MS/MS-Guided Isolation of Clarinoside, a New Anti-Inflammatory Pentalogin Derivative

 , ,
, , 

Abstract
:1. Introduction
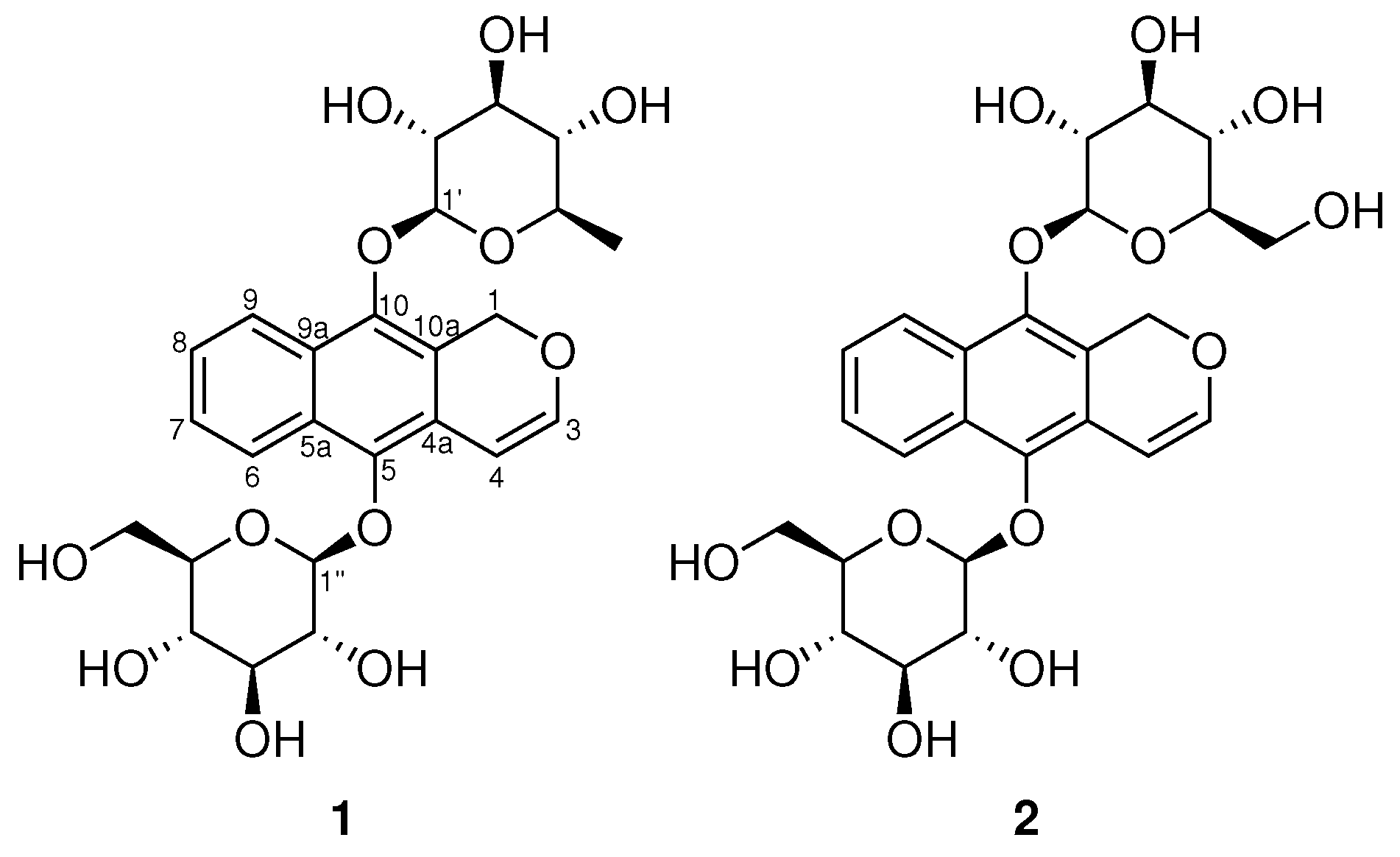
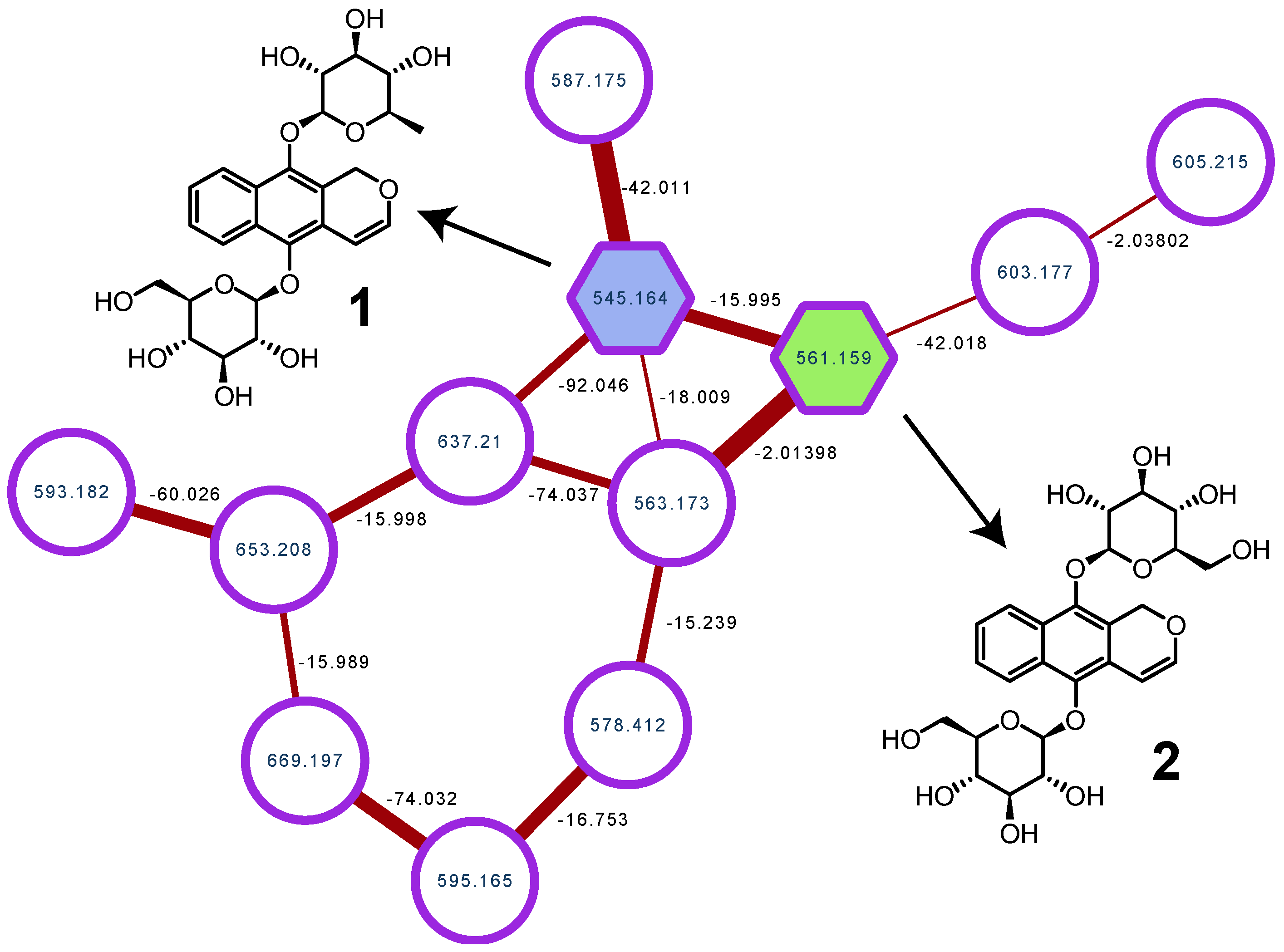
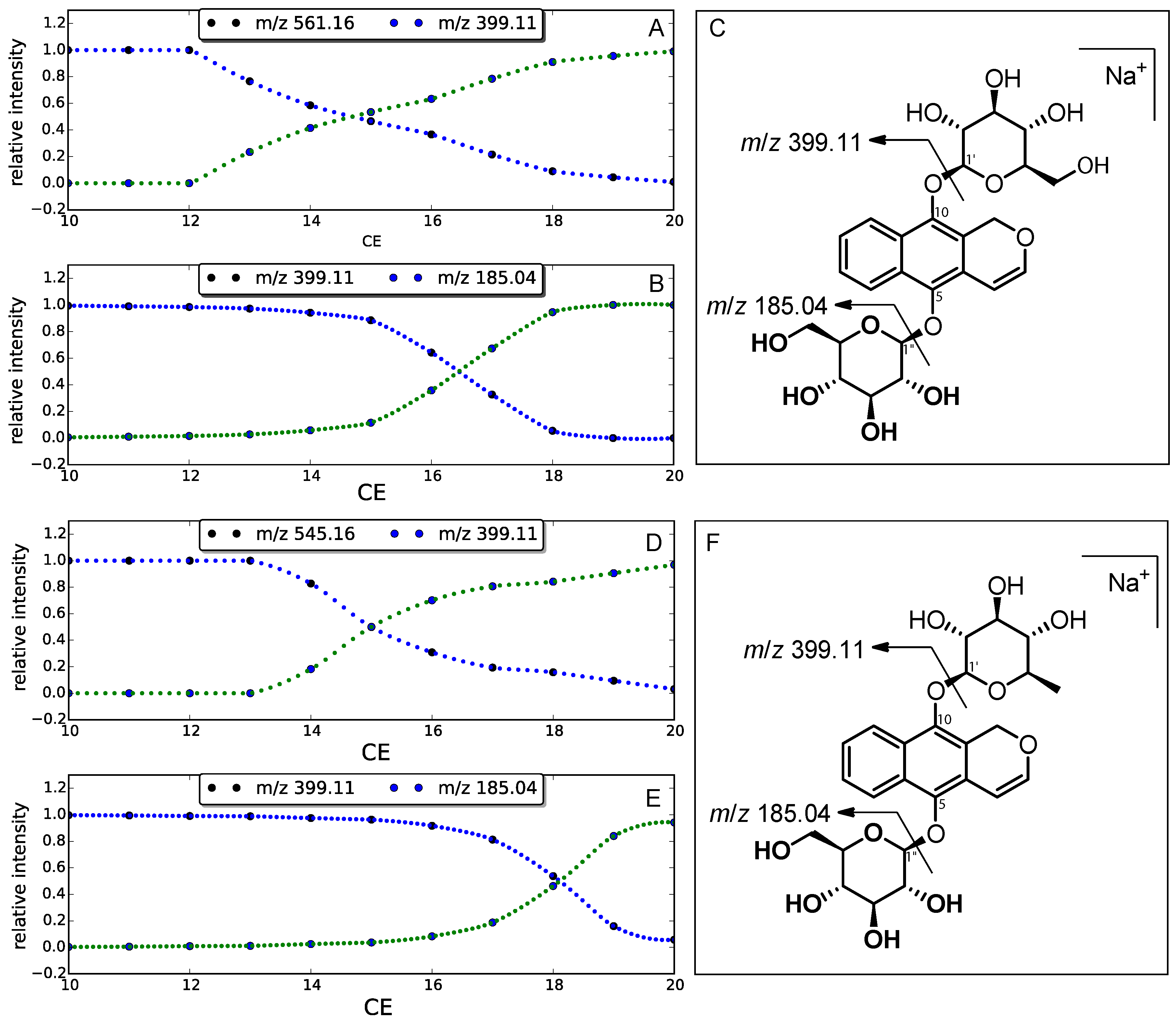
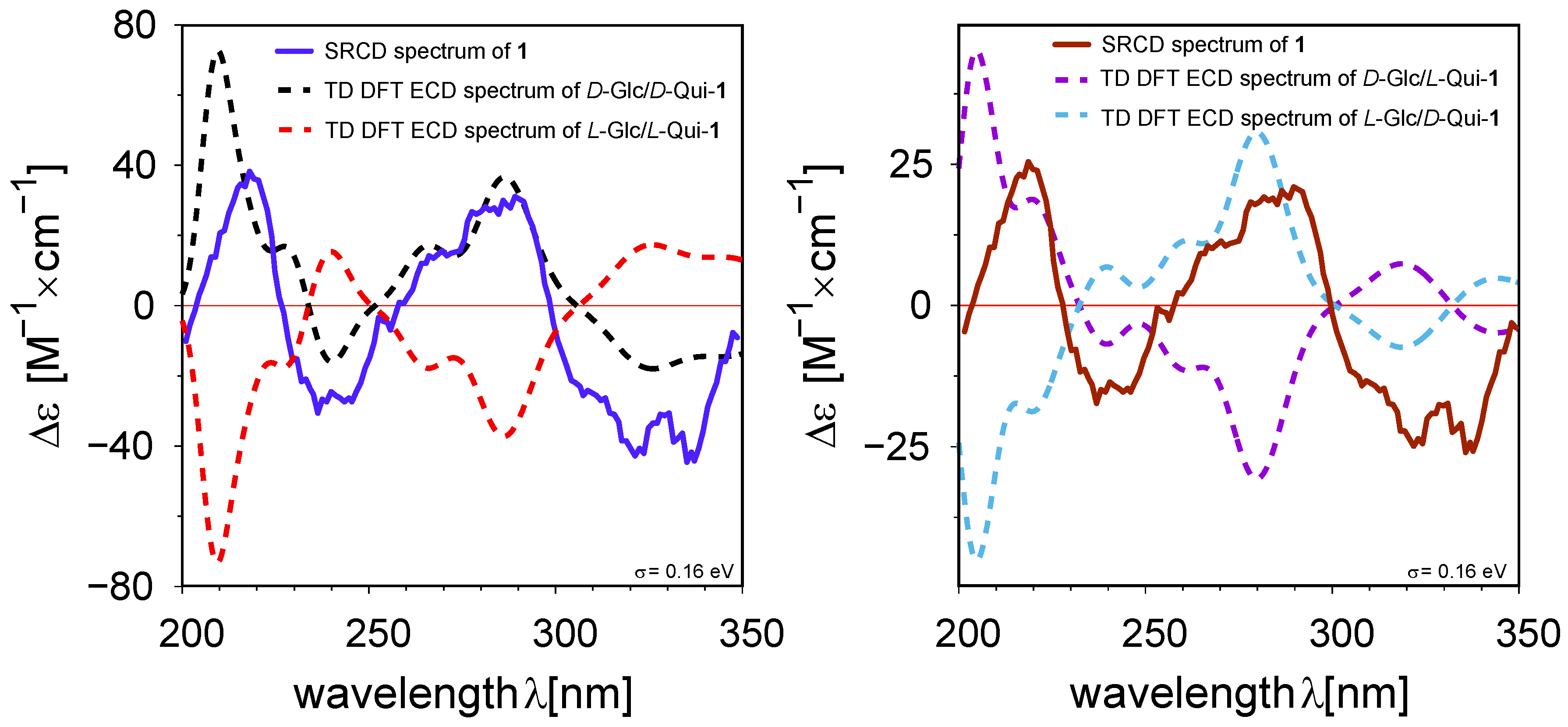
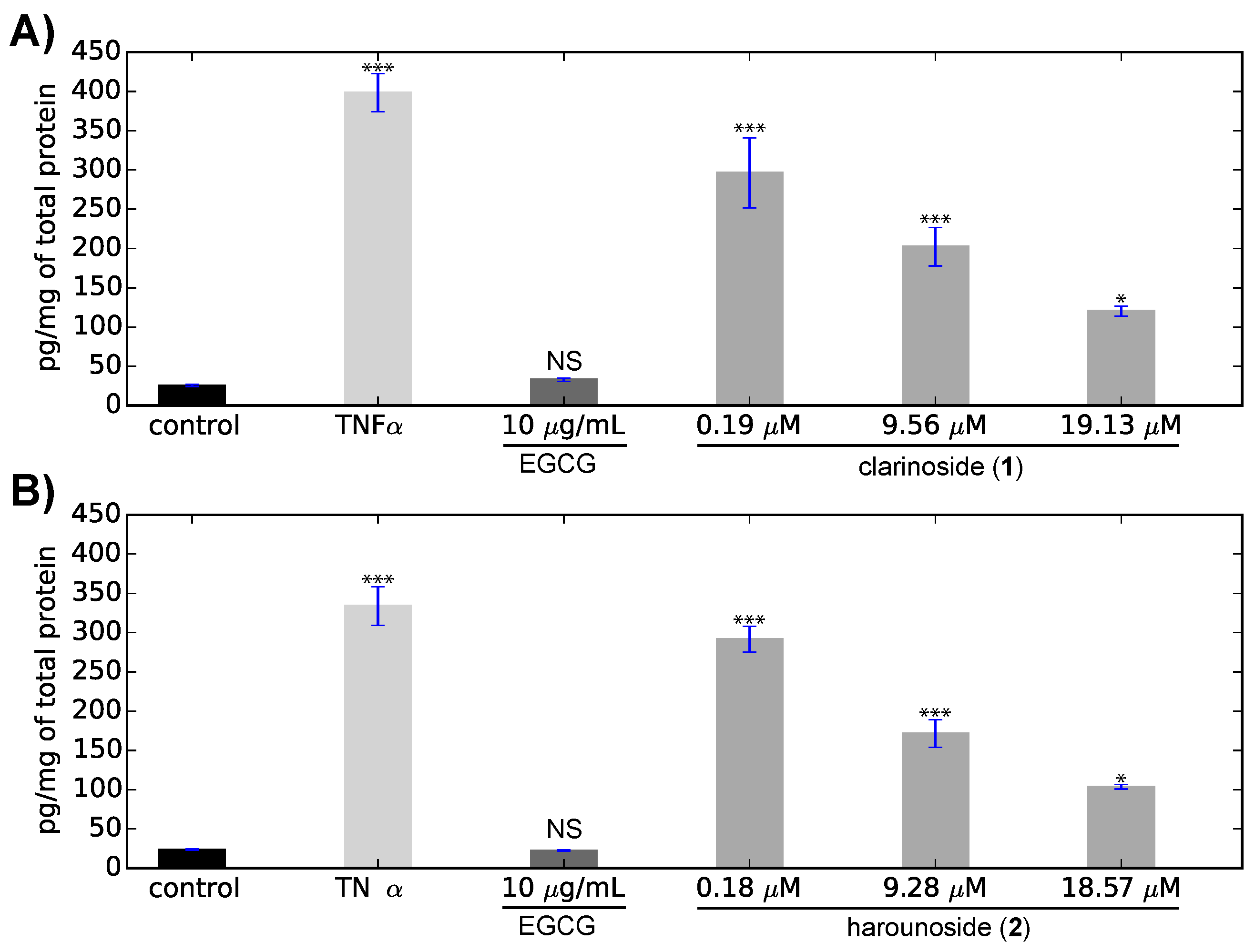
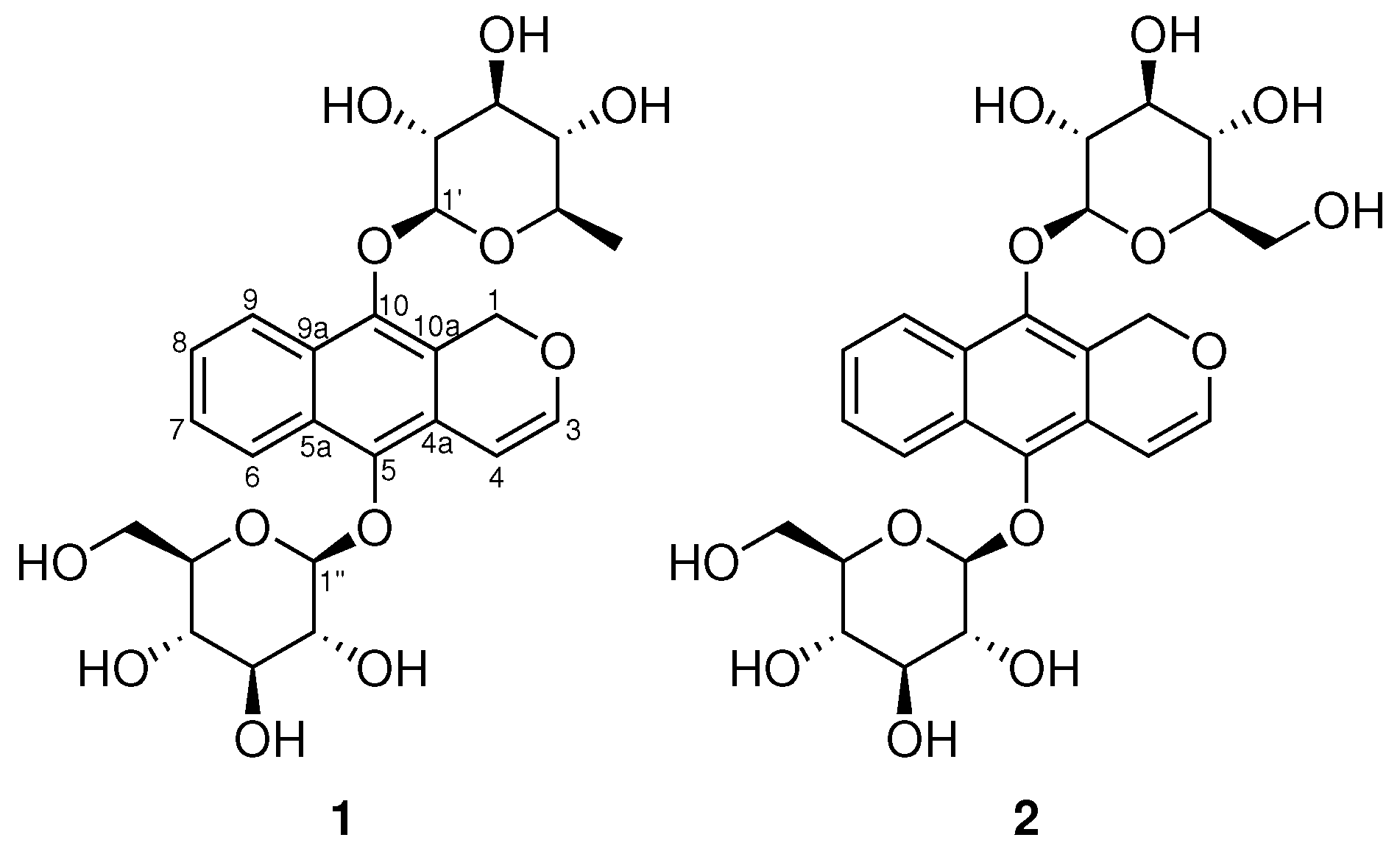
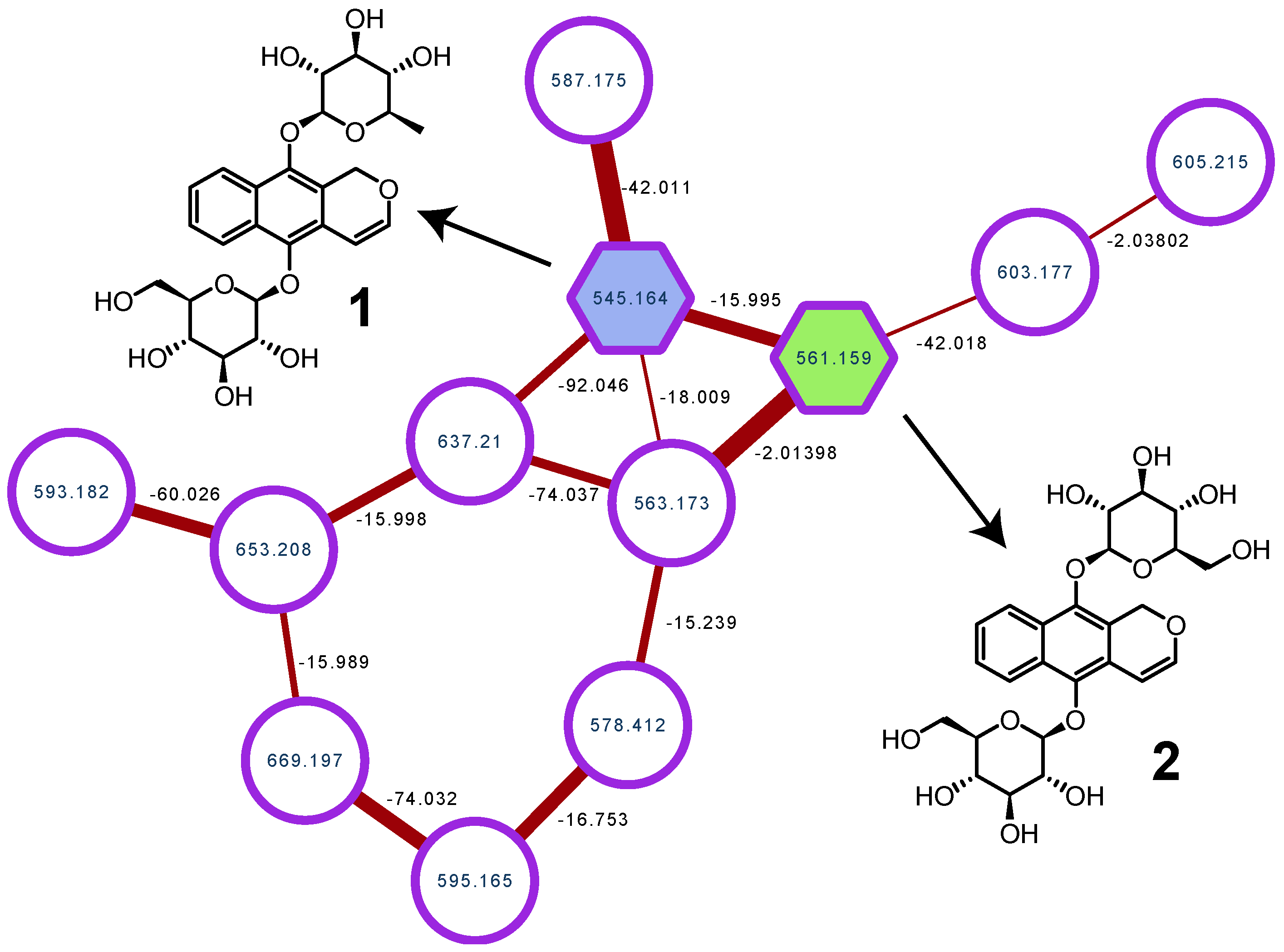
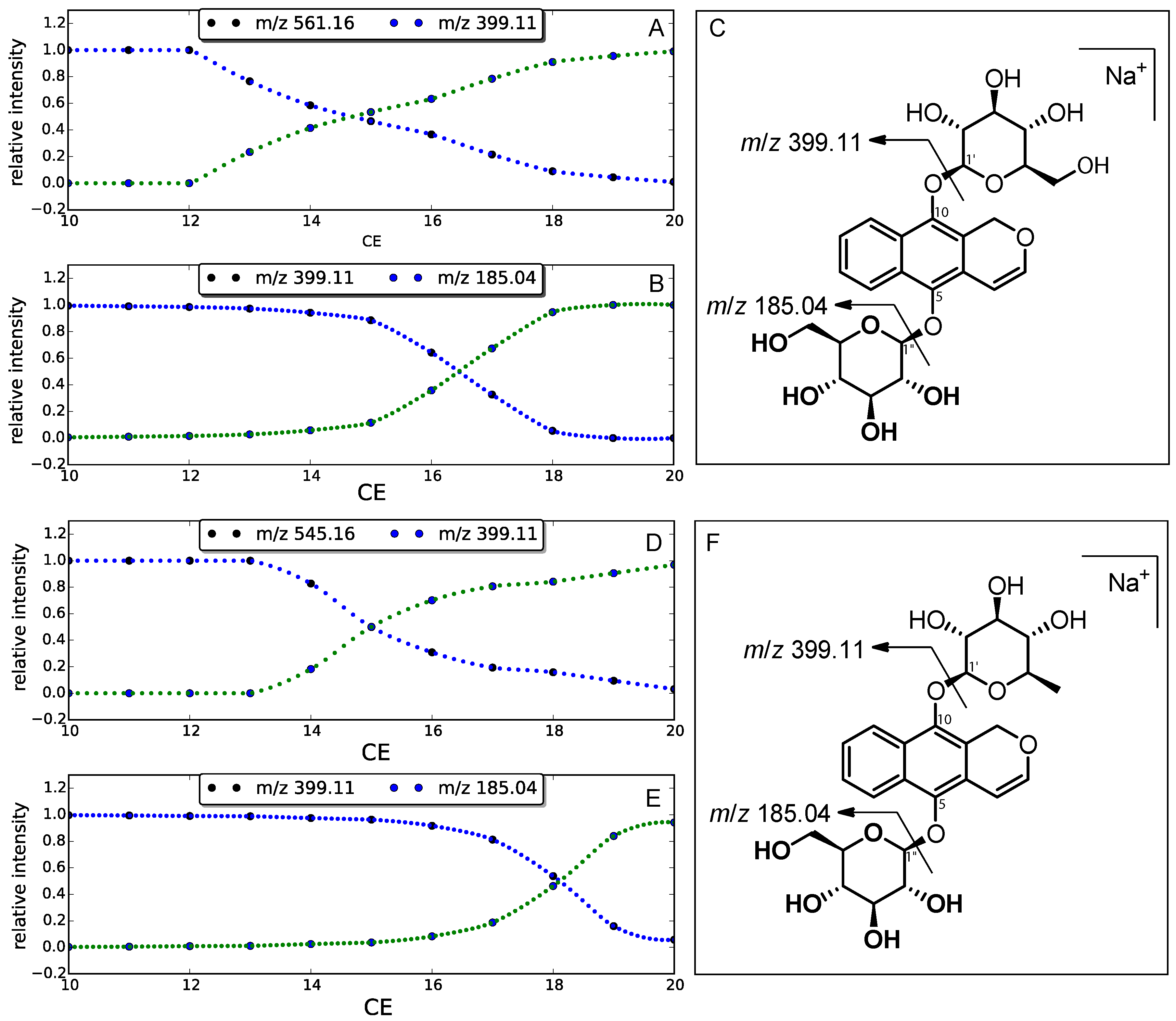
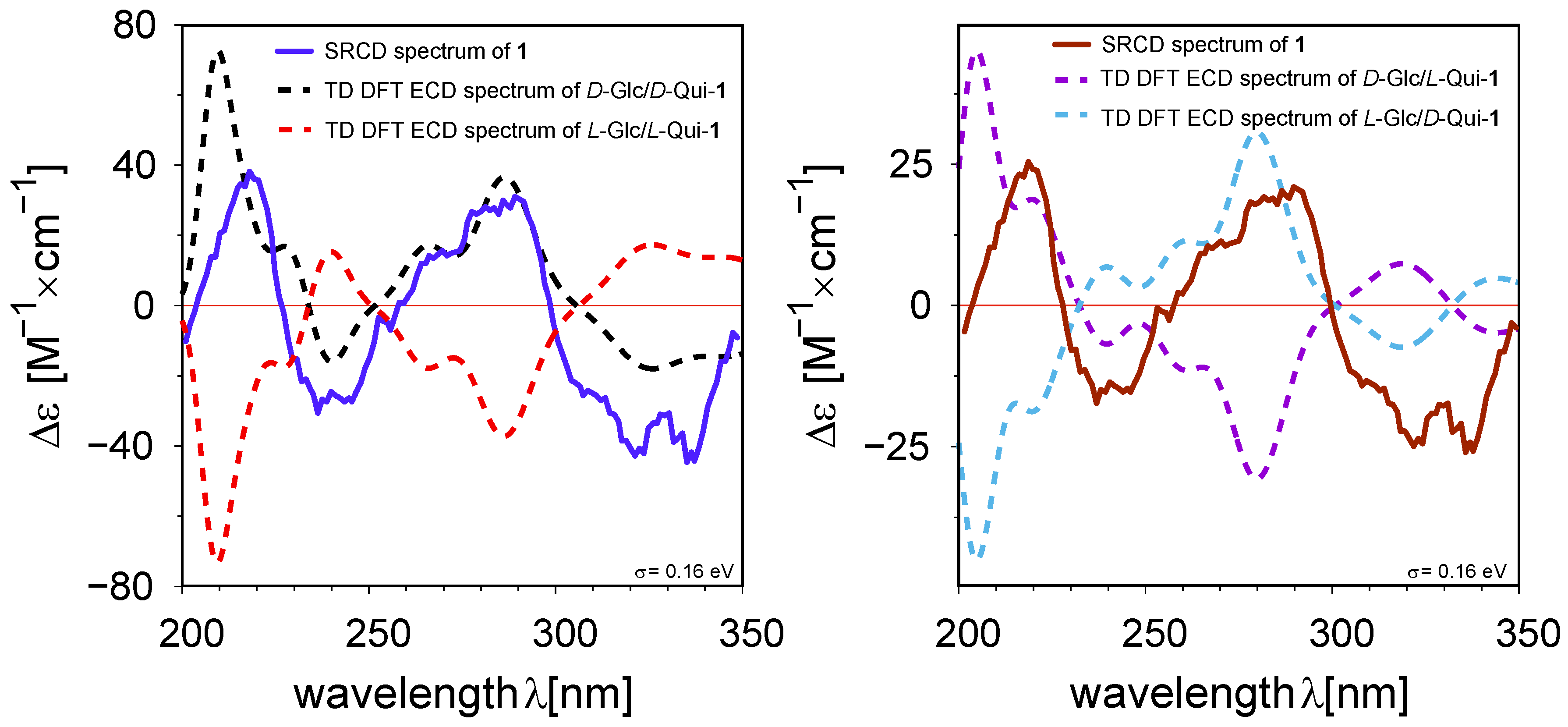
2. Results
3. Materials and Methods
3.1. General Procedure
3.2. Plant Material
3.3. Extraction and Purification
3.4. LC-MS Data Acquisition and Processing
3.5. Construction of the Molecular Network
3.6. Energy-Resolved Mass Spectrometry
3.7. Synchrotron Radiation Circular Dichroism
3.8. Computational Details
3.9. Cell Culture
3.10. Interleukine Release Measurement
3.11. Statistical Analyses
3.12. Compound Characterization
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References and Note
- Olivon, F.; Allard, P.M.; Koval, A.; Righi, D.; Genta-Jouve, G.; Neyts, J.; Apel, C.; Pannecouque, C.; Nothias, L.F.; Cachet, X.; et al. Bioactive Natural Products Prioritization Using Massive Multi-informational Molecular Networks. ACS Chem. Biol. 2017, 12, 2644–2651. [Google Scholar] [CrossRef] [PubMed]
- Nothias, L.F.; Nothias-Esposito, M.; da Silva, R.; Wang, M.; Protsyuk, I.; Zhang, Z.; Sarvepalli, A.; Leyssen, P.; Touboul, D.; Costa, J.; et al. Bioactivity-Based Molecular Networking for the Discovery of Drug Leads in Natural Product Bioassay-Guided Fractionation. J. Nat. Prod. 2018, 81, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Ekpendu, T.O.; Akah, P.A.; Adesomoju, A.A.; Okogun, J.I. Antiinflammatory and Antimicrobial Activities of Mitracarpus scaber Extracts. Int. J. Pharmacogn. 1994, 32, 191–196. [Google Scholar] [CrossRef]
- Bisignano, G.; Sanogo, R.; Marino, A.; Aquino, R.; D’angelo, V.; Germano, M.P.; De Pasquale, R.; Pizza, C. Antimicrobial activity of Mitracarpus scaber extract and isolated constituents. Lett. Appl. Microbiol. 2000, 30, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Gbaguidi, F.; Accrombessi, G.; Moudachirou, M.; Quetin-Leclercq, J. HPLC quantification of two isomeric triterpenic acids isolated from Mitracarpus scaber and antimicrobial activity on Dermatophilus congolensis. J. Pharm. Biomed. Anal. 2005, 39, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Okunade, A.L.; Clark, A.M.; Hufford, C.D.; Oguntimein, B.O. Azaanthraquinone: An Antimicrobial Alkaloid from Mitracarpus scaber. Planta Med. 1999, 65, 447–448. [Google Scholar] [CrossRef] [PubMed]
- Harouna, H.; Faure, R.; Elias, R.; Debrauwer, L.; Saadou, M.; Balansard, G.; Boudon, G. Harounoside a pentalongin hydroquinone diglycoside from Mitracarpus scaber. Phytochemistry 1995, 39, 1483–1484. [Google Scholar] [CrossRef]
- Pialat, J.P.; Hoffmann, P.; Moulis, C.; Fouraste, I.; Labidalle, S. Synthesis and Extraction of Pentalongin, A Naphthoquinoid From Mitracarpus Scaber. Nat. Prod. Lett. 1998, 12, 23–30. [Google Scholar] [CrossRef]
- Burgess, K.; Borutzki, Y.; Rankin, N.; Daly, R.; Jourdan, F. MetaNetter 2: A Cytoscape plugin for ab initio network analysis and metabolite feature classification. J. Chromatogr. B 2017, 1071, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Menachery, S.P.M.; Laprévote, O.; Nguyen, T.P.; Aravind, U.K.; Gopinathan, P.; Aravindakumar, C.T. Identification of position isomers by energy-resolved mass spectrometry. J. Mass Spectrom. 2015, 50, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Zhou, Y.; Liu, X.; Xiao, J.; Liu, Q.; Gu, Y.; Ding, L. Use of electrospray ionization ion-trap tandem mass spectrometry and principal component analysis to directly distinguish monosaccharides. Rapid Commun. Mass Spectrom. 2012, 26, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Giner, J.L.; Feng, J.; Kiemle, D.J. NMR Tube Degradation Method for Sugar Analysis of Glycosides. J. Nat. Prod. 2016, 79, 2413–2417. [Google Scholar] [CrossRef] [PubMed]
- Genta-Jouve, G.; Weinberg, L.; Cocandeau, V.; Maestro, Y.; Thomas, O.P.; Holderith, S. Revising the Absolute Configurations of Coatlines via Density Functional Theory Calculations of Electronic Circular Dichroism Spectra. Chirality 2013, 25, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Han, A.R.; Park, S.R.; Park, J.W.; Lee, E.Y.; Kim, D.M.; Kim, B.G.; Yoon, Y.J. Biosynthesis of Glycosylated Derivatives of Tylosin in Streptomyces venezuelae. J. Microbiol. Biotechnol. 2011, 21, 613–616. [Google Scholar] [CrossRef] [PubMed]
- De Castro, C.; Kenyon, J.J.; Cunneen, M.M.; Molinaro, A.; Holst, O.; Skurnik, M.; Reeves, P.R. The O-specific polysaccharide structure and gene cluster of serotype O:12 of the Yersinia pseudotuberculosis complex, and the identification of a novel l-quinovose biosynthesis gene. Glycobiology 2013, 23, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M.; Clark-Lewis, I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992, 307, 97–101. [Google Scholar] [CrossRef]
- Pease, J.E.; Sabroe, I. The Role of Interleukin-8 and its Receptors in Inflammatory Lung Disease. Am. J. Respir. Med. 2002, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- This concentration of EGCG has been measured on a previous experiment.
- Réfrégiers, M.; Wien, F.; Ta, H.P.; Premvardhan, L.; Bac, S.; Jamme, F.; Rouam, V.; Lagarde, B.; Polack, F.; Giorgetta, J.L.; et al. DISCO synchrotron-radiation circular-dichroism endstation at SOLEIL. J. Synchrotron Radiat. 2012, 19, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Jamme, F.; Rouam, V.; Wien, F.; Giorgetta, J.L.; Lagarde, B.; Chubar, O.; Bac, S.; Yao, I.; Rey, S.; et al. DISCO: A low-energy multipurpose beamline at synchrotron SOLEIL. J. Synchrotron Radiat. 2009, 16, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clarinoside (1) | Harounoside (2) [8] | |||||||
|---|---|---|---|---|---|---|---|---|
| No. | (Multiplicity, J) | No. | (Multiplicity, J) | No. | ||||
| 1 | 5.29 (dd, 40.7, 13.8 Hz) | 65.2 | 1 | 4.66 (d, 7.8) | 106.2 | 1 | 5.39; 5.30 | 65.4 |
| 3 | 6.67 (dd, 14.4, 5.9 Hz) | 147.8 | 2 | 3.61 (dd, 9.0, 7.8) | 75.9 | 3 | 6.68 | 147.8 |
| 4 | 6.66 (dd 14.4, 5.9 Hz) | 102.1 | 3 | 3.38 (t, 9.0) | 77.7 | 4 | 6.64 | 102.2 |
| 4a | 121.6 | 4 | 3.11 (t, 9.0) | 73.5 | 4a | 121.7 | ||
| 5 | 143.3 | 5 | 3.11 (m) | 77.8 | 5 | 143.4 | ||
| 5a | 131.0 | 6 | 1.21 (d, 5.4) | 18.1 | 5a | 131.0 | ||
| 6 | 8.43 (d, 8.2 Hz) | 124.7 | 1 | 4.79 (d, 7.8) | 106.9 | 6 | 8.42 | 124.7 |
| 7 | 7.44 (dt, 8.2, 1 Hz) | 127.0 | 2 | 3.65 (m) | 75.8 | 7 | 7.43 | 127.0 |
| 8 | 7.40 (dt, 8.2, 1 Hz) | 126.2 | 3 | 3.46 (m) | 71.5 | 8 | 7.39 | 126.3 |
| 9 | 8.41 (d, 8.2 Hz) | 123.7 | 4 | 3.14 (m) | 76.9 | 9 | 8.43 | 123.7 |
| 9a | 129.1 | 5 | 3.47 (t, 9.0) | 78.0 | 9a | 129.1 | ||
| 10 | 144.9 | 6 | 3.67 (m) | 62.7 | 10 | 145.0 | ||
| 10a | 122.6 | 10a | 122.7 | |||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Audoin, C.; Zampalégré, A.; Blanchet, N.; Giuliani, A.; Roulland, E.; Laprévote, O.; Genta-Jouve, G. MS/MS-Guided Isolation of Clarinoside, a New Anti-Inflammatory Pentalogin Derivative. Molecules 2018, 23, 1237. https://doi.org/10.3390/molecules23051237
Audoin C, Zampalégré A, Blanchet N, Giuliani A, Roulland E, Laprévote O, Genta-Jouve G. MS/MS-Guided Isolation of Clarinoside, a New Anti-Inflammatory Pentalogin Derivative. Molecules. 2018; 23(5):1237. https://doi.org/10.3390/molecules23051237
Chicago/Turabian StyleAudoin, Coralie, Adam Zampalégré, Natacha Blanchet, Alexandre Giuliani, Emmanuel Roulland, Olivier Laprévote, and Grégory Genta-Jouve. 2018. "MS/MS-Guided Isolation of Clarinoside, a New Anti-Inflammatory Pentalogin Derivative" Molecules 23, no. 5: 1237. https://doi.org/10.3390/molecules23051237
APA StyleAudoin, C., Zampalégré, A., Blanchet, N., Giuliani, A., Roulland, E., Laprévote, O., & Genta-Jouve, G. (2018). MS/MS-Guided Isolation of Clarinoside, a New Anti-Inflammatory Pentalogin Derivative. Molecules, 23(5), 1237. https://doi.org/10.3390/molecules23051237