Density Functional Theory Applied to Excited State Intramolecular Proton Transfer in Imidazole-, Oxazole-, and Thiazole-Based Systems

 and
and 
Abstract
: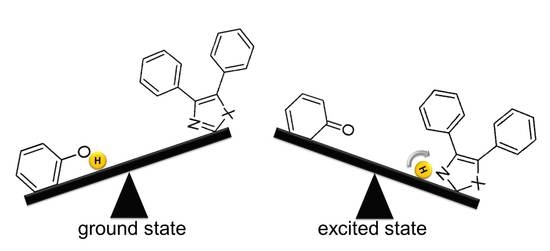
1. Introduction
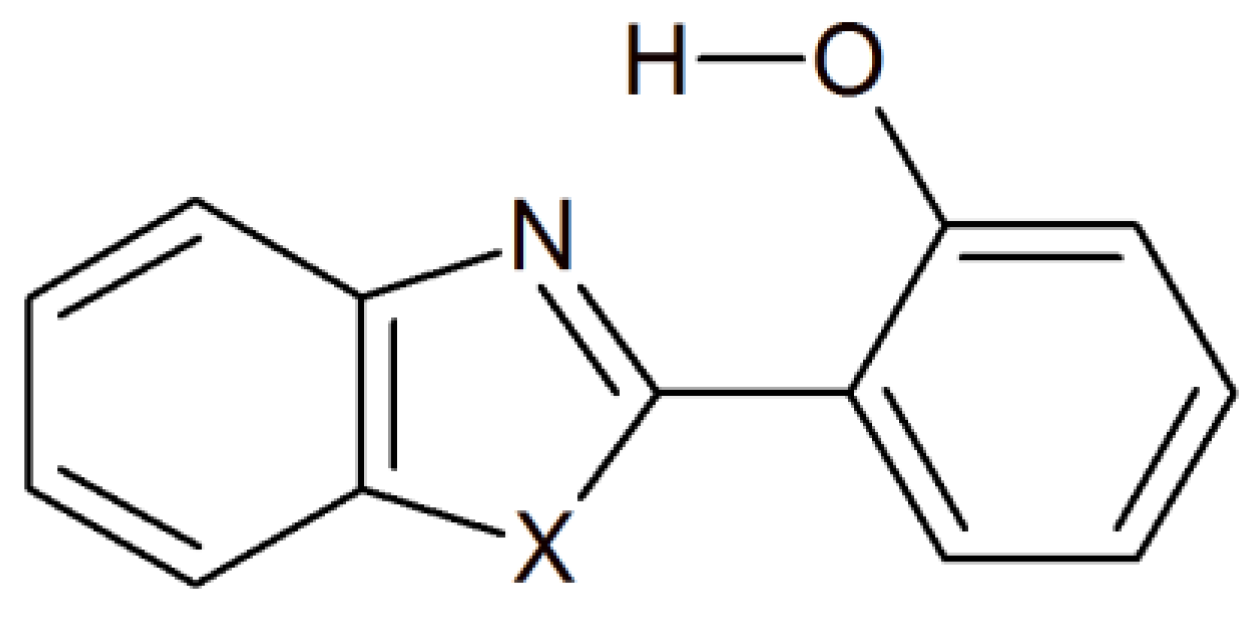
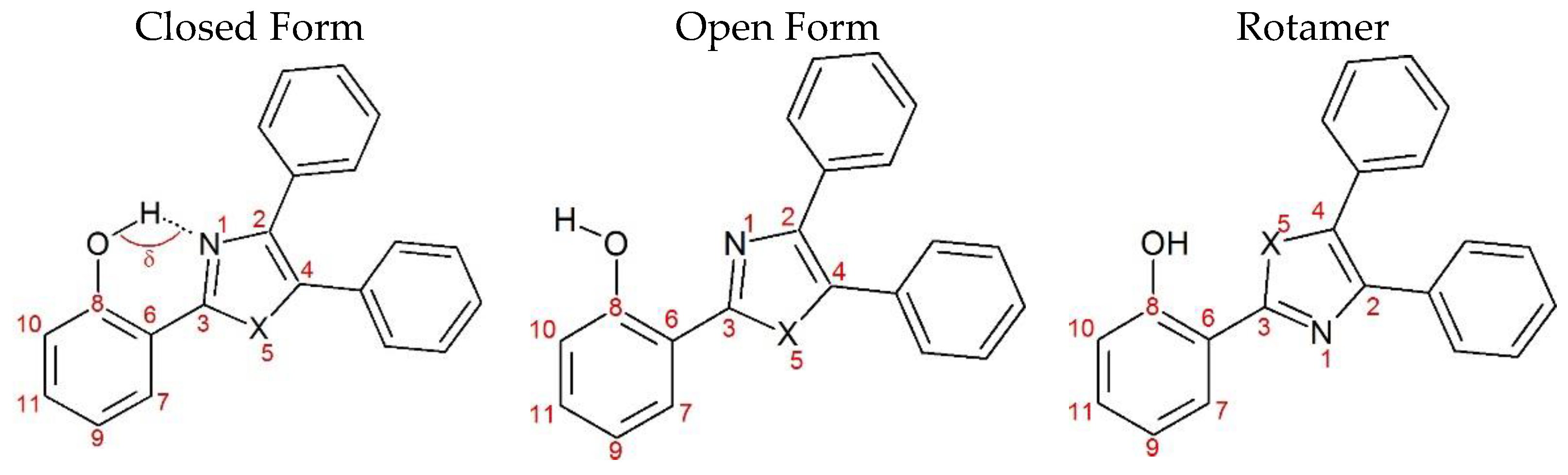
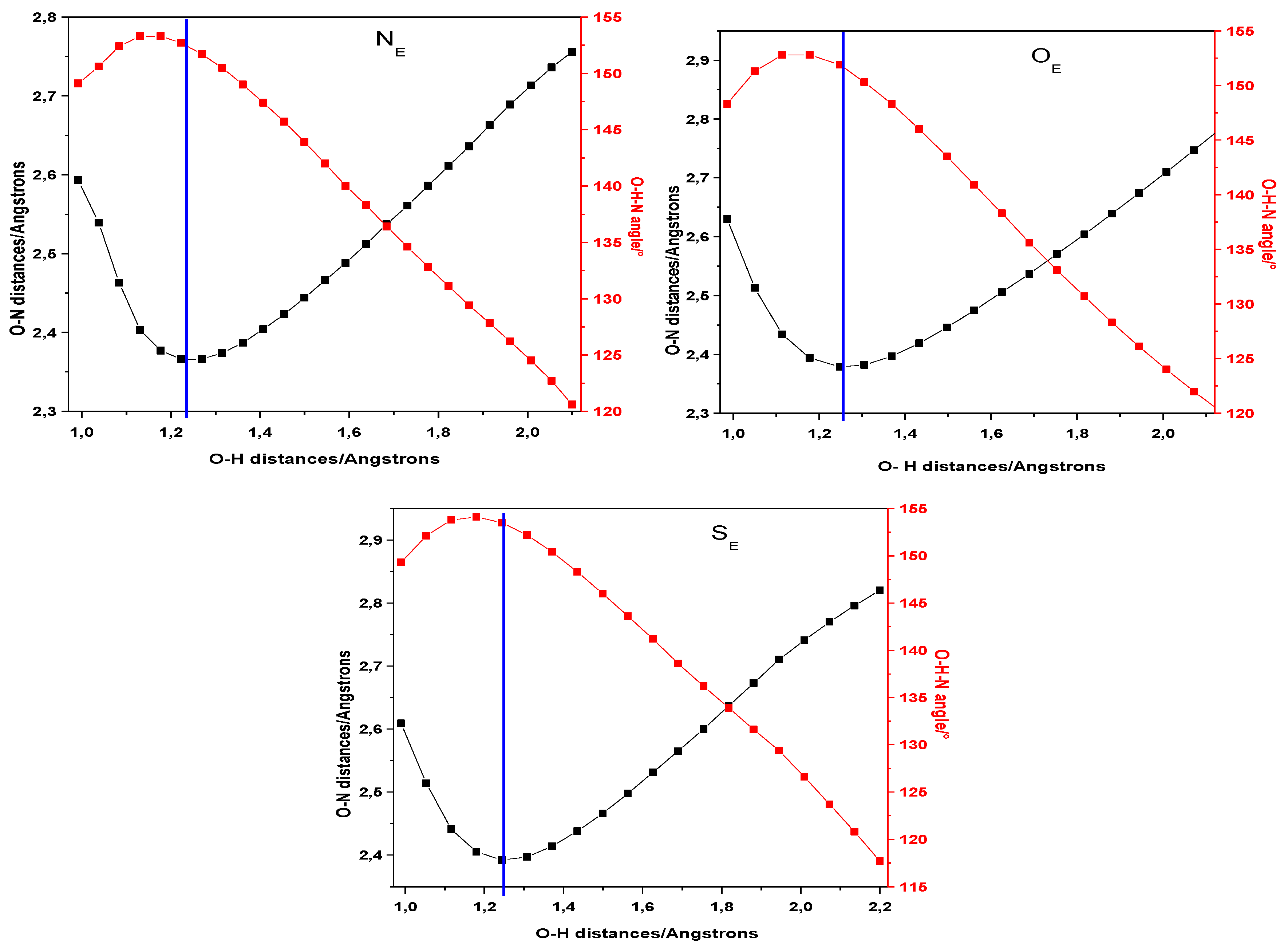
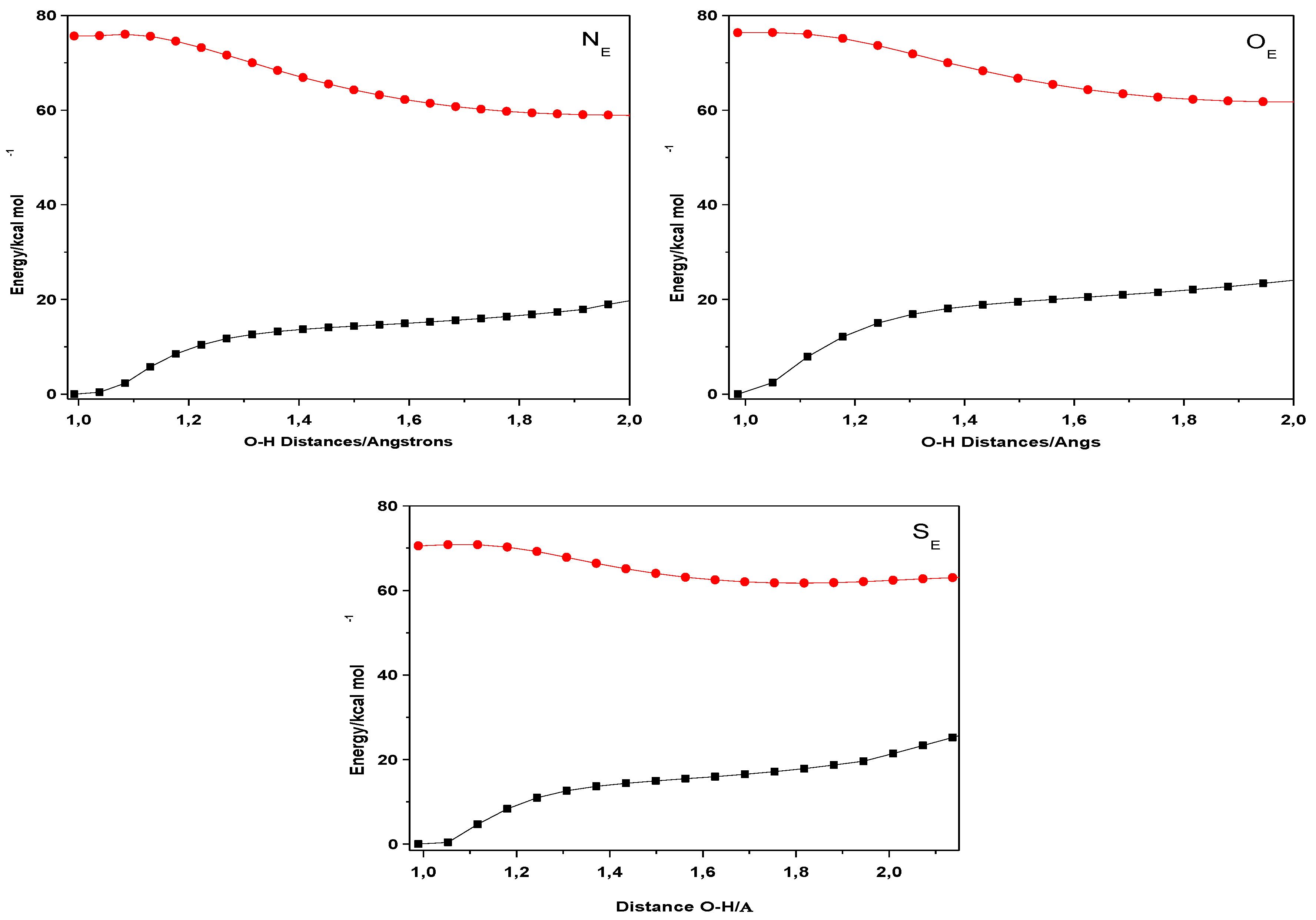
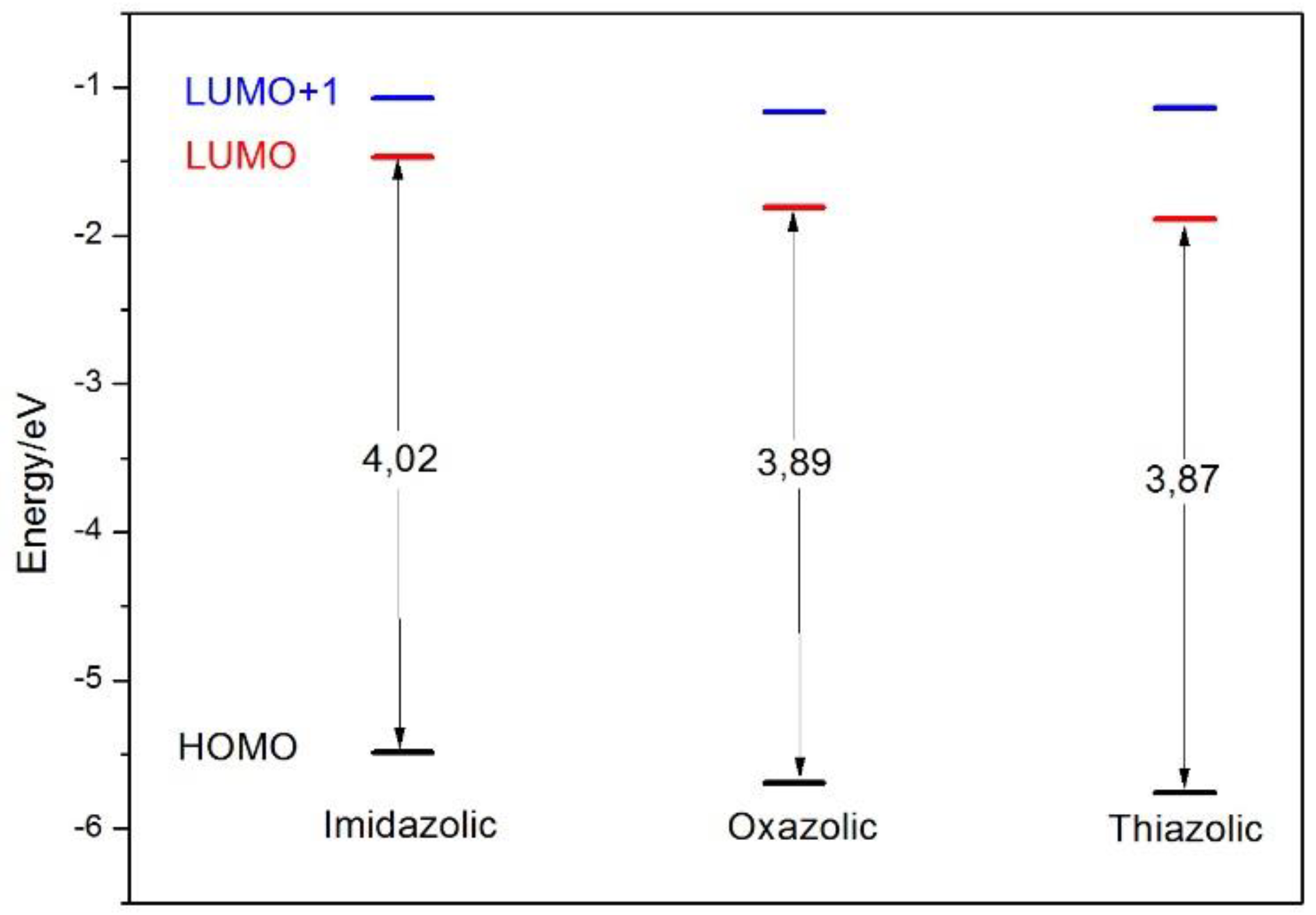
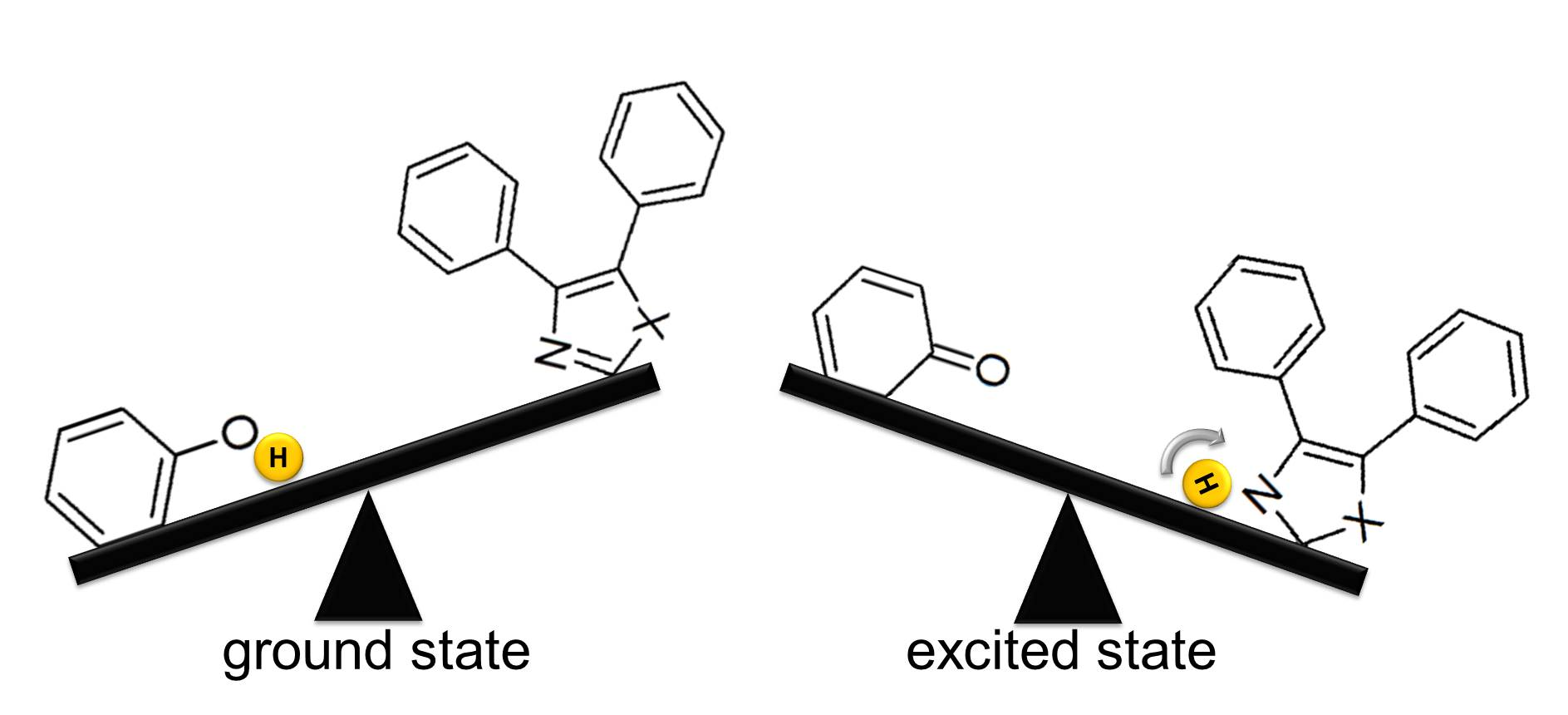
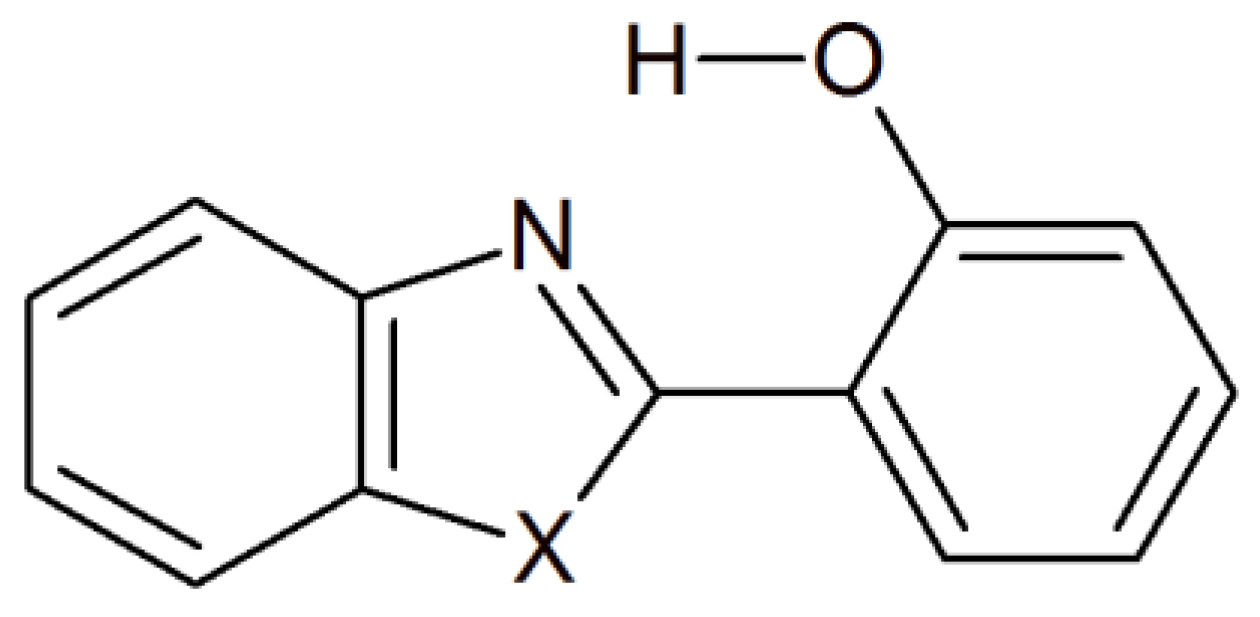
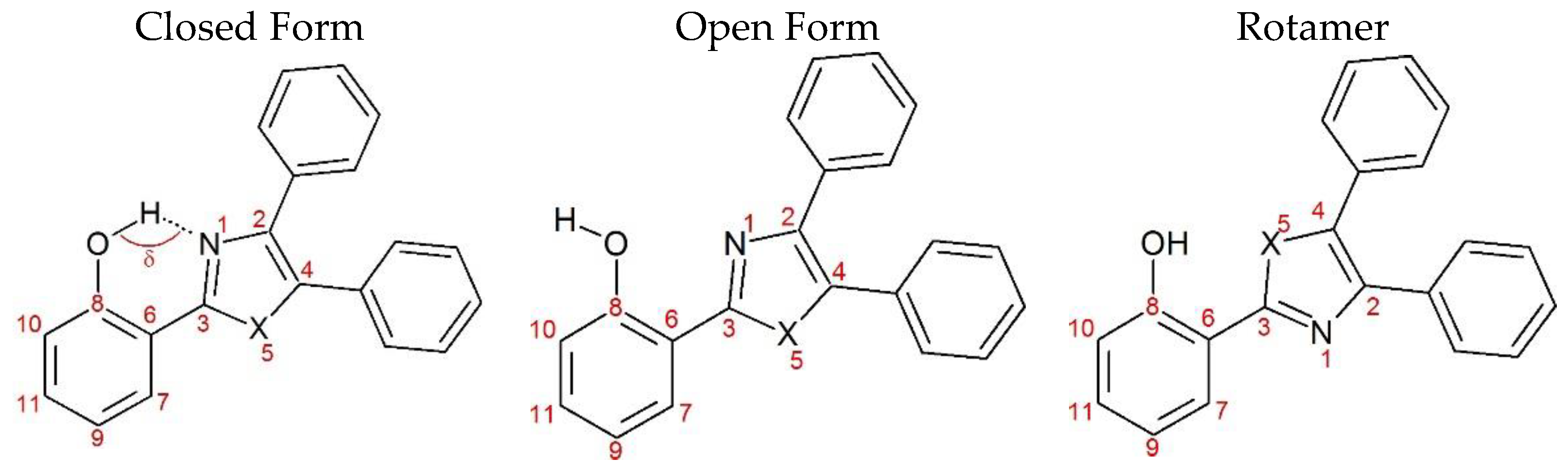
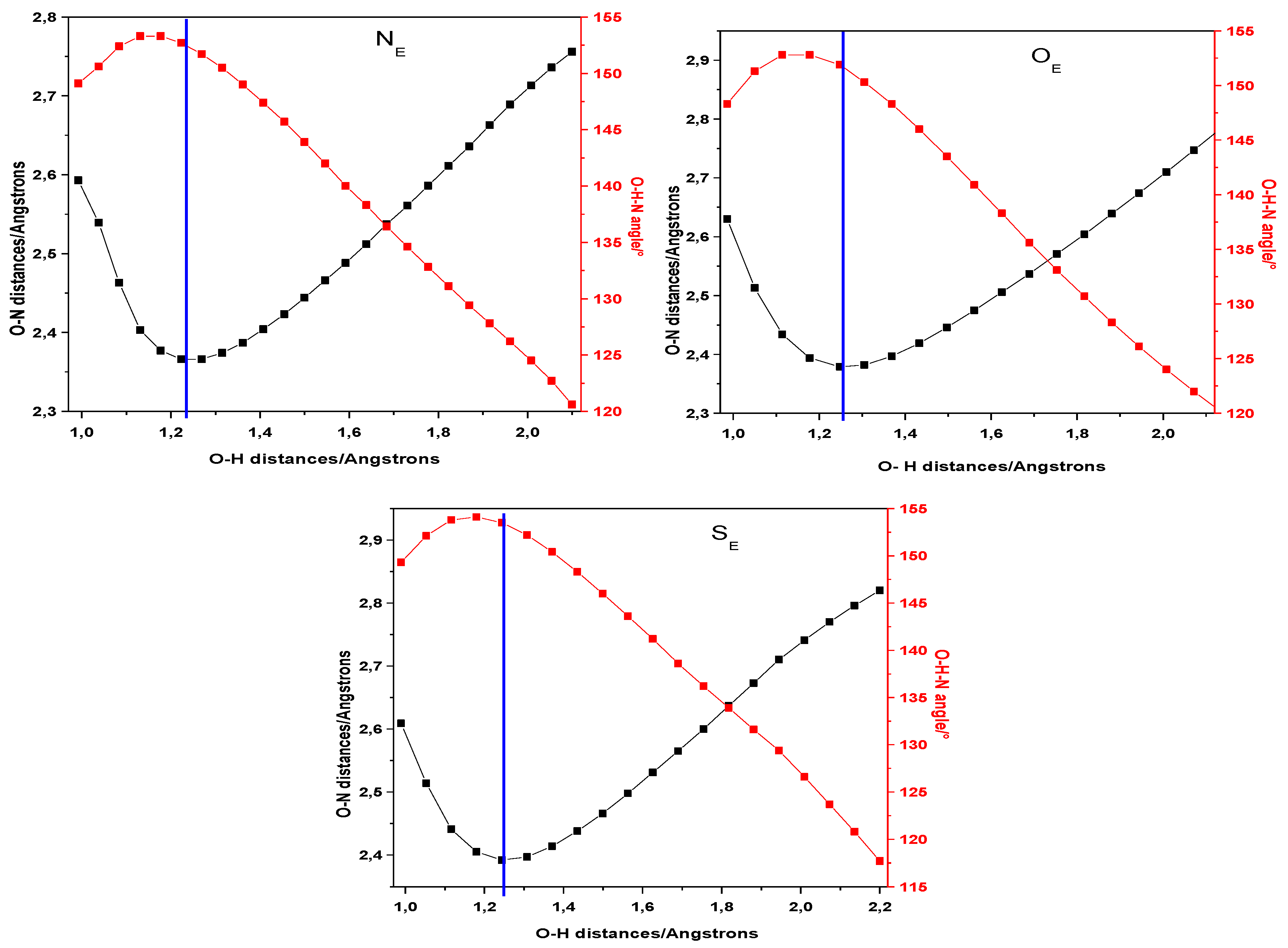
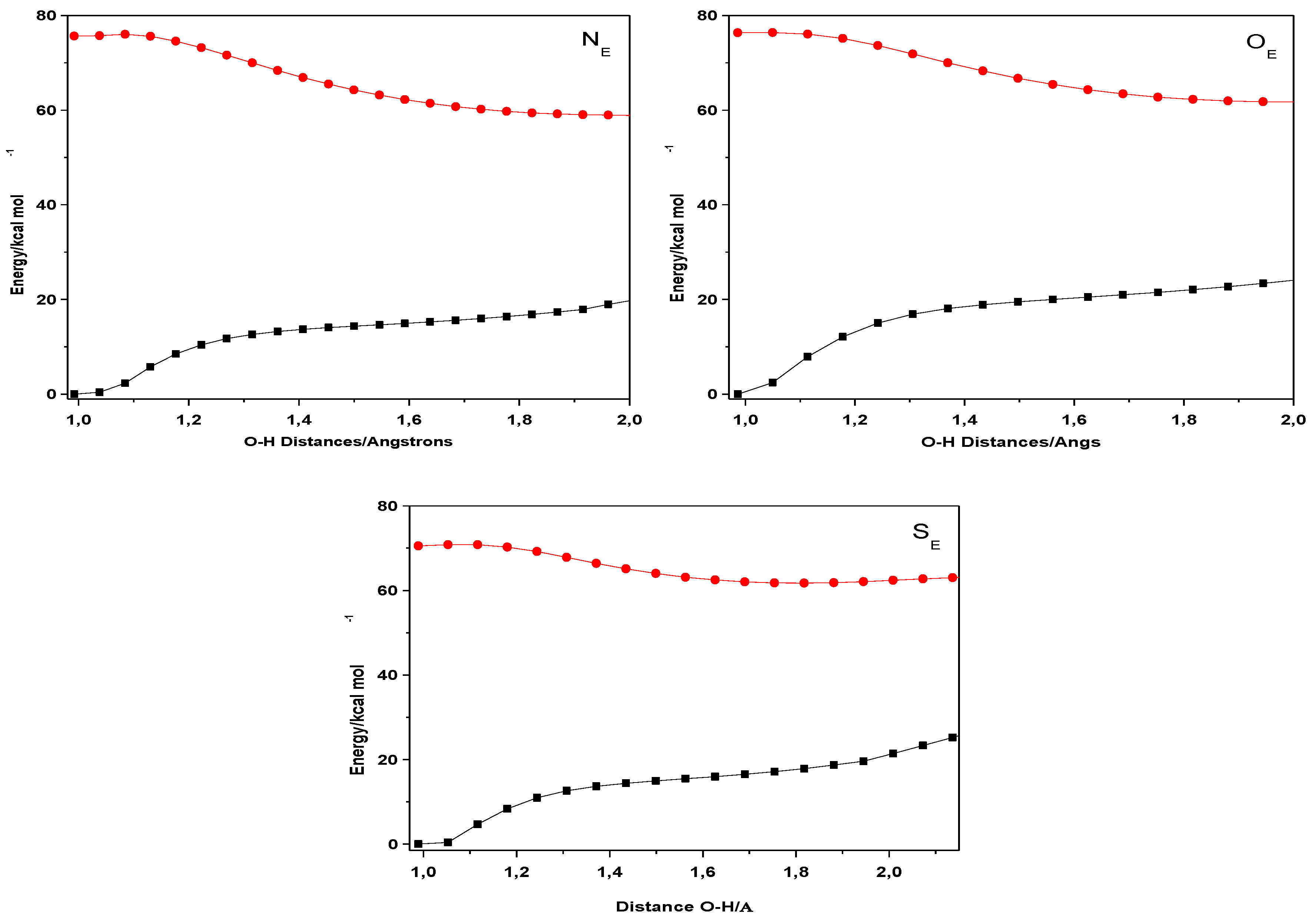
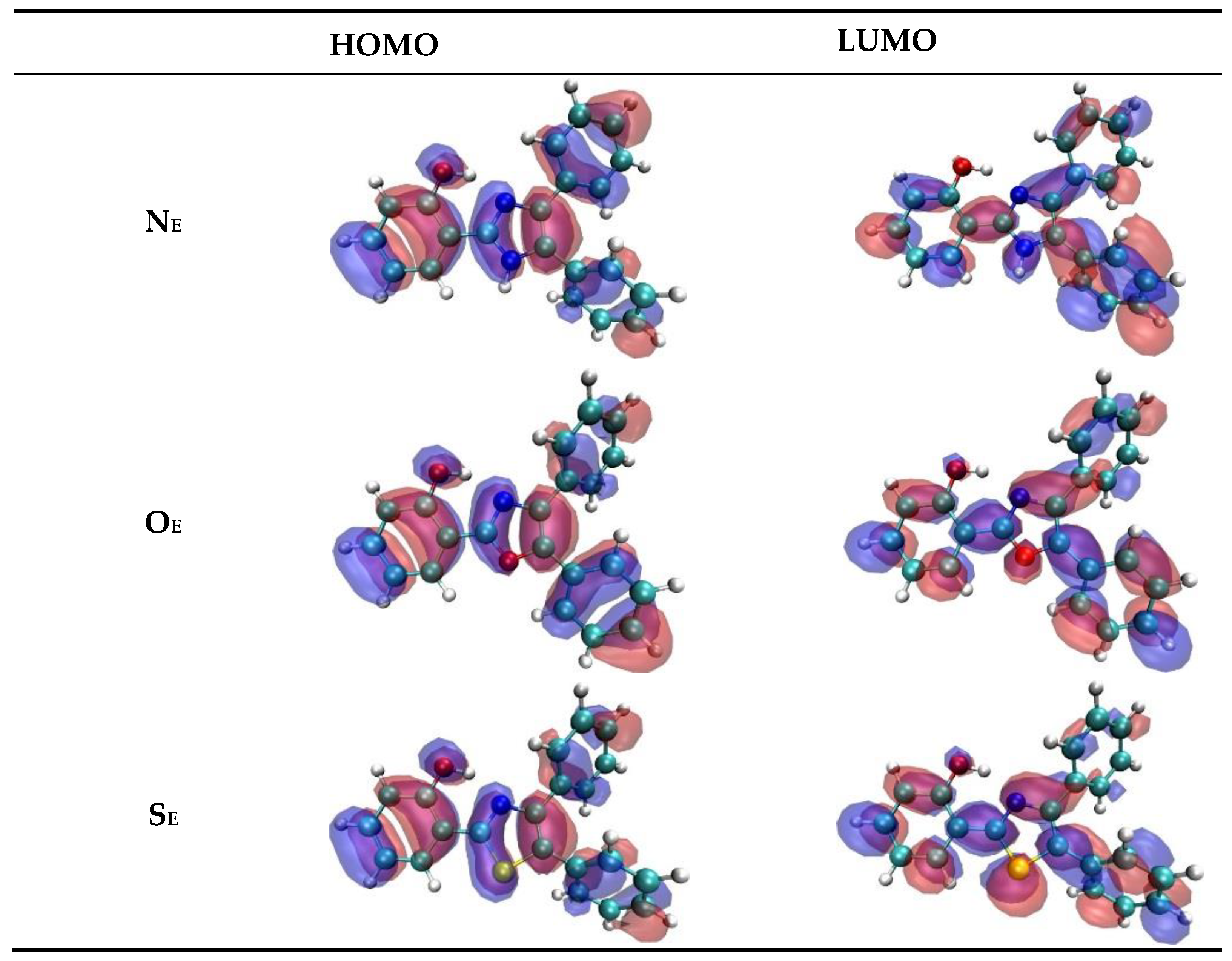
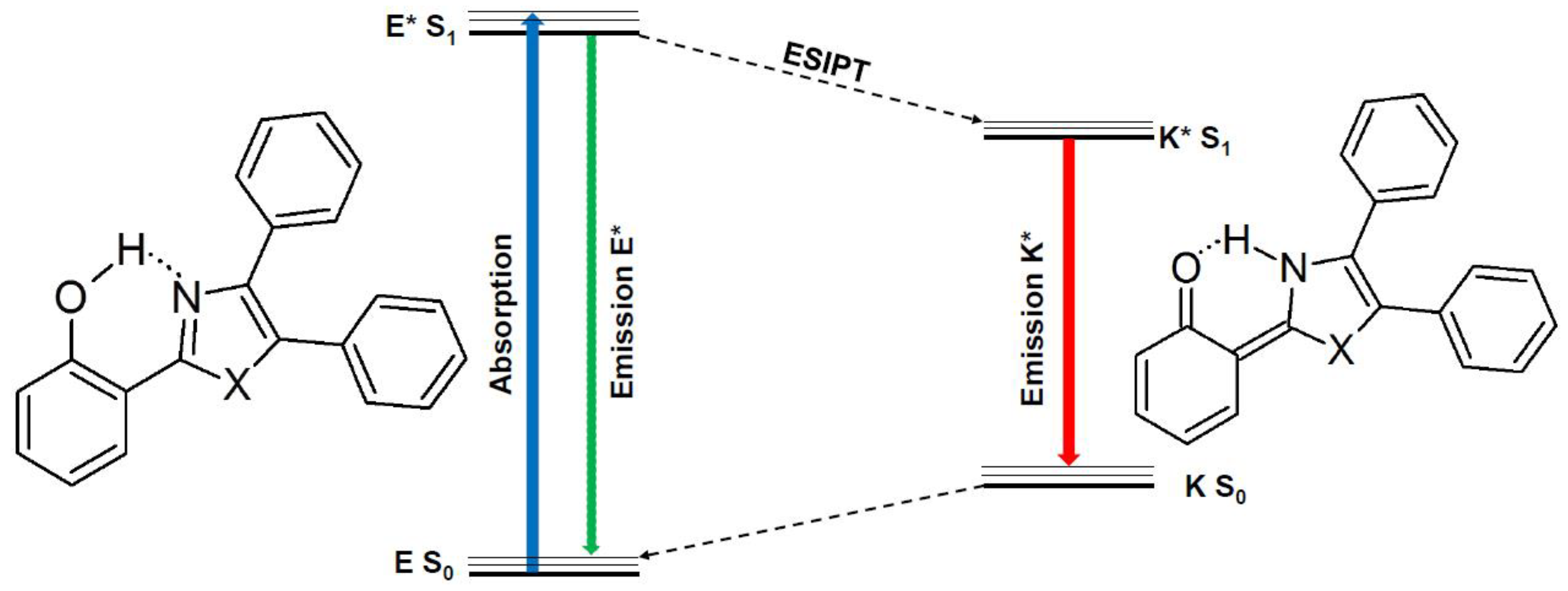
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Eisenberger, H.; Nickel, B.; Ruth, A.A.; Alsoufi, W.; Grellmann, K.H.; Novo, M. Keto-enol tautomerization of 2-(2’-hydroxyphenyl)benzoxazole and 2-(2’-hydroxy-4’-methylphenyl)benzoxazole in the triplet-state-hydrogen tunneling and isotope effects. 2. dual phosphorescence kinetics. J. Phys. Chem. 1991, 95, 10509–10518. [Google Scholar] [CrossRef]
- Arthenengeland, T.; Bultmann, T.; Ernsting, N.P.; Rodriguez, M.A.; Thiel, W. Singlet excited-state intramolecular proton-transfer in 2-(2’-hydroxyphenyl)benzoxazole—Spectroscopy at low temperatures, femtosecond transient absorption, and MNDO calculations. Chem. Phys. 1992, 163, 43–53. [Google Scholar] [CrossRef]
- Fernandez-Ramos, A.; Rodriguez-Otero, J.; Rios, M.A.; Soto, J. Intramolecular proton transfer in 2-(2’-hydroxyphenyl)benzoxazole: The reliability of ab initio calculations on simplified structures. J. Mol. Struct. 1999, 489, 255–262. [Google Scholar] [CrossRef]
- Iglesias, R.S.; Goncalves, P.F.B.; Livotto, P.R. Semi-empirical study of a set of 2-(2’-hydroxyphenyl)benzazoles using the polarizable continuum model. Chem. Phys. Lett. 2000, 327, 23–28. [Google Scholar] [CrossRef]
- Purkayastha, P.; Chattopadhyay, N. Role of rotamerisation and excited state intramolecular proton transfer in the photophysics of 2-(2’-hydroxyphenyl)benzoxazole, 2-(2’-hydroxyphenyl)benzimidazole and 2-(2’-hydroxyphenyl)benzothiazole: A theoretical study. Phys. Chem. Chem. Phys. 2000, 2, 203–210. [Google Scholar] [CrossRef]
- Purkayastha, P.; Chattopadhyay, N. Theoretical modelling for the ground state rotamerisation and excited state intramolecular proton transfer of 2-(2’-hydroxyphenyl)oxazole, 2-(2’-hydroxyphenyl)imidazole, 2-(2’-hydroxyphenyl)thiazole and their benzo analogues. Int. J. Mol. Sci. 2003, 4, 335–361. [Google Scholar] [CrossRef]
- Rodembusch, F.S.; Leusin, F.P.; Campo, L.F.; Stefani, V. Excited state intramolecular proton transfer in amino 2-(2’-hydroxyphenyl)benzazole derivatives: Effects of the solvent and the amino group position. J. Lumin. 2007, 126, 728–734. [Google Scholar] [CrossRef]
- Irgibaeva, I.S.; Birimzhanova, D.A.; Barashkov, N.N. Research of electronic absorption spectra of benzazols derivatives by Ab Initio calculations. Int. J. Quantum Chem. 2008, 108, 2700–2710. [Google Scholar] [CrossRef]
- Iijima, T.; Momotake, A.; Shinohara, Y.; Sato, T.; Nishimura, Y.; Arai, T. Excited-state intramolecular proton transfer of naphthalene-fused 2-(2’-Hydroxyaryl)benzazole Family. J. Phys. Chem. A 2010, 114, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Pang, Y. Excited-state intramolecular proton transfer in 2-(2’,6’-dihydroxyphenyl) benzoxazole: Effect of dual hydrogen bonding on the optical properties. Tetrahedron Lett. 2010, 51, 1914–1918. [Google Scholar] [CrossRef]
- Sepiol, J.; Grabowska, A.; Borowicz, P.; Kijak, M.; Broquier, M.; Jouvet, C.; Dedonder-Lardeux, C.; Zehnacker-Rentien, A. Excited-state intramolecular proton transfer reaction modulated by low-frequency vibrations: An effect of an electron-donating substituent on the dually fluorescent bis-benzoxazole. J. Chem. Phys. 2011, 135. [Google Scholar] [CrossRef] [PubMed]
- Jayabharathi, J.; Vimal, K.; Thanikachalam, V.; Kalaiarasi, V. Photophysical and excited-state intramolecular proton transfer of 2-(1-(3,5-dimethylphenyl)-1H-phenanthro [9,10-d] imidazol-2-yl)phenol: DFT analysis. Spectrochim. Acta A 2014, 125, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Houari, Y.; Chibani, S.; Jacquemin, D.; Laurent, A.D. TD-DFT Assessment of the excited state intramolecular proton transfer in hydroxyphenylbenzimidazole (HBI) dyes. J. Phys. Chem. B 2015, 119, 2180–2192. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P.; Ramalho, T.C. Probing the ESIPT process in 2-amino-1,4-naphthoquinone: Thermodynamics properties, solvent effect and chemometric analysis. Theor. Chem. Acc. 2016, 135, 39. [Google Scholar] [CrossRef]
- An, B.; Yuan, H.; Zhu, Q.; Li, Y.; Guo, X.; Zhang, J. Theoretical insight into the excited-state intramolecular proton transfer mechanisms of three amino-type hydrogen-bonding molecules. Spectrochim. Acta A 2017, 175, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Manojai, N.; Daengngern, R.; Kerdpol, K.; Ngaojampa, C.; Kungwan, N. Heteroatom effect on photophysical properties of 2-(2’-hydroxyphenyl) benzimidazole and its derivatives as fluorescent dyes: A TD-DFT study. J. Lumin. 2017, 188, 275–282. [Google Scholar] [CrossRef]
- Weller, A. Innermolekularer protonenubergang im angeregten zustand. Zeits. Elek. 1956, 60, 1144–1147. [Google Scholar]
- Weler, A. Fast reactions of excited molecules. Prog. React. Kinet. Mech. 1961, 1, 187. [Google Scholar]
- Chou, P.; Mcmorrow, D.; Aartsma, T.J.; Kasha, M. The proton-transfer laser-gain spectrum and amplification of spontaneous emission of 3-hydroxyflavone. J. Phys. Chem. 1984, 88, 4596–4599. [Google Scholar] [CrossRef]
- Kasha, M. Proton-transfer spectroscopy-perturbation of the tautomerization potential. J. Chem. Soc. Trans. II 1986, 82, 2379–2392. [Google Scholar] [CrossRef]
- Tarkka, R.M.; Zhang, X.J.; Jenekhe, S.A. Electrically generated intramolecular proton transfer: Electroluminescence and stimulated emission from polymers. J. Am. Chem. Soc. 1996, 118, 9438–9439. [Google Scholar] [CrossRef]
- Gormin, D.; Sytnik, A.; Kasha, M. Spectroscopy of amplified spontaneous emission laser spikes in polyhydroxyflavones. J. Phys. Chem. A 1997, 101, 672–677. [Google Scholar] [CrossRef]
- Costela, A.; Garcia-Moreno, I.; Mallavia, R.; Amat-Guerri, F.; Barroso, J.; Sastre, R. Proton-transfer lasers based on solid copolymers of modified 2-(2’-hydroxyphenyl)benzimidazoles with methacrylate monomers. Opt. Commun. 1998, 152, 89–95. [Google Scholar] [CrossRef]
- Garcia, O.; Garrido, L.; Sastre, R.; Costela, A.; Garcia-Moreno, I. Synthetic strategies for hybrid materials to improve properties for optoelectronic applications. Adv. Funct. Mater. 2008, 18, 2017–2025. [Google Scholar] [CrossRef]
- Vollmer, F.; Rettig, W. Fluorescence loss mechanism due to large-amplitude motions in derivatives of 2,2’-bipyridyl exhibiting excited-state intramolecular proton transfer and perspectives of luminescence solar concentrators. J. Photochem. Photobiol. A Chem. 1996, 95, 143–155. [Google Scholar] [CrossRef]
- Shynkar, V.V.; Klymchenko, A.S.; Piemont, E.; Demchenko, A.P.; Mely, Y. Dynamics of intermolecular hydrogen bonds in the excited states of 4’-dialkylamino-3-hydroxyflavones. On the pathway to an ideal fluorescent hydrogen bonding sensor. J. Phys. Chem. A 2004, 108, 8151–8159. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Hsieh, C.-C.; Cheng, Y.-M.; Lai, C.-H.; Chou, P.-T. Extensive spectral tuning of the proton transfer emission from 550 to 675 nm via a rational derivatization of 10-hydroxybenzo[h]quinoline. Chem. Commun. 2006, 42, 4395–4397. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.E.; Park, S.Y. Advanced organic optoelectronic materials: Harnessing excited-state intramolecular proton transfer (ESIPT) process. Adv. Mater. 2011, 23, 3615–3642. [Google Scholar] [CrossRef] [PubMed]
- Stueber, G.J.; Kieninger, M.; Schettler, H.; Busch, W.; Goeller, B.; Franke, J.; Kramer, H.E.A.; Hoier, H.; Henkel, S. Ultraviolet stabilizers of the 2-(2’-hydroxyphenyl)-1,3,5-triazine class-structural and spectroscopic characterization. J. Phys. Chem. 1995, 99, 10097–10109. [Google Scholar] [CrossRef]
- Keck, J.; Kramer, H.E.A.; Port, H.; Hirsch, T.; Fischer, P.; Rytz, G. Investigations on polymeric and monomeric intramolecularly hydrogen-bridged UV absorbers of the benzotriazole and triazine class. J. Phys. Chem. 1996, 100, 14468–14475. [Google Scholar] [CrossRef]
- Wu, J.; Liu, W.; Ge, J.; Zhang, H.; Wang, P. New sensing mechanisms for design of fluorescent chemosensors emerging in recent years. Chem. Soc. Rev. 2011, 40, 3483–3495. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Wolford, J.L.; O’Halloran, T.V. Emission ratiometric imaging of intracellular zinc: Design of a benzoxazole fluorescent sensor and its application in two-photon microscopy. J. Am. Chem. Soc. 2004, 126, 712–713. [Google Scholar] [CrossRef] [PubMed]
- Rodembusch, F.S.; Brand, F.R.; Correa, D.S.; Pocos, J.C.; Martinelli, M.; Stefani, V. Transition metal complexes from 2-(2’-hydroxyphenyl)benzoxazole: A spectroscopic and thermogravimetric stability study. Mater. Chem. Phys. 2005, 92, 389–393. [Google Scholar] [CrossRef]
- Orfão, R.B., Jr.; Alves, J.; Bartoloni, F.H. Spectroscopic studies on the interaction of metallic ions with an imidazolyl-phenolic system. J. Fluoresc. 2016, 26, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Orfão, R.B., Jr.; de Carvalho, F.; Homem-de-Mello, P.; Bartoloni, F.H. On the fluorescent, steric and electronic factors affecting the detection of metallic ions using an imidazolyl-phenolic derived fluorescent probe. J. Braz. Chem. Soc. 2017, 28, 1896–1904. [Google Scholar] [CrossRef]
- Das, K.; Sarkar, N.; Ghosh, A.K.; Majumdar, D.; Nath, D.N.; Bhattacharyya, K. Excited-state intramolecular proton-transfer in 2-(2’-hydroxyphenyl)benzimidazole and 2-(2’-hydroxyphenyl)-benzoxazole-effect of rotamerism and hydrogen-bonding. J. Phys. Chem. 1994, 98, 9126–9132. [Google Scholar] [CrossRef]
- Raymo, F.M.; Bartberger, M.D.; Houk, K.N.; Stoddart, J.F. The magnitude of [C-H∙∙∙O] hydrogen bonding in molecular and supramolecular assemblies. J. Am. Chem. Soc. 2001, 123, 9264–9267. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.H.; Wu, L.Z.; Zhang, L.P.; Chen, B. Supramolecular systems as microreactors: Control of product selectivity in organic phototransformation. Acc. Chem. Res. 2003, 36, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Glasbeek, M.; Zhang, H. Femtosecond studies of solvation and intramolecular configurational dynamics of fluorophores in liquid solution. Chem. Rev. 2004, 104, 1929–1954. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.; Smith, S.C. Bond selection in the photoisomerization reaction of anionic green fluorescent protein and kindling fluorescent protein chromophore models. J. Am. Chem. Soc. 2008, 130, 8677–8689. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Truhlar, D.G. A universal approach to solvation modeling. Acc. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chi, L.; Ji, S.; Wu, Y.; Song, P.; Han, K.; Guo, H.; James, T.D.; Zhao, J. Rational design of d-PeT phenylethynylated-carbazole monoboronic acid fluorescent sensors for the selective detection of alpha-hydroxyl carboxylic acids and monosaccharides. J. Am. Chem. Soc. 2009, 131, 17452–17463. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-J.; Han, K.-L. Hydrogen bonding in the electronic excited state. Acc. Chem. Res. 2012, 45, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Yang, D.; Sui, X.; Wang, D. Time-dependent density functional theory (TD-DFT) study on the excited-state intramolecular proton transfer (ESIPT) in 2-hydroxybenzoyl compounds: Significance of the intramolecular hydrogen bonding. Spectrochim. Acta A 2013, 102, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Johnson, E.R. Exchange-hole dipole moment and the dispersion interaction revisited. J. Chem. Phys. 2007, 127. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-H.G.; Sun, H.-L.S.; Tan, C.-J. TD-DFT study of the excited-state potential energy surfaces of 2-(2’-hydroxyphenyl)benzimidazole and its amino derivatives. J. Phys. Chem. A 2010, 114, 4065–4079. [Google Scholar] [CrossRef] [PubMed]
- Jayabharathi, J.; Kalaiarasi, V.; Thanikachalam, V.; Jayamoorthy, K. Dynamics of solvent controlled ESIPT of pi-expanded imidazole derivatives-pH Effect. J. Fluoresc. 2014, 24, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Mahanta, S.; Paul, B.K.; Singh, R.B.; Guchhait, N. Inequivalence of substitution pairs in hydroxynaphthaldehyde: A theoretical measurement by intramolecular hydrogen bond strength, aromaticity, and excited-state intramolecular proton transfer reaction. J. Comput. Chem. 2011, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Roohi, H.; Hejazi, F.; Mohtamedifar, N.; Jahantab, M. Excited state intramolecular proton transfer (ESIPT) in 2-(2’-hydroxyphenyl)benzoxazole and its naphthalene-fused analogs: A TD-DFT quantum chemical study. Spectrochim. Acta A 2014, 118, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Alarcos, N.; Gutierrez, M.; Liras, M.; Sanchez, F.; Moreno, M.; Douhal, A. Direct observation of breaking of the intramolecular H-bond, and slowing down of the proton motion and tuning its mechanism in an HBO derivative. Phys. Chem. Chem. Phys. 2015, 17, 14569–14581. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-C.; Cheng, Y.-M.; Hsu, C.-J.; Chen, K.-Y.; Chou, P.-T. Spectroscopy and femtosecond dynamics of excited-state proton transfer induced charge transfer reaction. J. Phys. Chem. A 2008, 112, 8323–8332. [Google Scholar] [CrossRef] [PubMed]
- Barboza, C.A.; Germino, J.C.; Santana, A.M.; Quites, F.J.; Muniz Vazquez, P.A.; Zambon Atvars, T.D. Structural correlations between luminescent properties and excited state internal proton transfer in some Zinc(II) N,N′-Bis(salicylidenes). J. Phys. Chem. C 2015, 119, 6152–6163. [Google Scholar] [CrossRef]
- Stenson, P. Crystal structure of 2-(ortho hydroxyphenyl) benzothiazole. Acta Chem. Scand. 1970, 24, 3729. [Google Scholar] [CrossRef]
- Catalan, J.; del Valle, J.C.; Palomar, J.; Diaz, C.; de Paz, J.L.G. The six-membered intramolecular hydrogen bond position as a switch for inducing an excited state intramolecular proton transfer (ESIPT) in esters of o-hydroxynaphthoic acids. J. Phys. Chem. A 1999, 103, 10921–10934. [Google Scholar] [CrossRef]
- Zhao, G.-J.; Liu, J.-Y.; Zhou, L.-C.; Han, K.-L. Site-selective photoinduced electron transfer from alcoholic solvents to the chromophore facilitated by hydrogen bonding: A new fluorescence quenching mechanism. J. Phys. Chem. B 2007, 111, 8940–8945. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-J.; Han, K.-L. Early time hydrogen-bonding dynamics of photoexcited coumarin 102 in hydrogen-donating solvents: Theoretical study. J. Phys. Chem. A 2007, 111, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zheng, R.; Wang, Y.; Lv, J. The ESIPT mechanism of dibenzimidazolo diimine sensor: A detailed TDDFT study. J. Phys. Org. Chem. 2016, 29, 161–165. [Google Scholar] [CrossRef]
- Beg, H.; Das, D.; Ash, S.; Misra, A. Computation of polarizability, hyper-polarizability and hardness as descriptor for enol-keto tautomerizations of 2-hydroxy pyridines. Comput. Theor. Chem. 2013, 1017, 200–207. [Google Scholar] [CrossRef]
- Roohi, H.; Mohtamedifar, N.; Hejazi, F. Intramolecular photoinduced proton transfer in 2-(2’-hydroxyphenyl)benzazole family: A TD-DFT quantum chemical study. Chem. Phys. 2014, 444, 66–76. [Google Scholar] [CrossRef]
- Li, C.; Yang, Y.; Ma, C.; Liu, Y. Effect of amino group on the excited-state intramolecular proton transfer (ESIPT) mechanisms of 2-(2’-hydroxyphenyl)benzoxazole and its amino derivatives. RSC Adv. 2016, 6, 5134–5140. [Google Scholar] [CrossRef]
- Crampton, M.R.; Robotham, I.A. Acidities of some substituted ammonium ions in dimethyl Sulfoxide[dagger]. J. Chem. Res. 1997, 22–23. [Google Scholar] [CrossRef]
- Zoltewicz, J.A.; Deady, L.W. Quaternization of heteroaromatic compounds: Quantitative aspects. Adv. Heterocyclic Chem. 1978, 22, 71–121. [Google Scholar] [CrossRef]
- Jayabharathi, J.; Thanikachalam, V.; Jayamoorthy, K.; Srinivasan, N. Synthesis, spectral studies and solvatochromism of some novel benzimidazole derivatives-ESIPT process. Spectrochim. Acta A 2013, 105, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, D.; Xu, K.; Li, J. Substituent and solvent effects on excited state intramolecular proton transfer in novel 2-(2’-hydroxyphenyl)benzothiazole derivatives. J. Photochem. Photobiol. A 2009, 205, 61–69. [Google Scholar] [CrossRef]
- Latha, V.; Annaraj, B.; Neelakantan, M.A. ESIPT inspired dual fluorescent probe (Z)-3-((4-(4-aminobenzyl) phenyl) amino)-1,3-diphenylprop-2-en-1-one: Experimental and DFT based approach to photophysical properties. Spectrochim. Acta A 2014, 133, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.L.; Domcke, W. Theoretical investigation of potential-energy surfaces relevant for excited-state hydrogen-transfer in o-hydroxybenzaldehyde. Chem. Phys. 1994, 184, 115–124. [Google Scholar] [CrossRef]
- Catalan, J.; Palomar, J.; DePaz, J.L.G. Intramolecular proton or hydrogen-atom transfer in the ground and excited states of 2-hydroxybenzoyl compounds. J. Phis. Chem. A 1997, 101, 7914–7921. [Google Scholar] [CrossRef]
- Maheshwari, S.; Chowdhury, A.; Sathyamurthy, N.; Mishra, H.; Tripathi, H.B.; Panda, M.; Chandrasekhar, J. Ground and excited state intramolecular proton transfer in salicylic acid: An ab initio electronic structure investigation. J. Phys. Chem. A 1999, 103, 6257–6262. [Google Scholar] [CrossRef]
- De Prasad, S.; Ash, S.; Dalai, S.; Misra, A. A DFT-based comparative study on the excited states intramolecular proton transfer in 1-hydroxy-2-naphthaldehyde and 2-hydroxy-3-naphthaldehyde. J. Mol. Struct. Theochem. 2007, 807, 33–41. [Google Scholar] [CrossRef]
- Beg, H.; De Prasad, S.; Ash, S.; Misra, A. Use of polarizability and chemical hardness to locate the transition state and the potential energy curve for double proton transfer reaction: A DFT based study. Comput. Theor. Chem. 2012, 984, 13–18. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. General performance of density functionals. J. Phys. Chem. A 2007, 111, 10439–10452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhao, J.; Cui, Y.; Wang, Q.; Dai, Y.; Song, P.; Xia, L. Theoretical study on the excited-state intramolecular proton-transfer reaction of 10-hydroxybenzo[h]quinoline in methanol and cyclohexane. J. Lumin. 2015, 161, 1–6. [Google Scholar] [CrossRef]
- Deshmukh, M.S.; Sekar, N. Photophysical properties of ESIPT inspired fluorescent 2-(2-hydroxyphenyl)-6-methylimidazo[4,5-f]isoindole-5,7(1H,6H)-dione and its derivative: Experimental and DFT based approach. Spectrochim. Acta A 2015, 135, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.K.; Guchhait, N. TD-DFT investigation of the potential energy surface for Excited-State Intramolecular Proton Transfer (ESIPT) reaction of 10-hydroxybenzo[h]quinoline: Topological (AIM) and population (NBO) analysis of the intramolecular hydrogen bonding interaction. J. Lumin. 2011, 131, 1918–1926. [Google Scholar] [CrossRef]
- Chai, S.; Cong, S.L. Excited state intramolecular proton transfer and substituent effect of 10-hydroxybenzo [h] quinoline: A time-dependent density functional theory study. Comput. Theor. Chem. 2014, 1034, 80–84. [Google Scholar] [CrossRef]
- Deshmukh, M.S.; Sekar, N. Chemiluminescence properties of luminol related o-hydroxybenzimidazole analogues: Experimental and DFT based approach to photophysical properties. Dyes Pigment. 2015, 113, 189–199. [Google Scholar] [CrossRef]
- Kataria, S.; Rhyman, L.; Ramasami, P.; Sekar, N. Comprehensive DFT and TD-DFT studies on the photophysical properties of 5,6-dichloro-1,3-Bis(2-Pyridylimino)-4,7-Dihydroxyisoindole: A new class of ESIPT fluorophore. J. Fluoresc. 2016, 26, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.J.; Han, K.L. Novel infrared spectra for intermolecular dihydrogen bonding of the phenol-borane-trimethylamine complex in electronically excited state. J. Chem. Phys. 2007, 127, 024306. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.J.; Han, K.L. Effects of hydrogen bonding on tuning photochemistry: Concerted hydrogen-bond strengthening and weakening. Chem. Phys. Chem. 2008, 9, 1842–1846. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.J.; Han, K.L. Time-dependent density functional theory study on hydrogen-bonded intramolecular charge-transfer excited state of 4-dimethylamino-benzonitrile in methanol. J. Comput. Chem. 2008, 29, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.J.; Northrop, B.H.; Han, K.L.; Stang, P.J. The effect of intermolecular hydrogen bonding on the fluorescence of a bimetallic platinum complex. J. Phys. Chem. A 2010, 114, 9007–9013. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Li, P.; Li, G.; Zhao, G.; Chu, T.; Han, K. A Near-IR reversible fluorescent probe modulated by selenium for monitoring peroxynitrite and imaging in living cells. J. Am. Chem. Soc. 2011, 133, 11030–11033. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.J.; Chen, R.K.; Sun, M.T.; Liu, J.Y.; Li, G.Y.; Gao, Y.L.; Han, K.L.; Yang, X.C.; Sun, L. Photoinduced intramolecular charge transfer and S-2 fluorescence in thiophene-pi-conjugated donor-acceptor systems: Experimental and TDDFT studies. Chem. Eur. J. 2008, 14, 6935–6947. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.M.; Becke, A.D. Basis-set-free local density-functional calculations of geometries of polyatomic-molecules. J. Chem. Phys. 1993, 99, 3898–3905. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Phys. Chem. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Feller, D.J. The role of databases in support of computational chemistry calculations. J. Comp. Chem 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
Sample Availability: not available. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Closed Form (kcal mol−1) | Open Form (kcal mol −1) | Rotamer (kcal mol−1) | |
|---|---|---|---|
| NE | 0 | 13.55 | 8.47 |
| OE | 0 | 12.55 | 5.77 |
| SE | 0 | 12.68 | 9.48 |
| NE | NE* | OE | OE* | SE | SE* | |
|---|---|---|---|---|---|---|
| O–H | 0.992 | 1.009 | 0.986 | 1.005 | 0.989 | 1.003 |
| H…N | 1.711 | 1.653 | 1.765 | 1.692 | 1.742 | 1.678 |
| X5–C3 | 1.364 | 1.382 | 1.351 | 1.360 | 1.751 | 1.776 |
| X5–C4 | 1.388 | 1.379 | 1.385 | 1.393 | 1.747 | 1.767 |
| δO–H∙∙∙N | 148.8 | 149.7 | 147.0 | 148.9 | 147.9 | 149.7 |
| 1H-Imidazole | 1,3-Oxazole | 1,3-Thiazole | |
|---|---|---|---|
| ΔΔG (kcal mol−1) | −230.61 | −213.79 | −219.25 |
| pKaH | 6.9 a | 0.8 b | 2.5 b |
| Atom | NE | NK | SE | SK | OE | OK |
|---|---|---|---|---|---|---|
| N | −0.4151 | −0.2140 | −0.4254 | −0.2456 | −0.3325 | −0.1723 |
| O | −0.5118 | −0.6061 | −0.4961 | −0.5698 | −0.4933 | −0.5735 |
| H | 0.3309 | 0.3004 | 0.3280 | 0.3167 | 0.3177 | 0.2896 |
| Main Composition | λabs (nm) | f | CI (%) | λemis (nm) | |
|---|---|---|---|---|---|
| NE | HOMO → LUMO HOMO → LUMO+1 | 337.5 310.6 | 0.3850 0.4288 | 94.2 86.6 | 523 |
| OE | HOMO → LUMO HOMO → LUMO+1 | 339.1 304.9 | 0.6724 0.2266 | 94.2 78.4 | 452 |
| SE | HOMO → LUMO HOMO → LUMO+1 | 346.5 302.8 | 0.5478 0.1188 | 93.6 86.3 | 490 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Carvalho, F.; Coutinho Neto, M.D.; Bartoloni, F.H.; Homem-de-Mello, P. Density Functional Theory Applied to Excited State Intramolecular Proton Transfer in Imidazole-, Oxazole-, and Thiazole-Based Systems. Molecules 2018, 23, 1231. https://doi.org/10.3390/molecules23051231
De Carvalho F, Coutinho Neto MD, Bartoloni FH, Homem-de-Mello P. Density Functional Theory Applied to Excited State Intramolecular Proton Transfer in Imidazole-, Oxazole-, and Thiazole-Based Systems. Molecules. 2018; 23(5):1231. https://doi.org/10.3390/molecules23051231
Chicago/Turabian StyleDe Carvalho, Fabricio, Maurício D. Coutinho Neto, Fernando H. Bartoloni, and Paula Homem-de-Mello. 2018. "Density Functional Theory Applied to Excited State Intramolecular Proton Transfer in Imidazole-, Oxazole-, and Thiazole-Based Systems" Molecules 23, no. 5: 1231. https://doi.org/10.3390/molecules23051231
APA StyleDe Carvalho, F., Coutinho Neto, M. D., Bartoloni, F. H., & Homem-de-Mello, P. (2018). Density Functional Theory Applied to Excited State Intramolecular Proton Transfer in Imidazole-, Oxazole-, and Thiazole-Based Systems. Molecules, 23(5), 1231. https://doi.org/10.3390/molecules23051231