Identification and Expression of miRNAs Related to Female Flower Induction in Walnut (Juglans regia L.)


Abstract
1. Introduction
2. Results
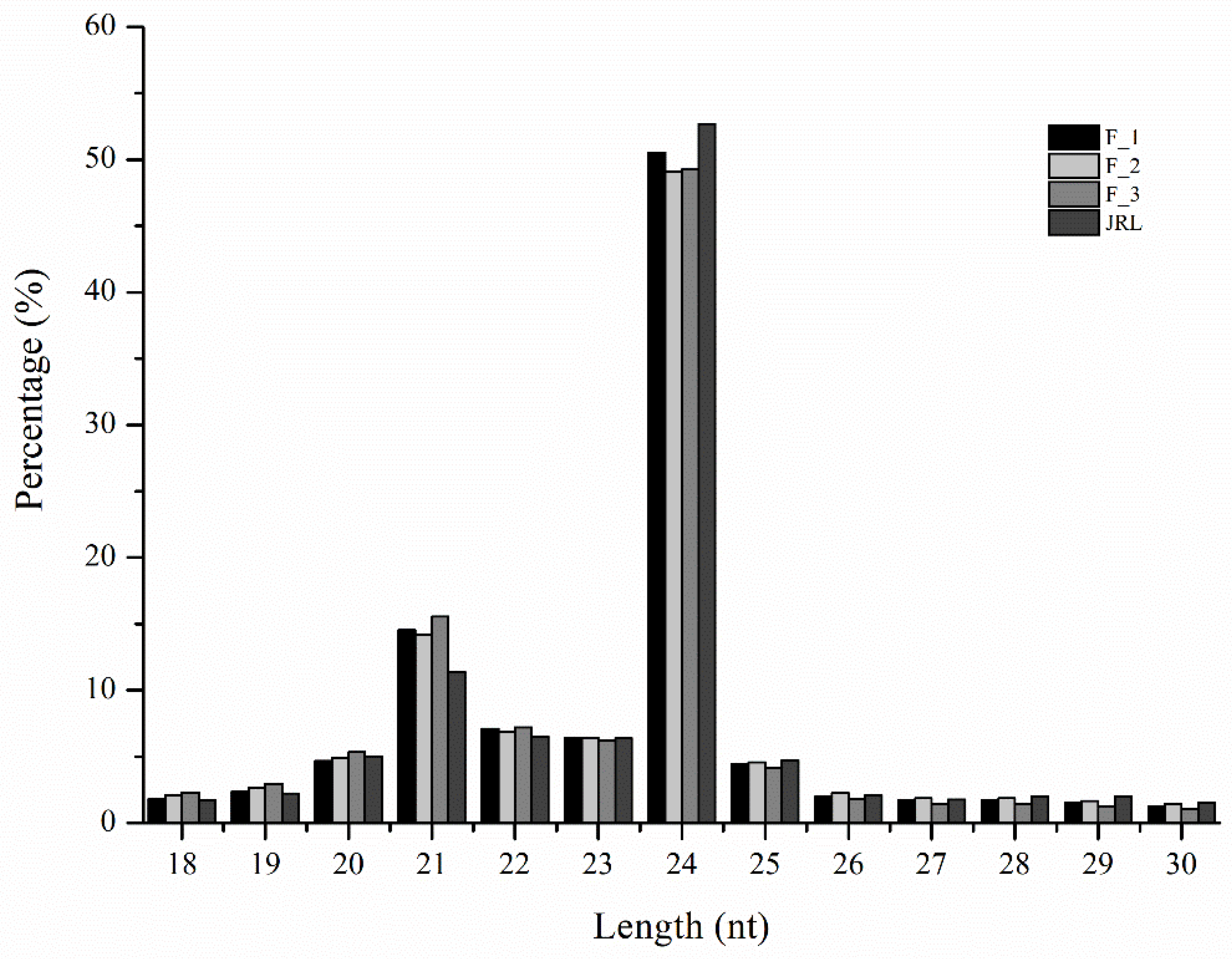
2.1. Analysis of miRNA Sequences
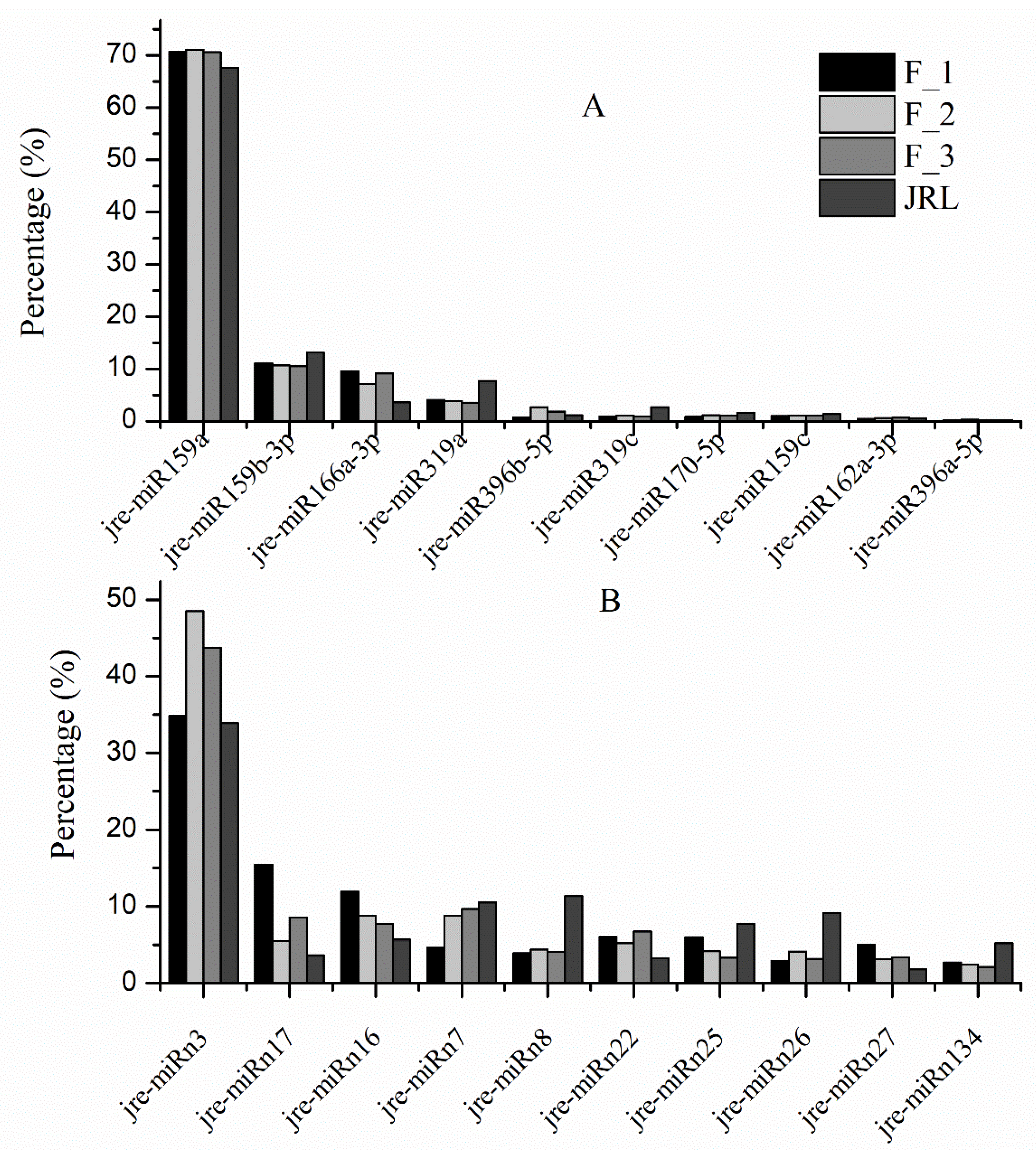
2.2. Identification of Known miRNAs in J. regia
2.3. Identification of Novel miRNAs in J. regia
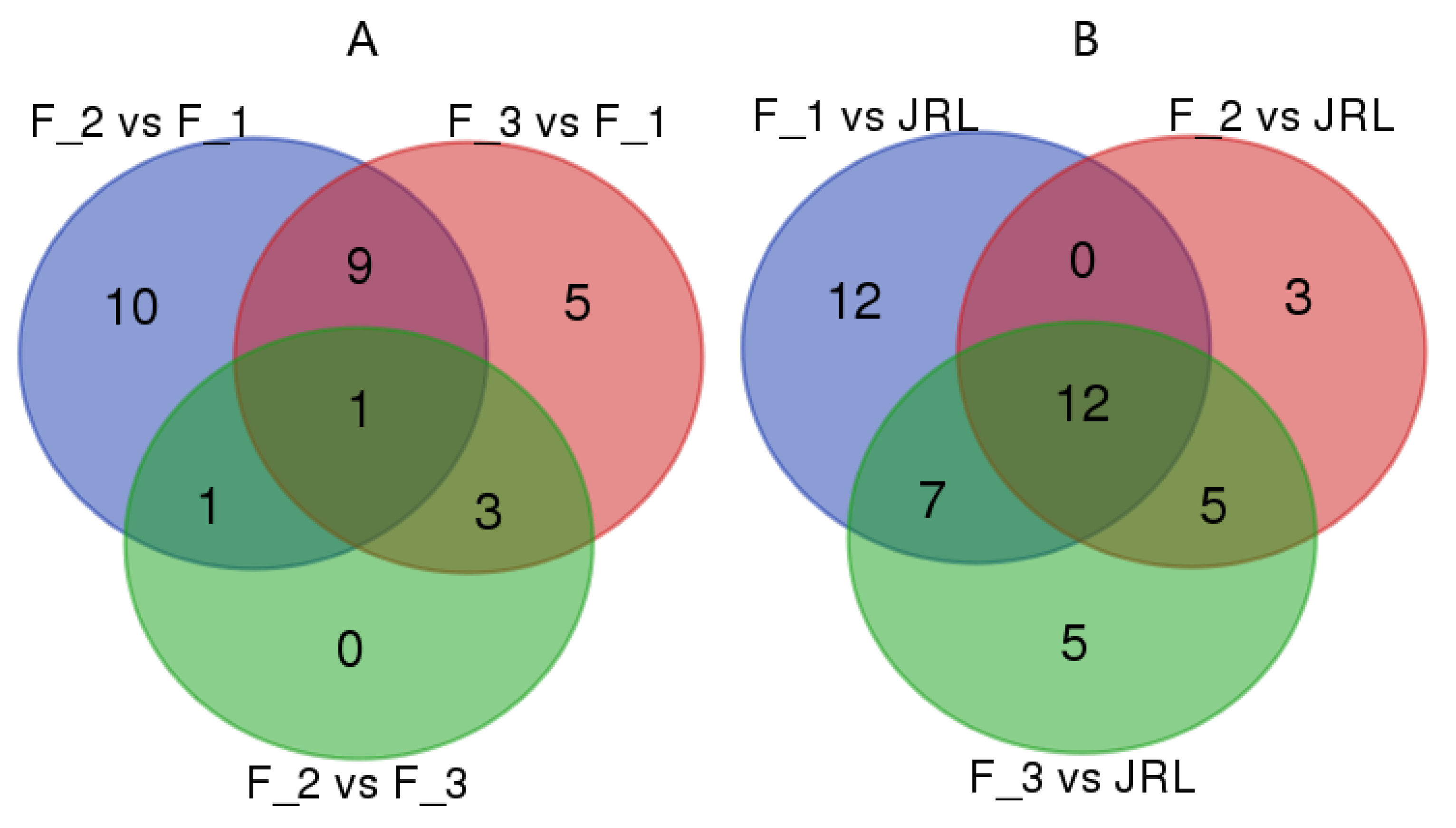
2.4. Differential Expression of miRNAs in J. regia
2.5. RT-qPCR Validation of J. regia miRNAs
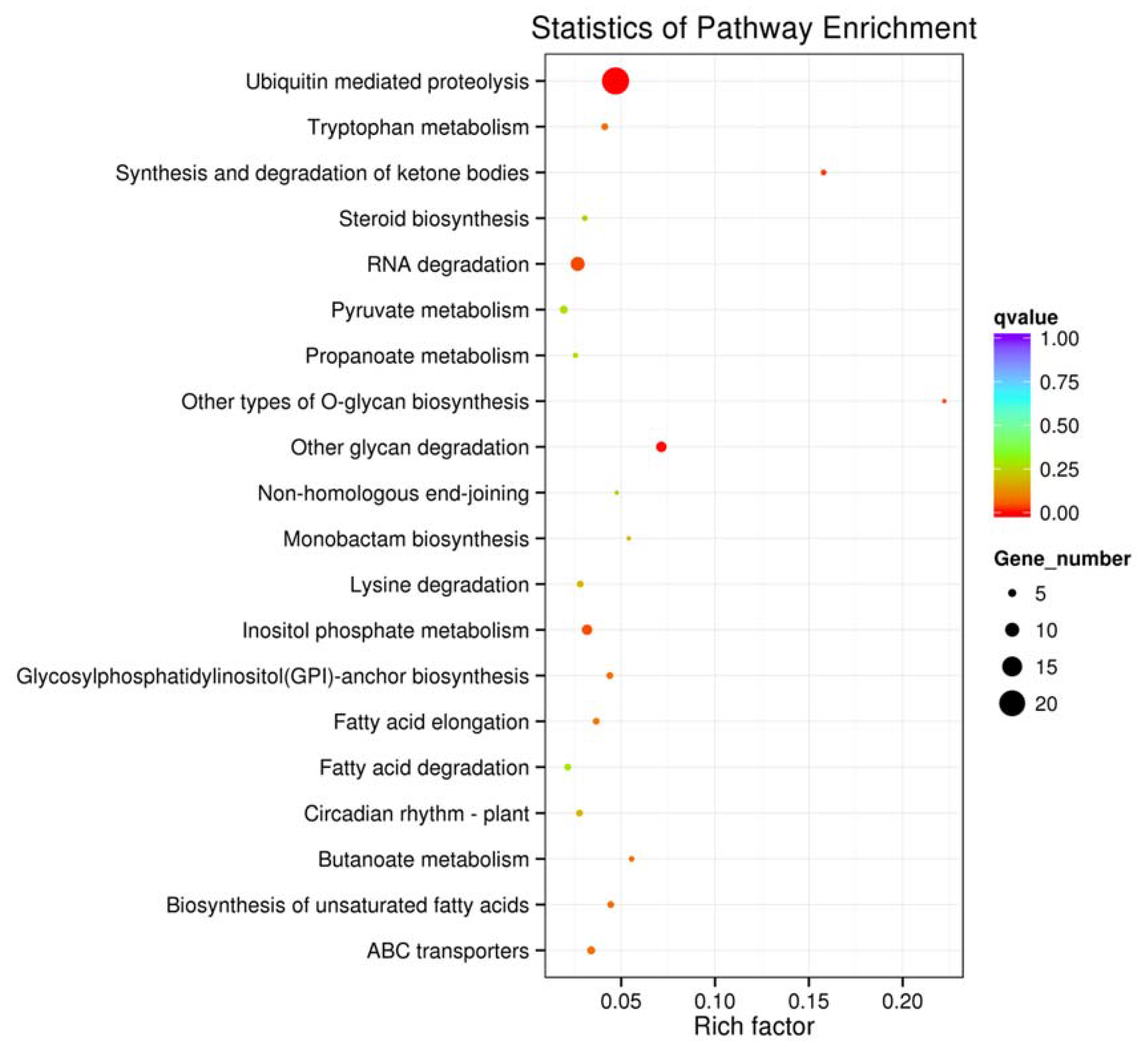
2.6. Target Prediction and Function Analysis of miRNAs in J. regia
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Small RNA Library Construction and Deep Sequencing
4.3. Bioinformatics Analysis of Small RNAs
4.4. Expression Analysis of miRNA
4.5. Validation and Expression of miRNAs by RT-qPCR
4.6. Target Gene Prediction and Function Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Aradhya, M.K.; Potter, D.; Simon, C.J. Cladistic biogeography of Juglans (Juglandaceae) based on chloroplast DNA intergenic spacer sequences. In Darwins Harvest New Approaches to the Origins; Columbia University Press: New York, NY, USA, 2004. [Google Scholar]
- Xi, R. Discussion on the origin of walnut in China. Acta Hortic. 1990, 284, 353–361. [Google Scholar]
- Valiente, J.I.; Albrigo, L.G. Flower bud induction of sweet orange trees [Citrus Sinensis (L.) Osbeck]: Effect of low temperatures, crop load, and bud age. J. Am. Soc. Hortic. Sci. 2004, 129, 158–164. [Google Scholar]
- Ben-Tal, Y. Flowering: Its control by vegetative growth inhibition. In V International Symposium on Growth Regulators in Fruit Production; International Society for Horticultural Science: Leuven, Belgium, 1986; pp. 329–336. [Google Scholar]
- Xia, X.; Xi, R. The periods of physiological and morphological differentiation of pistillate flower buds in walnut (Juglans regia L.). J. Agric. Univ. Hebei 1989, 18–21. [Google Scholar]
- Khan, M.R.G.; Ai, X.Y.; Zhang, J.Z. Genetic regulation of flowering time in annual and perennial plants. Wiley Interdiscip. Rev. RNA 2014, 5, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843. [Google Scholar]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Eleonora, S.; Stephen, J. The role of microRNAs in the control of flowering time. J. Exp. Bot. 2014, 65, 365–380. [Google Scholar]
- Chen, X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Chuck, G.; Meeley, R.; Irish, E.; Sakai, H.; Hake, S. The maize tasselseed4 microRNA controls sex determination and meristem cell fate by targeting Tasselseed6/indeterminate spikelet1. Nat. Genet. 2007, 39, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Terzi, L.C.; Simpson, G.G. Regulation of flowering time by RNA processing. Curr. Top. Microbiol. Immunol. 2008, 326, 201–218. [Google Scholar] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Montgomery, T.A.; Howell, M.D.; Allen, E.; Dvorak, S.K.; Alexander, A.L.; Carrington, J.C. Regulation of auxin response factor3 by TAS3 ta-siRNA affects developmental timing and patterning in Arabidopsis. Curr. Biol. 2006, 16, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Ahn, H.J.; Chiou, T.J.; Ahn, J.H. The role of the miR399-PHO2 module in the regulation of flowering time in response to different ambient temperatures in Arabidopsis thaliana. Mol. Cells 2011, 32, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Ai, X.Y.; Guo, W.W.; Peng, S.A.; Deng, X.X.; Hu, C.G. Identification of miRNAs and their target genes using deep sequencing and degradome analysis in Trifoliate orange [Poncirus trifoliate (L.) Raf]. Mol. Biotechnol. 2012, 51, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Huang, J.Q.; Huang, Y.J.; Li, Z.; Zheng, B.S. Discovery and profiling of novel and conserved microRNAs during flower development in Carya cathayensis via deep sequencing. Planta 2012, 236, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, L.; Wang, T.; Tian, Q.; Zhang, W.H. Identification of drought-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genom. 2011, 12, 367. [Google Scholar]
- Li, X.; Jin, F.; Jin, L.; Jackson, A.; Ma, X.; Shu, X.; Wu, D.; Jin, G. Characterization and comparative profiling of the small RNA transcriptomes in two phases of flowering in Cymbidium ensifolium. BMC Genom. 2015, 16, 622. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Chen, W.; Zhang, C.; Korir, N.K.; Yu, H.; Ma, Z.; Fang, J. Deep sequencing discovery of novel and conserved microRNAs in Trifoliate orange (Citrus trifoliata). BMC Genom. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Kou, S.J.; Wu, X.M.; Liu, Z.; Liu, Y.L.; Xu, Q.; Guo, W.W. Selection and validation of suitable reference genes for miRNA expression normalization by quantitative RT-PCR in citrus somatic embryogenic and adult tissues. Plant Cell Rep. 2012, 31, 2151–2163. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Zhang, D.; Li, Y.; Zhao, C.; Zhang, S.; Shen, Y.; An, N.; Han, M. Genome-wide identification of vegetative phase transition-associated microRNAs and target predictions using degradome sequencing in Malus hupehensis. BMC Genom. 2014, 15, 1125. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, D.; Liu, Y.; Wang, L.; Qiao, X.; Zhang, S. Identification of miRNAs involved in pear fruit development and quality. BMC Genom. 2014, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Paola, D.D.; Zuluaga, D.L.; Sonnante, G. The miRNAome of durum wheat: Isolation and characterisation of conserved and novel microRNAs and their target genes. BMC Genom. 2016, 17, 505. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Wall, P.K.; Diloreto, S.; Depamphilis, C.W.; Carlson, J.E. Conservation and divergence of microRNAs in Populus. BMC Genom. 2007, 8, 481. [Google Scholar] [CrossRef] [PubMed]
- Kulcheski, F.R.; Oliveira, L.F.D.; Molina, L.G.; Almerão, M.P.; Rodrigues, F.A.; Marcolino, J.; Barbosa, J.F.; Stolf-Moreira, R.; Nepomuceno, A.L.; Marcelino-Guimarães, F.C. Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genom. 2011, 12, 307. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.; Meng, X.; Ma, C.; Yi, G. Identification of miRNAs involved in fruit ripening in cavendish bananas by deep sequencing. BMC Genom. 2015, 16, 776. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Benham, A.L.; Zhu, H.; Khan, M.F.; Reid, J.G.; Nagaraja, A.K.; Fountain, M.D., Jr.; Dziadek, O.; Han, D.; Ma, L. Discovery of novel microRNAs in female reproductive tract using next generation sequencing. PLoS ONE 2010, 5, e9637. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.T.; Wang, M.; Fu, S.X.; Yang, W.C.; Qi, C.K.; Wang, X.J. Small RNA profiling in two Brassica napus cultivars identifies microRNAs with oil production-and development-correlated expression and new small RNA classes. Plant Physiol. 2012, 158, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Ru, P.; Xu, L.; Ma, H.; Huang, H. Plant fertility defects induced by the enhanced expression of microRNA167. Cell Res. 2006, 16, 457. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.F.; Tian, Q.; Reed, J.W. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 2006, 133, 4211–4218. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kim, B.; Song, S.K.; Heo, J.O.; Yu, N.I.; Lee, S.A.; Kim, M.; Kim, D.G.; Sohn, S.O.; Lim, C.E.; et al. Large-scale analysis of the gras gene family in Arabidopsis thaliana. Plant Mol. Biol. 2008, 67, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Moxon, S.; Schwach, F.; Dalmay, T.; Maclean, D.; Studholme, D.J.; Moulton, V. A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics 2008, 24, 2252–2253. [Google Scholar] [CrossRef] [PubMed]
- Ming, W.; Yang, S.; Shi, S.; Tian, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 1–10. [Google Scholar]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L. Integrated profiling of microRNAs and mRNAs: MicroRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. Degseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ma, Y.K.; Chen, T.; Wang, M.; Wang, X.J. Psrobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. Kegg for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds F_1, F_2, F_3 and JRL are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F_1 | F_2 | F_3 | JRL | |
|---|---|---|---|---|
| Raw reads | 12,151,175 | 11,933,426 | 11,798,939 | 11,579,086 |
| Clean reads | 11,968,359 | 11,705,783 | 11,580,851 | 11,324,267 |
| sRNA reads with 18–30 nt | 10,765,695 | 10,020,557 | 10,094,699 | 8,440,666 |
| Mapped sRNA reads | 4,873,510 | 4,702,765 | 4,608,652 | 3,525,507 |
| Known miRNA | 964,182 | 828,091 | 952,787 | 603,457 |
| Novel miRNA | 129,206 | 87,699 | 95,201 | 58,900 |
| rRNA, tRNA, snRNA, snoRNA, ta-siRNA | 368,813 | 378,828 | 344,701 | 222,502 |
| Others | 3,411,309 | 3,408,147 | 3,215,963 | 2,640,648 |
| MiRNA | Target Genes | Gene Description |
|---|---|---|
| jre-miRn69 | Cluster-14922.52553, Cluster-14922.56228, Cluster-14922.99553, Cluster-14922.71416, Cluster-14922.41338, Cluster-14922.41337, Cluster-14922.50813, Cluster-14922.76833 | Ethylene-responsive transcription factor RAP2-7 OS = Arabidopsis thaliana GN = RAP2-7 PE = 2 SV = 2 |
| Cluster-14922.29861, Cluster-14922.29860, Cluster-14922.35646, Cluster-14922.29863 | Floral homeotic protein APETALA 2 OS = Arabidopsis thaliana GN = AP2 PE = 1 SV = 1 | |
| jre-miR157a-5p | Cluster-14922.56642, Cluster-14922.98238, Cluster-14922.32383, Cluster-14922.41178 | Squamosa promoter-binding protein 1 OS = Antirrhinum majus GN = SBP1 PE = 2 SV = 1 |
| Cluster-14922.65142, Cluster-14922.65141, Cluster-14922.63070, Cluster-14922.39484 | Squamosa promoter-binding-like protein 18 OS = Oryza sativa subsp. Japonica GN = SPL18 PE = 2 SV = 1 | |
| Cluster-14922.54371, Cluster-14922.56749, Cluster-14922.54372 | Squamosa promoter-binding-like protein 9 OS = Arabidopsis thaliana GN = SPL9 PE = 2 SV = 2 | |
| Cluster-14922.69694 | Squamosa promoter-binding-like protein 13A OS = Arabidopsis thaliana GN = SPL13A PE = 2 SV = 1 | |
| Cluster-14922.29571, Cluster-14922.29570, Cluster-14922.53662, Cluster-14922.43359 | Squamosa promoter-binding-like protein 6 OS = Arabidopsis thaliana GN = SPL6 PE = 2 SV = 2 | |
| Cluster-14922.44489, Cluster-14922.44491, Cluster-14922.47505 | Squamosa promoter-binding-like protein 7 OS = Oryza sativa subsp. Indica GN = SPL7 PE = 2 SV = 1 | |
| Cluster-14922.22729, Cluster-14922.24187, Cluster-14922.24188, Cluster-14922.35309, Cluster-14922.65226 | Squamosa promoter-binding-like protein 16 OS = Oryza sativa subsp. Japonica GN = SPL16 PE = 2 SV = 1 | |
| Cluster-14922.60789, Cluster-14922.63041 | Squamosa promoter-binding-like protein 4 OS = Arabidopsis thaliana GN = SPL4 PE = 1 SV = 1 | |
| jre-miR160a-5p | Cluster-14922.45926, Cluster-14922.65024, Cluster-14922.86555, Cluster-14922.65022, Cluster-14922.94990, Cluster-14922.67646 Cluster-14922.58894, Cluster-14922.92805, Cluster-14922.45431, Cluster-14922.86030, Cluster-14922.85483 | Auxin response factor 18 OS = Oryza sativa subsp. Japonica GN = ARF18 PE = 2 SV = 1 |
| jre-miR167a-5p | Cluster-14922.60064, Cluster-14922.57359, Cluster-14922.61850, Cluster-14922.62563, Cluster-14922.54061 | Auxin response factor 8 OS = Arabidopsis thaliana GN = ARF8 PE = 2 SV = 2 |
| jre-miR171b-3p, jre-miRn46, jre-miRn49 | Cluster-14922.65200 | Scarecrow-like protein 6 OS = Arabidopsis thaliana GN = SCL6 PE = 1 SV = 1 |
| jre-miRn49 | Cluster-14922.90636 | Probable indole-3-acetic acid-amido synthetase GH3.1 OS = Arabidopsis thaliana GN = GH3.1 PE = 2 SV = 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Quan, S.; Xu, H.; Ma, L.; Niu, J. Identification and Expression of miRNAs Related to Female Flower Induction in Walnut (Juglans regia L.). Molecules 2018, 23, 1202. https://doi.org/10.3390/molecules23051202
Zhou L, Quan S, Xu H, Ma L, Niu J. Identification and Expression of miRNAs Related to Female Flower Induction in Walnut (Juglans regia L.). Molecules. 2018; 23(5):1202. https://doi.org/10.3390/molecules23051202
Chicago/Turabian StyleZhou, Li, Shaowen Quan, Hang Xu, Li Ma, and Jianxin Niu. 2018. "Identification and Expression of miRNAs Related to Female Flower Induction in Walnut (Juglans regia L.)" Molecules 23, no. 5: 1202. https://doi.org/10.3390/molecules23051202
APA StyleZhou, L., Quan, S., Xu, H., Ma, L., & Niu, J. (2018). Identification and Expression of miRNAs Related to Female Flower Induction in Walnut (Juglans regia L.). Molecules, 23(5), 1202. https://doi.org/10.3390/molecules23051202