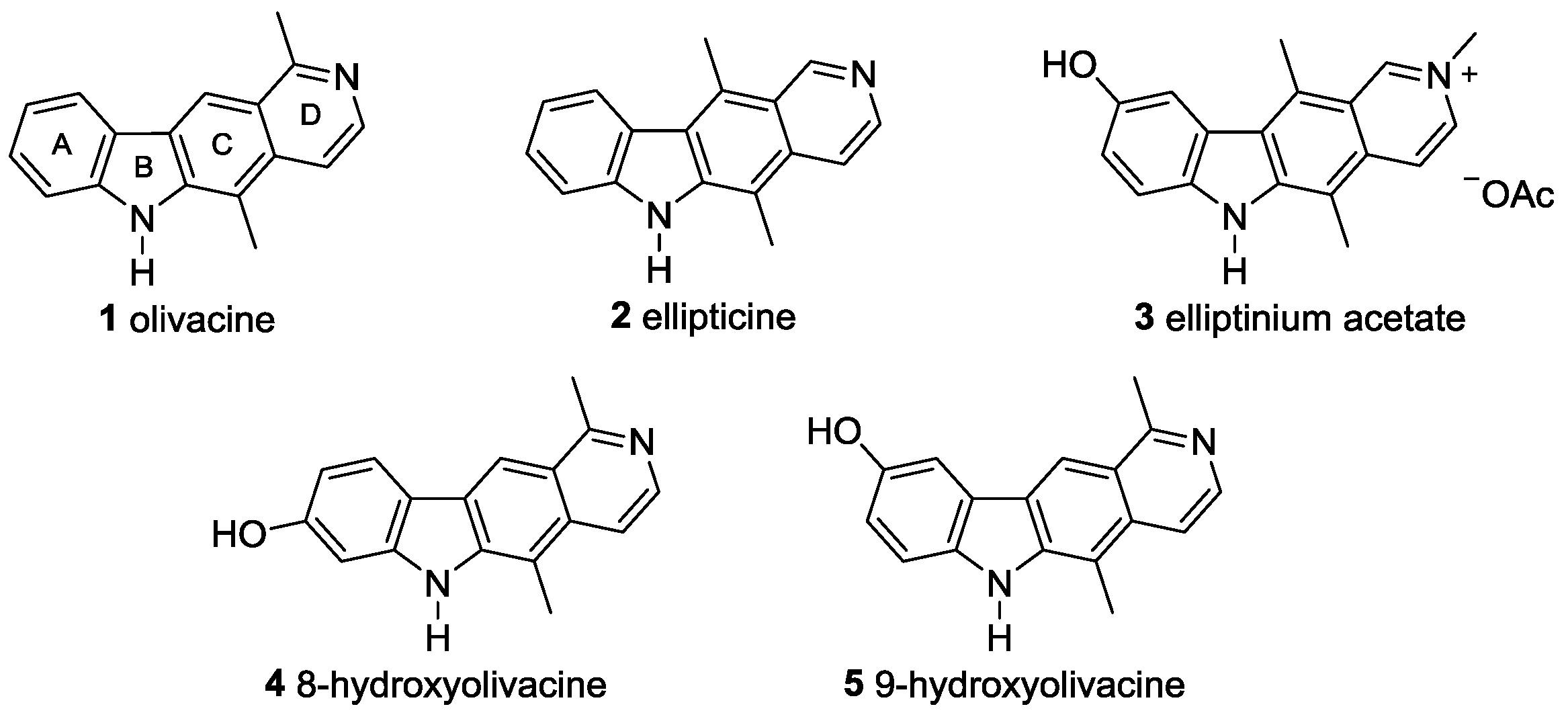
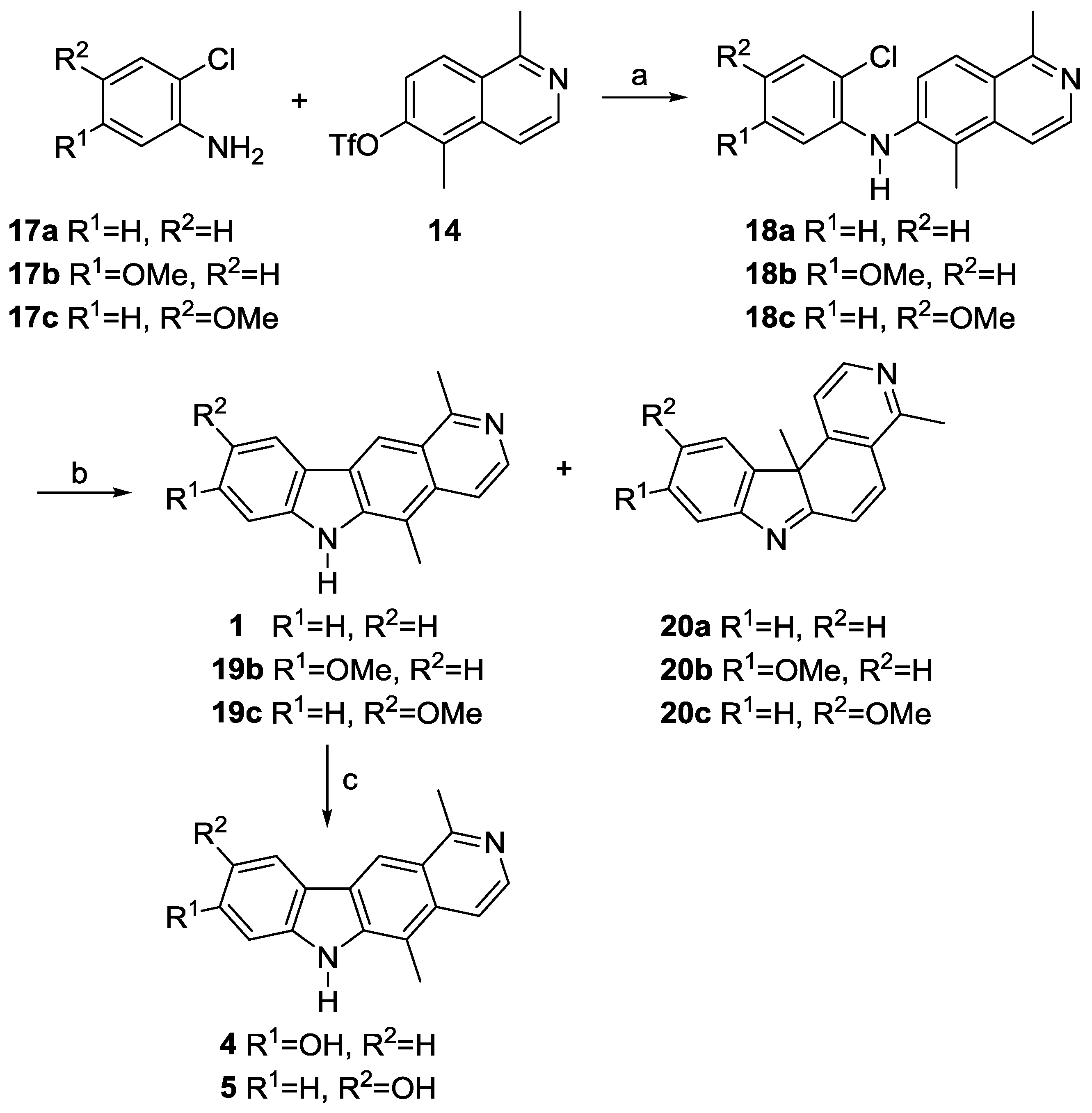
N-(2-Chlorophenyl)-1,5-dimethylisoquinolin-6-amine (18a). 2-Chloroaniline (17a, 78 µL, 0.74 mmol) was added dropwise to a solution of 1,5-dimethylisoquinolin-6-yl trifluoromethanesulfonate (14, 0.15 g, 0.49 mmol), palladium(II) acetate (8.3 mg, 37 µmol), XPhos (35 mg, 74 µmol), and cesium carbonate (224 mg, 0.688 mmol) in toluene (12 mL). The mixture was heated at reflux for 1.5 h. After cooling to room temperature, the reaction mixture was filtered over a short pad of Hyflo (ethyl acetate), and the solvent was evaporated. Purification of the residue by column chromatography (silica gel, dichloromethane/ethyl acetate, 9:1 to 0:1, each + 1% ethanol) provided N-(2-chlorophenyl)-1,5-dimethylisoquinolin-6-amine (18a, 0.130 g, 0.460 mmol, 94%) as brownish crystals. M.p. 194–198 °C; UV (MeOH): λ = 221, 249, 278, 320 nm; fluorescence (MeOH): λex = 221, λem = 229 (sh), 334 nm; IR (ATR): ν = 3189, 3078, 2955, 2919, 2850, 1589, 1567, 1542, 1501, 1474, 1452, 1396, 1367, 1307, 1294, 1267, 1225, 1198, 1129, 1058, 1033, 996, 933, 862, 843, 822, 793, 751, 706 cm–1; 1H-NMR (500 MHz, CDCl3): δ = 2.35 (s, 3H), 2.93 (s, 3H), 6.15 (br s, 1H), 6.85 (td, J = 7.7, 3.0 Hz, 1H), 6.95 (dd, J = 8.2, 1.4 Hz, 1H), 7.10–7.14 (m, 1H), 7.40 (dd, J = 8.0, 1.4 Hz, 1H), 7.51 (d, J = 9.0 Hz, 1H), 7.63 (d, J = 6.1 Hz, 1H), 7.96 (d, J = 9.0 Hz, 1H), 8.39 (d, J = 6.1 Hz, 1H); 13C-NMR (125 MHz, CDCl3): δ = 12.58 (CH3), 22.58 (CH3), 115.47 (CH), 116.05 (CH), 120.90 (CH), 121.87 (C), 122.07 (CH), 122.83 (C), 124.52 (C), 124.73 (CH), 127.51 (CH), 129.80 (CH), 136.78 (C), 140.07 (C), 140.19 (C), 142.35 (CH), 158.61 (C); MS (EI): m/z (%) = 282 (100, [M]+), 247 (74), 232 (29), 204 (15), 171 (12), 115 (10), 75 (11); MS (ESI, +25 V): m/z = 283.2 [M + H]+.
Crystal data: C17H15ClN2, crystal size 0.22 × 0.20 × 0.06 mm3, M = 282.76 g mol−1, monoclinic, space group: Cc, a = 11.700(2), b = 9.117(2), c = 14.024(3) Å, β = 110.73(3)°, V = 1399.1(5) Å3, Z = 4, ρcalcd. = 1.342 g cm−3, µ = 0.264 mm−1, T = 198(2) K, λ = 0.71073 Å, θ range: 3.11–27.00°, 20817 reflections collected, 3047 independent (Rint = 0.0534), 187 parameters. The structure was solved by direct methods and refined by the full-matrix least-squares method on F2; 2296 reflections observed, R1 = 0.0407, wR2 = 0.0805 [I > 2 σ(I)]; maximal residual electron density: 0.276 e Å−3. CCDC 1838728.
N-(2-Chloro-5-methoxyphenyl)-1,5-dimethylisoquinolin-6-amine (18b). 2-Chloro-5-methoxyaniline (17b, 92 µL, 0.74 mmol) was added dropwise to a solution of 1,5-dimethylisoquinolin-6-yl trifluoromethanesulfonate (14, 0.15 g, 0.49 mmol), palladium(II) acetate (8.3 mg, 37 µmol), XPhos (35 mg, 74 µmol), and cesium carbonate (224 mg, 0.688 mmol) in toluene (12 mL). The mixture was heated at reflux for five hours. After cooling to room temperature, the reaction mixture was filtered over a short pad of Hyflo (ethyl acetate), and the solvent was evaporated. Purification of the residue by column chromatography (silica gel, dichloromethane/ethyl acetate, 1:1 to 0:1, each + 1% ethanol) provided N-(2-chloro-5-methoxyphenyl)-1,5-dimethylisoquinolin-6-amine (18b, 0.141 g, 0.451 mmol, 92%) as a beige solid. M.p. 135–138 °C; UV (MeOH): λ = 224, 277, 322 nm; fluorescence (MeOH): λex = 224, λem = 301 (sh), 336 nm; IR (ATR): ν = 3416, 3068, 2998, 2929, 2853, 1596, 1508, 1447, 1421, 1383, 1343, 1312, 1287, 1230, 1207, 1170, 1138, 1069, 1027, 924, 820, 732, 671, 640 cm–1; 1H-NMR (500 MHz, CDCl3): δ = 2.53 (s, 3H), 2.94 (s, 3H), 3.68 (s, 3H), 6.13 (br s, 1H), 6.40 (dd, J = 8.8, 2.8 Hz, 1H), 6.47 (d, J = 2.8 Hz, 1H), 7.28 (d, J = 8.8 Hz, 1H), 7.53 (d, J = 9.0 Hz, 1H), 7.63 (d, J = 6.1 Hz, 1H), 7.97 (d, J = 9.0 Hz, 1H), 8.39 (d, J = 6.1 Hz, 1H); 13C-NMR (125 MHz, CDCl3): δ = 12.66 (CH3), 22.61 (CH3), 55.46 (CH3), 101.82 (CH), 105.94 (CH), 113.40 (C), 115.53 (CH), 122.55 (CH), 123.53 (C), 124.67 (C), 124.76 (CH), 130.02 (CH), 136.78 (C), 139.77 (C), 141.06 (CH), 142.38 (C), 158.64 (C), 159.20 (C); MS (EI): m/z (%) = 312 (100, [M]+), 277 (80), 262 (76), 247 (13), 233 (18), 219 (12), 139 (10), 117 (16), 63 (10); MS (ESI, +10 V): m/z = 313.3 [M + H]+.
N-(2-Chloro-4-methoxyphenyl)-1,5-dimethylisoquinolin-6-amine (18c). A solution of 2-chloro-4-methoxyaniline (17c, 127 mg, 0.806 mmol) in toluene (4 mL) was added dropwise to a solution of 1,5-dimethylisoquinolin-6-yl trifluoromethanesulfonate (14, 164 mg, 0.537 mmol), palladium(II) acetate (9 mg, 0.04 mmol), XPhos (38 mg, 81 µmol), and cesium carbonate (245 mg, 0.752 mmol) in toluene (10 mL). The mixture was heated at reflux for one hour. After cooling to room temperature, the reaction mixture was filtered over a short pad of Hyflo (ethyl acetate), and the solvent was evaporated. Purification of the residue by column chromatography (silica gel, dichloromethane/ethyl acetate, 9:1 to 0:1, each + 1% ethanol) provided N-(2-chloro-4-methoxyphenyl)-1,5-dimethylisoquinolin-6-amine (18c, 139 mg, 0.444 mmol, 83%) as a beige solid. M.p. 104–107 °C; UV (MeOH): λ = 226, 255, 318 nm; fluorescence (MeOH): λex = 255, λem = 422 nm; IR (ATR): ν = 3229, 3074, 2993, 2948, 2832, 1731, 1633, 1606, 1562, 1485, 1451, 1436, 1387, 1341, 1308, 1283, 1211, 1182, 1112, 1046, 936, 894, 864, 822, 789, 773, 689, 664 cm–1; 1H-NMR (500 MHz, CDCl3): δ = 2.51 (s, 3H), 2.93 (s, 3H), 3.80 (s, 3H), 5.87 (br s, 1H), 6.79 (dd, J = 8.7, 2.8 Hz, 1H), 7.02 (d, J = 2.8 Hz, 1H), 7.09 (d, J = 8.9 Hz, 1H), 7.29 (d, J = 9.1 Hz, 1H), 7.62 (d, J = 6.2 Hz, 1H), 7.90 (d, J = 9.1 Hz, 1H), 8.31 (d, J = 6.2 Hz, 1H); 13C-NMR (125 MHz, CDCl3): δ = 12.01 (CH3), 21.92 (CH3), 55.84 (CH3), 113.76 (CH), 115.29 (CH), 115.33 (CH), 117.69 (C), 118.89 (CH), 122.01 (CH), 123.18 (C), 124.98 (CH), 126.33 (C), 132.26 (C), 136.87 (C), 140.79 (CH, HSQC), 142.90 (C, HMBC), 155.61 (C), 158.06 (C); MS (EI): m/z (%) = 312 (100, [M]+), 297 (44), 277 (12), 262 (14), 233 (17), 169 (12), 155 (11), 128 (14), 116 (15); MS (ESI, +10 V): m/z = 313.2 [M + H]+; elemental analysis: calcd. for C18H17ClN2O: C: 69.12, H: 5.48, N: 8.96; found: C: 68.62, H: 5.72, N: 9.30.
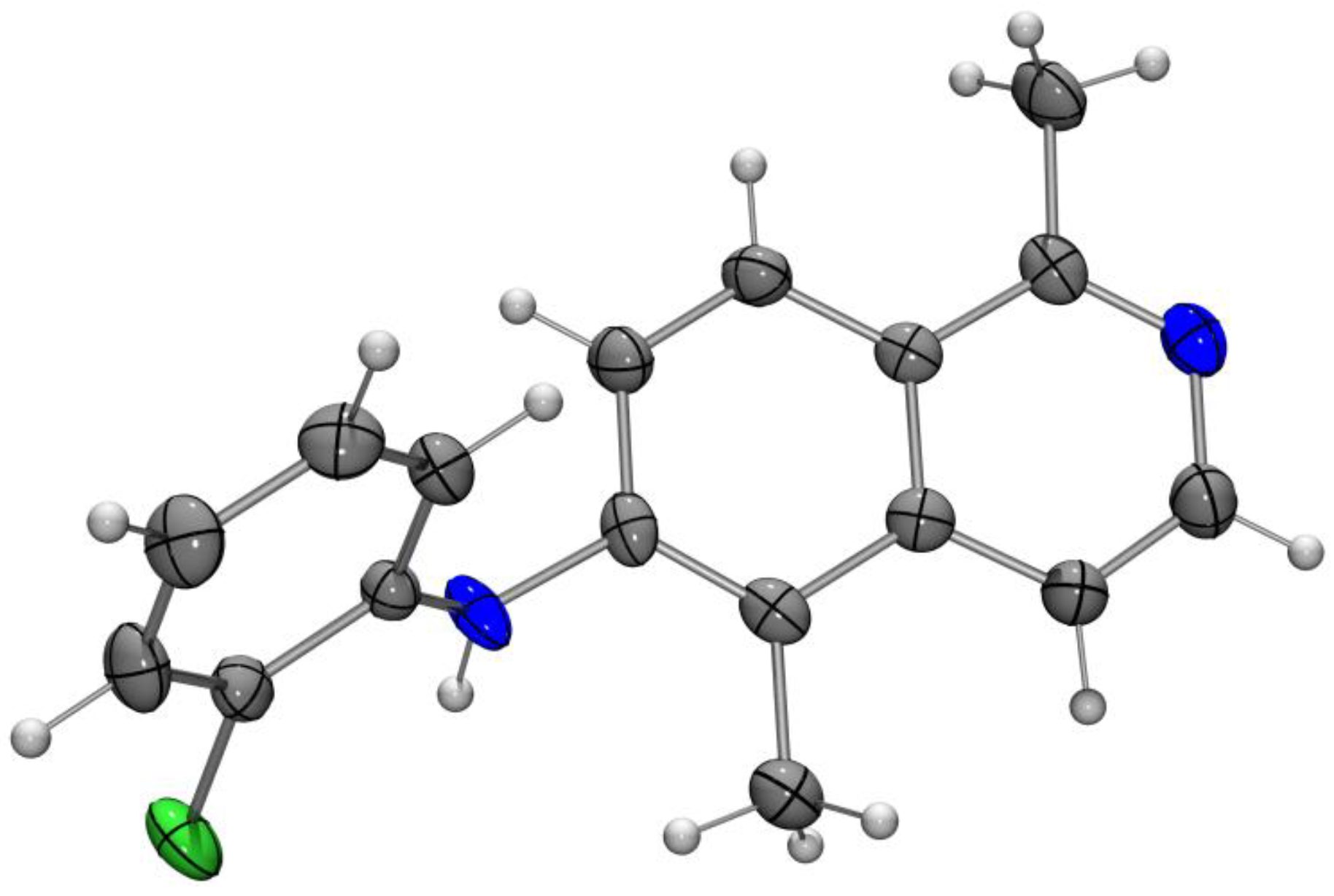
Olivacine (1). N-(2-Chlorophenyl)-1,5-dimethylisoquinolin-6-amine (18a, 20 mg, 71 µmol), palladium(II) acetate (4.8 mg, 21 µmol), tri-tert-butylphosphonium tetrafluoroborate (8.1 mg, 42 µmol), and potassium carbonate (39.1 mg, 0.283 mmol) were dissolved in DMF (0.5 mL). The reaction mixture was placed in a preheated oil bath at 140 °C and stirred for 30 min. After filtration over a short pad of Celite (CH2Cl2), the halogenated solvent was evaporated, and the residue was dissolved in ethyl acetate, and washed three times with water, and then with brine. The aqueous layer was extracted with ethyl acetate, and the combined organic layers were dried (sodium sulfate). The solvent was evaporated, and the residue was purified by column chromatography (silica gel, dichloromethane/ethyl acetate, 9:1 to 0:1, each + 5% ethanol) to provide olivacine (1, 12.4 mg, 50.3 µmol, 71%) as brown crystals. M.p. 320–324 °C; UV (MeOH): λ = 223, 237, 275, 285, 292, 327, 342, 374, 391 nm; fluorescence (MeOH): λex = 285, λem = 431 nm; IR (ATR): ν = 3058, 2965, 2909, 2765, 1674, 1597, 1479, 1467, 1407, 1334, 1311, 1280, 1252, 1222, 1196, 1150, 1108, 1064, 942, 862, 813, 765, 739, 695, 640 cm–1; 1H-NMR (500 MHz, methanol-d4): δ = 2.85 (s, 3H), 3.07 (t, JHD = 1.1 Hz) and 3.09 (s, 3H), 7.24–7.27 (m, 1H), 7.49–7.54 (m, 2H), 7.89 (d, J = 6.3 Hz, 1H), 8.18 (d, J = 6.3 Hz, 1H), 8.27–8.29 (m, 1H), 8.87 (s, 1H); 13C-NMR (125 MHz, methanol-d4): δ = 12.42 (CH3), 22.35 (CH3), 111.86 (CH), 112.42 (C), 116.05 (CH), 116.64 (CH), 120.58 (CH), 122.14 (CH), 123.57 (C), 124.41 (C), 127.25 (C), 128.93 (CH), 134.41 (C), 138.84 (CH), 142.80 (C), 144.34 (C), 160.29 (C); MS (EI): m/z (%) = 246 (100, [M]+), 229 (7), 217 (7), 204 (9), 123 (7); MS (ESI, +10 V): m/z = 247.1 [M + H]+; HRMS (ESI): calcd. for C17H14N2: 246.1157, found: 246.11537; elemental analysis: calcd. for C17H14N2: C: 82.90, H: 5.73, N: 11.37; found: C: 83.20, H: 5.81, N: 11.42.
Crystal data: C17H14N2·CH3OH, crystal size 0.45 × 0.12 × 0.07 mm3, M = 278.34 g mol−1, orthorhombic, space group: Pbca, a = 4.860(1), b = 21.337(5), c = 28.048(6) Å, V = 2908.5(11) Å3, Z = 8, ρcalcd. = 1.271 g cm−3, µ = 0.080 mm−1, T = 198(2) K, λ = 0.71073 Å, θ range: 3.48–25.40°, 60682 reflections collected, 2656 independent (Rint = 0.0501), 198 parameters. The structure was solved by direct methods and refined by the full-matrix least-squares method on F2; 1934 reflections observed, R1 = 0.0463, wR2 = 0.1044 [I > 2 σ(I)]; maximal residual electron density: 0.204 e Å−3. CCDC 1838729.
4,11b-Dimethyl-11bH-pyrido[3,4-c]carbazole (20a, 2.1 mg, 8.5 µmol, 12%), dark brown oil, less polar side product. UV (MeOH): λ = 250, 282 (sh), 359 nm; fluorescence (MeOH): λex = 250, λem = 417 nm; IR (ATR): ν = 3348, 2924, 2853, 2487, 1630, 1594, 1545, 1446, 1200, 1116, 950, 811, 772, 749, 679 cm–1; 1H-NMR (600 MHz, methanol-d4): δ = 1.65 (s, 3H), 2.76 (s, 3H), 7.01 (d, J = 10.0 Hz, 1H), 7.49 (t, J = 7.4 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.63 (d, J = 10.0 Hz, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.84 (d, J = 5.1 Hz, 1H), 8.09 (d, J = 7.3 Hz, 1H), 8.40 (d, J = 5.1 Hz, 1H); 13C-NMR (150 MHz, methanol-d4): δ = 21.70 (CH3), 33.28 (CH3), 59.3 (C, HMBC), 119.63 (CH), 122.46 (CH), 123.20 (CH), 125.25 (CH), 127.3 (C, HMBC), 127.89 (CH), 129.93 (CH), 136.11 (CH), 140.89 (C), 149.15 (CH), 153.38 (C), 155.0 (C, HMBC), 158.0 (C, HMBC), 184.5 (C, HMBC); MS (EI): m/z (%) = 246 (100, [M]+), 231 (24), 204 (12), 176 (7); MS (ESI, +50 V): m/z = 247.1 [M + H]+, 493.5 [2M + H]+.
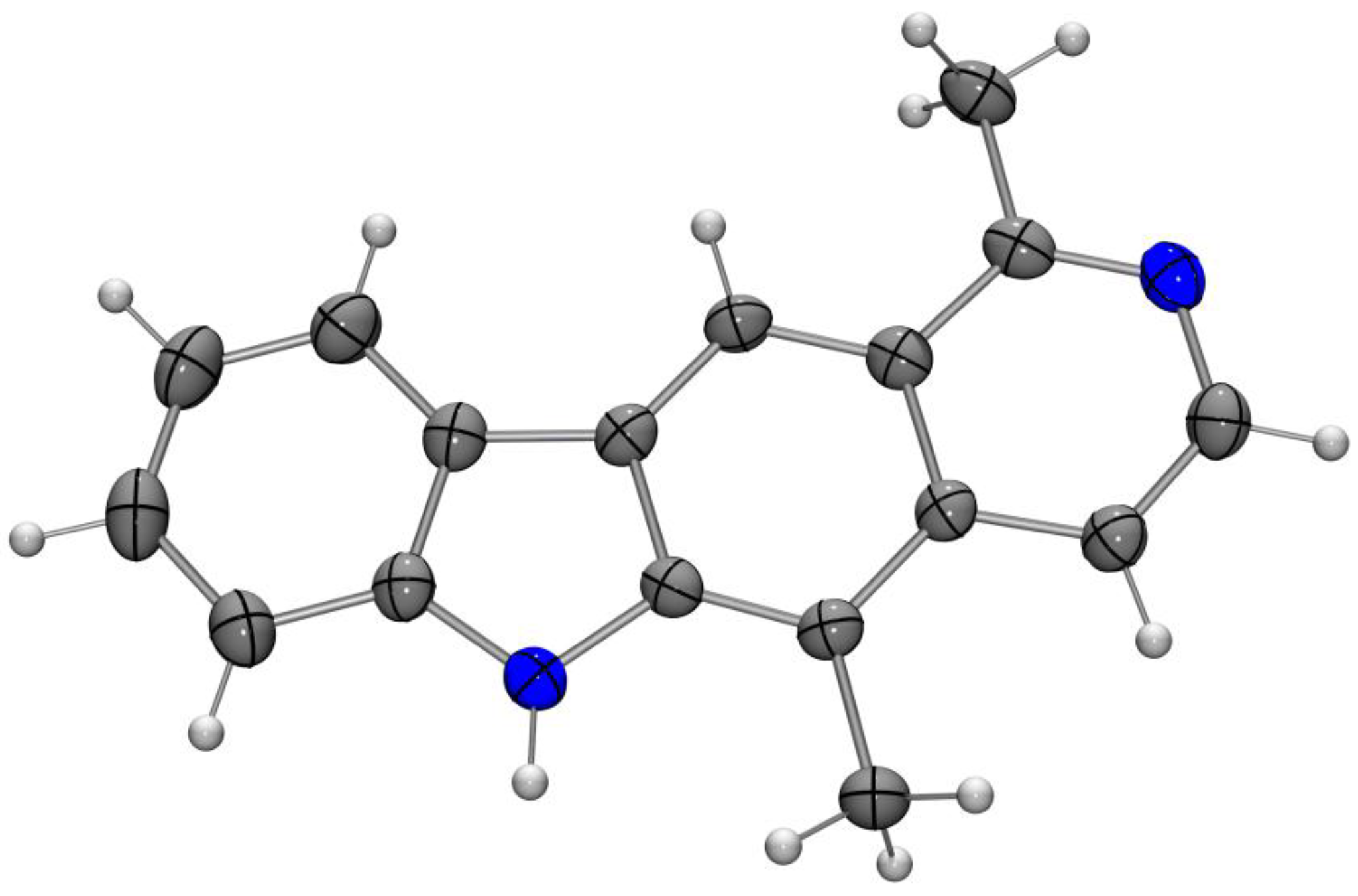
8-Methoxyolivacine (19b). N-(2-Chloro-5-methoxyphenyl)-1,5-dimethylisoquinolin-6-amine (18b, 14 mg, 45 µmol), palladium(II) acetate (3.0 mg, 13 µmol), tri-tert-butylphosphonium tetrafluoroborate (5.1 mg, 27 µmol) and potassium carbonate (24.7 mg, 0.179 mmol) were dissolved in DMF (0.5 mL). The reaction mixture was placed in a preheated oil bath at 140 °C and stirred for 20 min. After filtration over a short pad of Celite (CH2Cl2), the halogenated solvent was evaporated, and the residue was dissolved in ethyl acetate, washed three times with water, and then with brine. The aqueous layer was extracted with ethyl acetate, and the combined organic layers were dried (sodium sulfate). The solvent was evaporated and the residue was purified by column chromatography (silica gel, dichloromethane/ethyl acetate, 9:1 to 0:1, each + 5% ethanol) to provide 8-methoxyolivacine (19b, 8.0 mg, 29 µmol, 65%) as yellow crystals. M.p. 280–283 °C; UV (MeOH): λ = 227, 271, 281, 300, 316, 351 nm; fluorescence (MeOH): λex = 300, λem = 430, 515 nm; IR (ATR): ν = 3141, 3046, 2993, 2886, 2821, 2713, 1622, 1595, 1563, 1493, 1472, 1460, 1412, 1388, 1335, 1315, 1297, 1267, 1216, 1197, 1160, 1137, 1099, 1068, 1030, 996, 942, 916, 870, 810, 753 cm–1; 1H-NMR (500 MHz, DMSO-d6): δ = 2.79 (s, 3H), 3.01 (s, 3H), 3.89 (s, 3H), 6.85 (dd, J = 8.6, 2.2 Hz, 1H), 7.00 (d, J = 2.2 Hz, 1 H), 7.78 (d, J = 6.1 Hz, 1H), 8.23 (d, J = 6.1 Hz, 1H), 8.24 (d, J = 8.6 Hz, 1 H), 8.77 (s, 1H), 11.26 (s, 1H); 13C-NMR (125 MHz, DMSO-d6): δ = 12.36 (CH3), 22.97 (CH3), 55.32 (CH3), 94.84 (CH), 107.55 (CH), 110.68 (C), 113.55 (CH), 114.78 (CH), 116.19 (C), 122.00 (C), 122.28 (CH), 125.00 (C), 131.74 (C), 139.10 (CH), 140.79 (C), 144.13 (C), 158.26 (C), 159.96 (C); MS (EI): m/z (%) = 276 (100, [M]+), 261 (14), 233 (49), 138 (8), 116 (10); MS (ESI, +10 V): m/z = 277.1 [M + H]+; HRMS (ESI): calcd. for C18H16N2O: 276.1263, found: 276.1261.
Crystal data: C18H16N2O·CH3OH, crystal size 0.160 × 0.080 × 0.060 mm3, M = 308.37 g mol−1, orthorhombic, space group: Pbca, a = 4.9253(3), b = 21.4925(15), c = 29.523(2) Å, V = 3125.3(4) Å3, Z = 8, ρcalcd. = 1.311 g cm−3, µ = 0.685 mm−1, T = 150(2) K, λ = 1.54178 Å, θ range: 2.993–68.188°, 31382 reflections collected, 2811 independent (Rint = 0.0544), 230 parameters. The structure was solved by direct methods and refined by the full-matrix least-squares method on F2; 2283 reflections observed, R1 = 0.0387, wR2 = 0.1001 [I > 2 σ(I)]; maximal residual electron density: 0.221 e Å−3. CCDC 1838730.
9-Methoxy-4,11b-dimethyl-11bH-pyrido[3,4-c]carbazole (20b, 1.1 mg, 4.0 µmol, 9%), brown oil, less polar side product. UV (MeOH): λ = 221, 300, 325 nm; fluorescence (MeOH): λex = 221, λem = 296, 339 nm; IR (ATR): ν = 3414, 3058, 2924, 2855, 1734, 1655, 1632, 1593, 1535, 1484, 1459, 1437, 1377, 1334, 1276, 1231, 1182, 1149, 1129, 1074, 935, 826, 740, 683 cm–1; 1H-NMR (600 MHz, methanol-d4): δ = 1.62 (s, 3H), 2.74 (s, 3H), 3.94 (s, 3H), 6.98 (d, J = 10.0 Hz, 1H), 7.04 (dd, J = 8.2, 1.7 Hz, 1H), 7.26 (s, 1H), 7.61 (d, J = 10.0 Hz, 1H), 7.78 (d, J = 4.9 Hz, 1H), 7.94 (d, J = 8.2 Hz, 1H), 8.38 (d, J = 4.9 Hz, 1H); 13C-NMR (150 MHz, methanol-d4): δ = 21.67 (CH3), 33.43 (CH3), 56.12 (CH3), 58.80 (C), 108.13 (CH), 113.67 (CH), 119.73 (CH), 123.16 (CH), 125.46 (CH), 127.23 (C), 132.78 (C, HMBC), 136.16 (CH), 149.15 (CH), 153.81 (C), 156.57 (C, HMBC), 158.04 (C), 162.22 (C), 186.18 (C); MS (EI): m/z (%) = 276 (85, [M]+), 261 (100), 233 (25), 218 (52), 190 (16); MS (ESI, +50 V): m/z = 277.2 [M + H]+.
9-Methoxyolivacine (19c). N-(2-Chloro-4-methoxyphenyl)-1,5-dimethylisoquinolin-6-amine (18c, 55.0 mg, 176 µmol), palladium(II) acetate (11.8 mg, 53 µmol), tri-tert-butylphosphonium tetrafluoroborate (20.1 mg, 106 µmol), and potassium carbonate (97.2 mg, 0.703 mmol) were dissolved in DMF (1.4 mL). The reaction mixture was placed in a preheated oil bath at 140 °C and stirred for 35 min. After filtration over a short pad of Celite (CH2Cl2), the halogenated solvent was evaporated, and the residue was dissolved in ethyl acetate, and then washed three times with water, and then with brine. The aqueous layer was extracted with ethyl acetate, and the combined organic layers were dried (sodium sulfate). The solvent was evaporated, and the residue was purified by column chromatography (silica gel, dichloromethane/ethyl acetate, 9:1 to 0:1, each + 5% ethanol) to provide 9-methoxyolivacine (19c, 30.1 mg, 109 µmol, 62%) as a yellow solid. M.p. 273–274 °C; UV (MeOH): λ = 224, 242, 272, 296, 332, 394 nm; fluorescence (MeOH): λex = 296, λem = 471 nm; IR (ATR): ν = 3143, 2914, 1632, 1600, 1485, 1436, 1405, 1380, 1330, 1306, 1265, 1206, 1175, 1104, 1030, 935, 879, 862, 838, 809, 767, 735, 698 cm–1; 1H-NMR (500 MHz, DMSO-d6): δ = 2.80 (s, 3H), 3.04 (s, 3H), 3.90 (s, 3H), 7.14 (dd, J = 10.0, 2.5 Hz, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.79 (d, J = 6.1 Hz, 1H), 8.01 (d, J = 2.5 Hz, 1H), 8.24 (d, J = 6.1 Hz, 1H), 8.96 (s, 1H), 11.16 (s, 1H); 13C-NMR (125 MHz, DMSO-d6): δ = 12.80 (CH3), 23.45 (CH3), 56.11 (CH3), 104.87 (CH), 111.36 (C), 112.00 (CH), 115.18 (CH), 115.71 (CH), 117.04 (CH), 122.00 (C), 123.65 (C), 125.36 (C), 132.64 (C), 137.53 (C), 139.69 (CH), 141.61 (C), 153.78 (C), 159.21 (C); MS (EI): m/z (%) = 276 (100, [M]+), 261 (90), 233 (27), 116 (10); MS (ESI, +10 V): m/z = 277.1 [M + H]+; HRMS (ESI): calcd. for C18H16N2O: 276.1263, found: 276.1269.
10-Methoxy-4,11b-dimethyl-11bH-pyrido[3,4-c]carbazole (20c, 1.4 mg, 5.0 µmol, 3%), brown oil, less polar side product. UV (MeOH): λ = 260, 291 (sh), 381 nm; fluorescence (MeOH): λex = 260, λem = 349 (sh), 434 nm; IR (ATR): ν = 3389, 2924, 2854, 1733, 1655, 1624, 1590, 1536, 1466, 1434, 1380, 1335, 1295, 1275, 1240, 1218, 1165, 1065, 1030, 952, 865, 822, 744, 677 cm–1; 1H-NMR (600 MHz, methanol-d4): δ = 1.63 (s, 3H), 2.74 (s, 3H), 4.00 (s, 3H), 6.95 (d, J = 10.0 Hz, 1H), 7.10 (dd, J = 8.5, 2.1 Hz, 1H), 7.55 (d, J = 10.0 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.66 (d, J = 2.1 Hz, 1H), 7.83 (d, J = 5.1 Hz, 1H), 8.38 (d, J = 5.1 Hz, 1H); 13C-NMR (150 MHz, methanol-d4): δ = 21.69 (CH3), 33.33 (CH3), 56.44 (CH3), 59.25 (C), 112.13 (CH), 114.60 (CH), 119.52 (CH), 122.93 (CH), 123.26 (CH), 127.5 (C, HMBC), 134.87 (CH), 142.7 (C, HMBC), 147.3 (C, HMBC), 148.89 (CH), 153.2 (C, HMBC), 157.8 (C, HMBC), 160.73 (C), 182.4 (C, HMBC); MS (EI): m/z (%) = 276 (100, [M]+), 261 (42), 246 (24), 233 (46), 218 (31), 190 (13); MS (ESI, +50 V): m/z = 277.2 [M + H]+.
8-Hydroxyolivacine (4). 8-Methoxyolivacine (19b, 17.0 mg, 61.5 µmol) was dissolved in 48% aqueous HBr (1.1 mL), and the mixture was heated at reflux for 24 h. After cooling to room temperature, the mixture was carefully neutralized using a 25% aqueous solution of ammonia. The mixture was extracted with ethyl acetate until the aqueous layer was completely colorless. Evaporation of the organic solvent led to a yellow solid, which was purified by chromatography (Alox N, 5% H2O, CH2Cl2/methanol, 1:1) to provide 8-hydroxyolivacine (4, 13.5 mg, 51.5 µmol, 84%) as a yellow solid. An additional purification by preparative HPLC provided very pure 4 (8.5 mg, 32 µmol) for biological testing. M.p. 239 °C; UV (MeOH): λ = 239, 301, 317 nm; fluorescence (MeOH): λex = 301, λem = 434, 520 nm; IR (ATR): ν = 3505, 3279, 3198, 2827, 1660, 1619, 1474, 1433, 1407, 1341, 1190, 1166, 1138, 1102, 840, 800, 722, 633 cm–1; 1H-NMR (500 MHz, methanol-d4): δ = 2.94 (s, 3H), 3.34 (s, 3H), 6.90 (dd, J = 8.5, 2.1 Hz, 1H), 7.01 (d, J = 2.1 Hz, 1H), 8.18 (d, J = 7.0 Hz, 1H), 8.20 (d, J = 8.5 Hz, 1H), 8.37 (d, J = 7.0 Hz, 1H), 9.00 (s, 1H); 13C-NMR (125 MHz, methanol-d4): δ = 12.41 (CH3), 18.63 (CH3), 98.31 (CH), 111.38 (CH), 113.55 (C), 115.93 (C), 116.93 (CH), 119.58 (CH), 121.73 (C), 123.95 (CH), 127.12 (CH), 130.23 (C), 134.44 (C), 146.42 (2C), 157.30 (C), 161.11 (C); MS (EI): m/z (%) = 262 (100, [M]+), 180 (10); MS (ESI, +10 V): m/z = 263.1 [M + H]+, 547 [2M + Na]+; HRMS (ESI): calcd. for C17H14N2O: 262.1106, found: 262.1104.
9-Hydroxyolivacine (5). 9-Methoxyolivacine (19c, 38.0 mg, 138 µmol) was dissolved in 48% aqueous HBr (2.3 mL), and the mixture was heated at reflux for 24 h. After cooling to room temperature, the mixture was carefully neutralized using a 25% aqueous solution of ammonia. The mixture was extracted with ethyl acetate until the aqueous layer was colorless. The combined organic layers were washed with water and brine, and then dried (sodium sulfate), and the solvent was evaporated. The residue was purified by column chromatography (silica gel, CH2Cl2/THF, 4:1 to 2:3) to provide 9-hydroxyolivacine (5, 25.2 mg, 96.1 µmol, 70%) as a yellow solid. An additional purification by preparative HPLC provided very pure 5 (6.1 mg, 23 µmol) for biological testing. M.p. 249 °C; UV (MeOH): λ = 245, 274, 311, 355, 375 nm; fluorescence (MeOH): λex = 311, λem = 482 nm; IR (ATR): ν = 3220, 2921, 2853, 1734, 1666, 1611, 1425, 1328, 1288, 1185, 1127, 975, 840, 799, 721 cm–1; 1H-NMR (500 MHz, methanol-d4): δ = 2.96 (s, 3H), 3.36 (s, 3H, HSQC), 7.22 (dd, J = 8.6, 2.3 Hz, 1H), 7.51 (d, J = 8.6 Hz, 1H), 7.82 (d, J = 2.3 Hz, 1H), 8.19 (d, J = 7.1 Hz, 1H), 8.38 (d, J = 7.1 Hz, 1H), 9.17 (s, 1H); 13C-NMR (125 MHz, methanol-d4): δ = 12.42 (CH3), 18.67 (CH3), 107.97 (CH), 113.13 (CH), 113.96 (C), 119.43 (CH), 119.48 (CH), 119.54 (CH), 121.00 (C), 124.35 (C), 127.40 (CH), 129.67 (C), 134.68 (C), 138.24 (C), 146.56 (C), 153.52 (C), 158.18 (C); MS (EI): m/z (%) = 262 (100, [M]+), 131 (12); MS (ESI, +10 V): m/z = 263.1 [M + H]+; HRMS (ESI): calcd. for C17H14N2O: 262.1106, found: 262.1107.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}