
Detecting Differential Transcription Factor Activity from ATAC-Seq Data



Abstract
1. Introduction
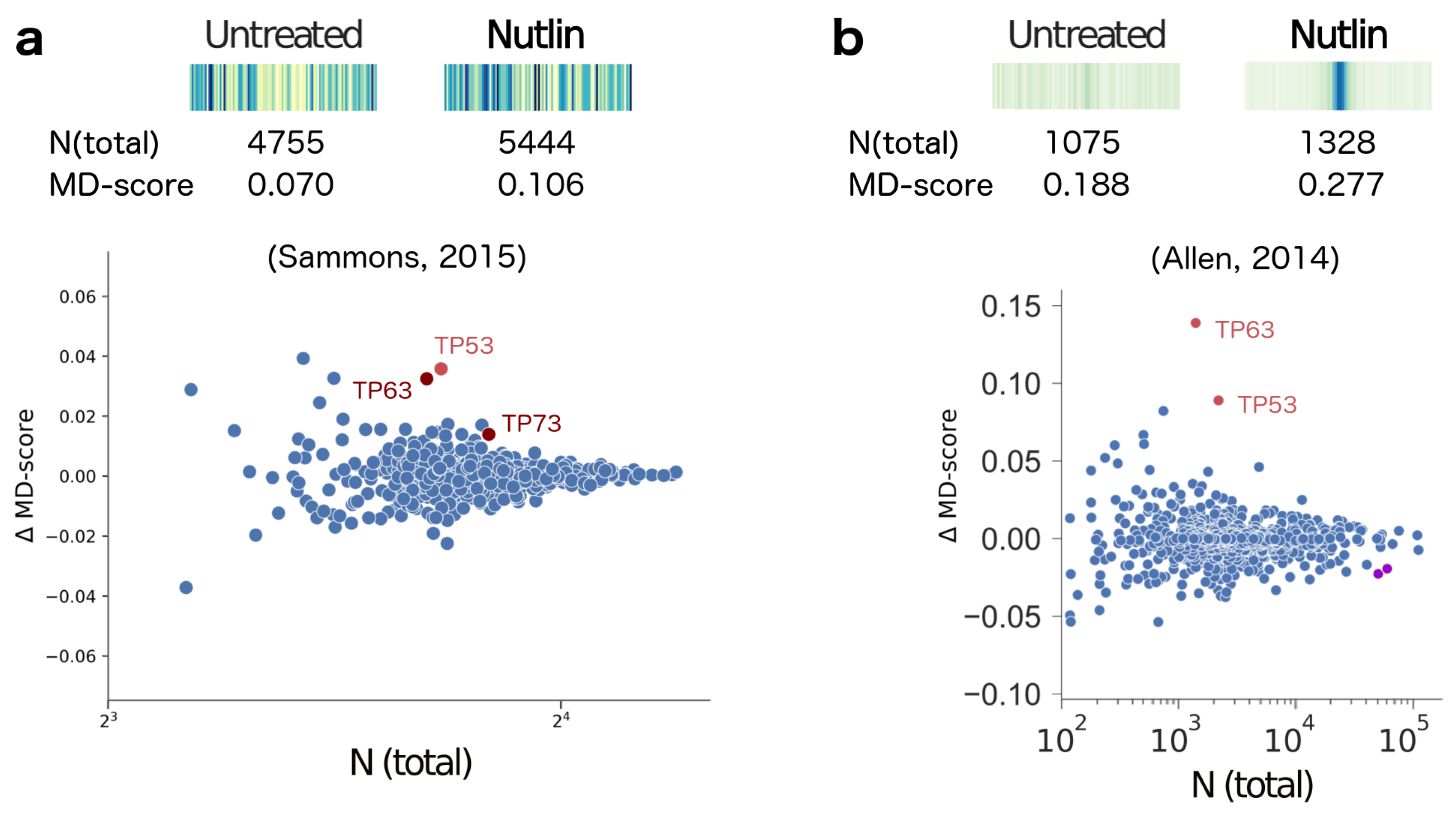
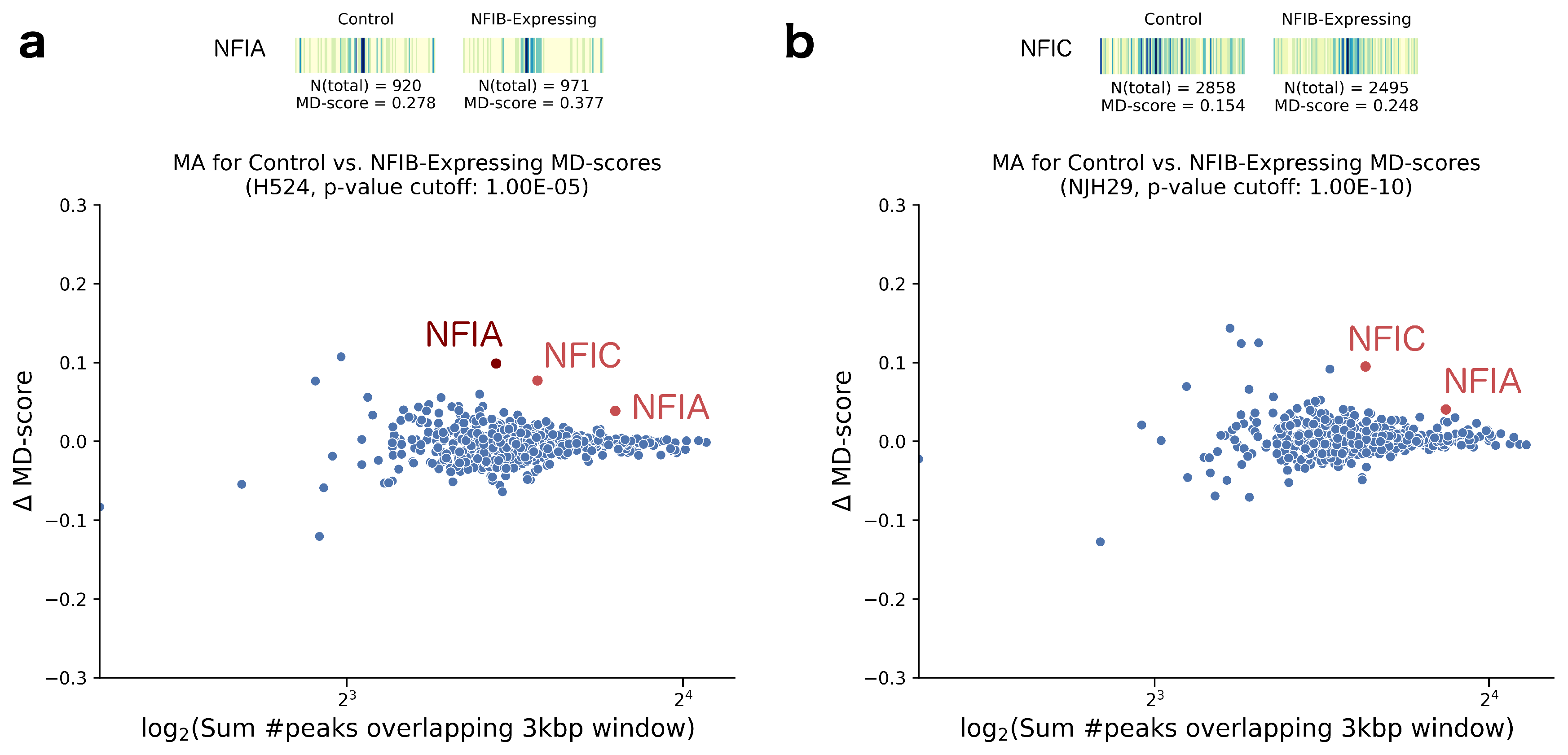
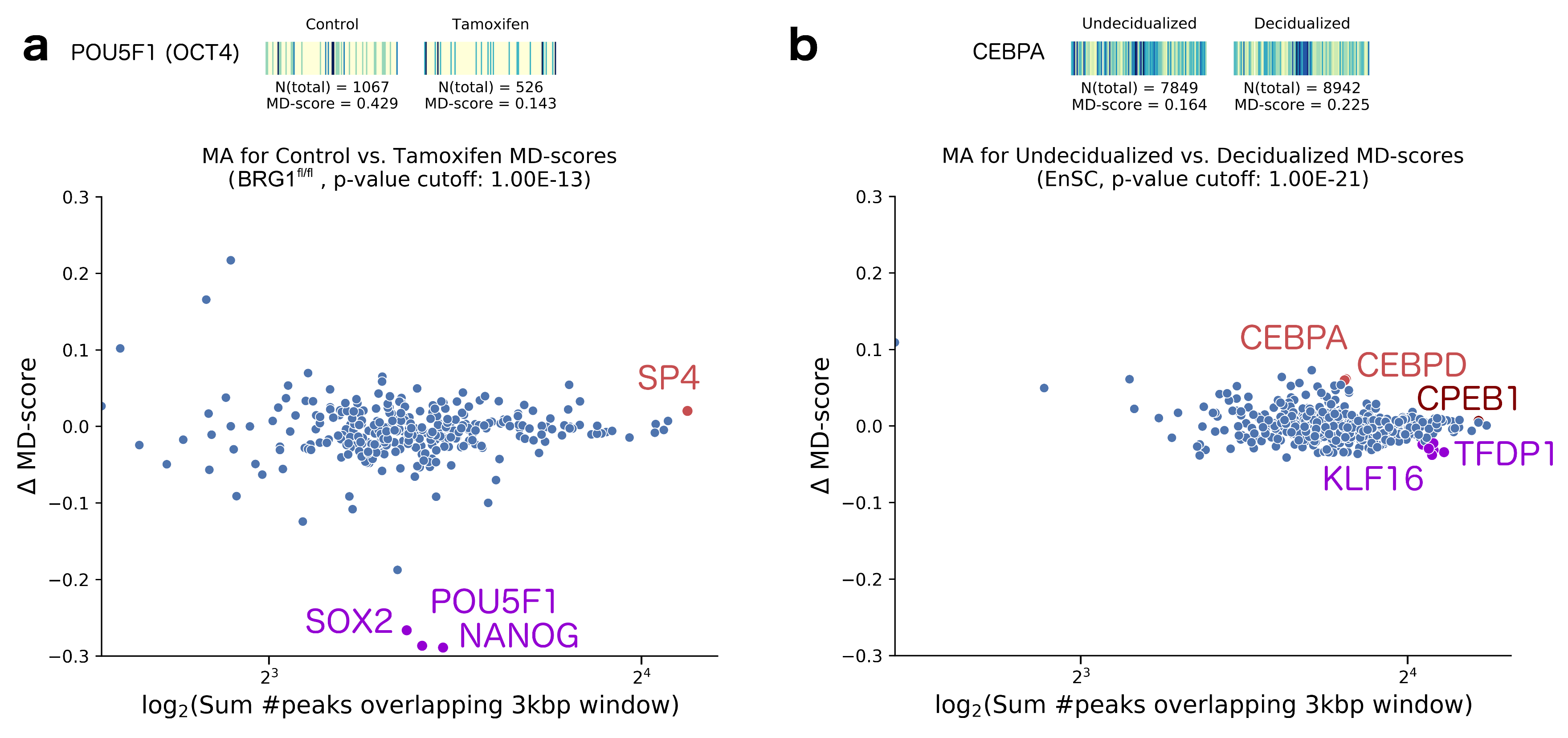
2. Results
3. Discussion
4. Materials and Methods
4.1. Processing Pipeline
4.2. Public Datasets
4.3. DAStk Software
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| TF | transcription factor |
| ATAC | assay for transposase-accessible Chromatin |
| SNP | single nucleotide polymorphisms |
| ChIP | chromatin immunoprecipitation |
| eRNA | enhancer RNA |
| MD-score | motif displacement score |
| SCLC | small cell lung carcinoma |
References
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Latchman, D.S. Transcription-Factor Mutations and Disease. N. Engl. J. Med. 1996, 334, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, M.C.J.; Kuiper, R.P.; Carmichael, C.L.; Wilkins, E.J.; Dors, N.; Carmagnac, A.; Schouten-van Meeteren, A.Y.N.; Li, X.; Stankovic, M.; Kamping, E.; et al. Novel RUNX1 mutations in familial platelet disorder with enhanced risk for acute myeloid leukemia: Clues for improved identification of the FPD/AML syndrome. Leukemia 2009, 24, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.J.; Liu, H.; Carlson, J.C.; Shaffer, J.R.; Feingold, E.; Wehby, G.; Laurie, C.A.; Jain, D.; Laurie, C.C.; Doheny, K.F.; et al. A Genome-wide Association Study of Nonsyndromic Cleft Palate Identifies an Etiologic Missense Variant in GRHL3. Am. J. Hum. Genet. 2016, 98, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Kelley, P.; Kenyon, J.; Hoover, D. Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. J. Med. Genet. 2000, 37, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Tassabehji, M.; Newton, V.E.; Read, A.P. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat. Genet. 1994, 8, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Marini, M.; Bocciardi, R.; Gimelli, S.; Di Duca, M.; Divizia, M.T.; Baban, A.; Gaspar, H.; Mammi, I.; Garavelli, L.; Cerone, R.; et al. A spectrum of LMX1B mutations in Nail-Patella syndrome: New point mutations, deletion, and evidence of mosaicism in unaffected parents. Genet. Med. 2010, 12, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Stiles, A.R.; Simon, M.T.; Stover, A.; Eftekharian, S.; Khanlou, N.; Wang, H.L.; Magaki, S.; Lee, H.; Partynski, K.; Dorrani, N.; et al. Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol. Genet. Metab. 2016, 119, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.M.; Rajakumar, G. Genetics of Congenital Heart Defects: The NKX2-5 Gene, a Key Player. Genes 2016, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Newbury-Ecob, R.A.; Terrett, J.A.; Wilson, D.I.; Curtis, A.R.J.; Yi, C.H.; Gebuhr, T.; Bullen, P.J.; Robson, S.C.; Strachan, T.; et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 1997, 15, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Narumi, Y.; Nishina, S.; Tokimitsu, M.; Aoki, Y.; Kosaki, R.; Wakui, K.; Azuma, N.; Murata, T.; Takada, F.; Fukushima, Y.; et al. Identification of a novel missense mutation of MAF in a Japanese family with congenital cataract by whole exome sequencing: A clinical report and review of literature. Am. J. Med. Genet. Part A 2014, 164, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. Pitt-Hopkins Syndrome: Intellectual disability due to loss of TCF4-regulated gene transcription. Exp. Mol. Med. 2013, 45, e21. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Young, R.A. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Haugen, E.; Sandstrom, R.; Vierstra, J.; Shafer, A.; Kaul, R.; Stamatoyannopoulos, J.A. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 2015, 47, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Corradin, O.; Scacheri, P.C. Enhancer variants: Evaluating functions in common disease. Genome Med. 2014, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; Ewels, P.A.; et al. Mapping long-range promoter contacts in human cells with high- resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.T.; Li, W.; Rosenfeld, M.G.; Glass, C.K. Enhancer {RNAs} and regulated transcriptional programs. Trends Biochem. Sci. 2014, 39, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2011, 40, D930. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Farnham, P.J. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 2009, 10, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Gade, P.; Kalvakolanu, D.V. Chromatin Immunoprecipitation Assay as a Tool for Analyzing Transcription Factor Activity. Methods Mol. Biol. (Clifton, N. J.) 2012, 809, 85–104. [Google Scholar]
- Whitfield, T.W.; Wang, J.; Collins, P.J.; Partridge, E.C.; Aldred, S.F.; Trinklein, N.D.; Myers, R.M.; Weng, Z. Functional analysis of transcription factor binding sites in human promoters. Genome Biol. 2012, 13, R50. [Google Scholar] [CrossRef] [PubMed]
- Spivakov, M. Spurious transcription factor binding: Non-functional or genetically redundant? BioEssays 2014, 36, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Cusanovich, D.A.; Pavlovic, B.; Pritchard, J.K.; Gilad, Y. The Functional Consequences of Variation in Transcription Factor Binding. PLoS Genet. 2014, 10, e1004226. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Consortium, R.E.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Steinhauser, S.; Kurzawa, N.; Eils, R.; Herrmann, C. A comprehensive comparison of tools for differential ChIP-seq analysis. Brief. Bioinf. 2016, 17, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Hesselberth, J.R.; Chen, X.; Zhang, Z.; Sabo, P.J.; Sandstrom, R.; Reynolds, A.P.; Thurman, R.E.; Neph, S.; Kuehn, M.S.; Noble, W.S.; et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods 2009, 6, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hoffman, M.M.; Bilmes, J.A.; Hesselberth, J.R.; Noble, W.S. A dynamic Bayesian network for identifying protein-binding footprints from single molecule-based sequencing data. Bioinformatics 2010, 26, i334–i342. [Google Scholar] [CrossRef] [PubMed]
- Neph, S.; Vierstra, J.; Stergachis, A.B.; Reynolds, A.P.; Haugen, E.; Vernot, B.; Thurman, R.E.; John, S.; Sandstrom, R.; Johnson, A.K.; et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012, 489, 83–90. [Google Scholar] [CrossRef] [PubMed]
- He, H.H.; Meyer, C.A.; Hu, S.S.; Chen, M.W.; Zang, C.; Liu, Y.; Rao, P.K.; Fei, T.; Xu, H.; Long, H.; et al. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat. Methods 2013, 11, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.; Goldstein, I.; Hager, G.L. Bivariate Genomic Footprinting Detects Changes in Transcription Factor Activity. Cell Rep. 2017, 19, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Kulakovskiy, I.V.; Medvedeva, Y.A.; Schaefer, U.; Kasianov, A.S.; Vorontsov, I.E.; Bajic, V.B.; Makeev, V.J. HOCOMOCO: A comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 2013, 41, D195–D202. [Google Scholar] [CrossRef] [PubMed]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef] [PubMed]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC® and its module TRANSCompel®: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed]
- Portales-Casamar, E.; Thongjuea, S.; Kwon, A.T.; Arenillas, D.; Zhao, X.; Valen, E.; Yusuf, D.; Lenhard, B.; Wasserman, W.W.; Sandelin, A. JASPAR 2010: The greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010, 38, D105–D110. [Google Scholar] [CrossRef] [PubMed]
- Pique-Regi, R.; Degner, J.F.; Pai, A.A.; Gaffney, D.J.; Gilad, Y.; Pritchard, J.K. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011, 21, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, R.I.; Hashimoto, T.; O’Donnell, C.W.; Lewis, S.; Barkal, A.A.; van Hoff, J.P.; Karun, V.; Jaakkola, T.; Gifford, D.K. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nature Biotechnol. 2014, 32, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Segal, E.; Shapira, M.; Regev, A.; Pe’er, D.; Botstein, D.; Koller, D.; Friedman, N. Module networks: Identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 2003, 34, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Regev, A.; Roy, S. Comparative Analysis of Gene Regulatory Networks: From Network Reconstruction to Evolution. Annu. Rev. Cell Dev. Biol. 2015, 31, 399–428. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, S.; Hughes, J.D.; Campbell, M.J.; Cho, R.J.; Church, G.M. Systematic determination of genetic network architecture. Nat. Genet. 1999, 22, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Pilpel, Y.; Sudarsanam, P.; Church, G.M. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat. Genet. 2001, 29, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Balwierz, P.J.; Pachkov, M.; Arnold, P.; Gruber, A.J.; Zavolan, M.; van Nimwegen, E. ISMARA: Automated modeling of genomic signals as a democracy of regulatory motifs. Genome Res. 2014, 24, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.N.; Therneau, T.M.; Zhang, Y.; Poland, G.A.; Kocher, J.P. Calculating Sample Size Estimates for RNA Sequencing Data. J. Comput. Biol. 2013, 20, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Core, L.J.; Waterfall, J.J.; Lis, J.T. Nascent RNA Sequencing Reveals Widespread Pausing and Divergent Initiation at Human Promoters. Science 2008, 322, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.; Fuda, N.J.; Core, L.J.; Lis, J.T. Precise Maps of RNA Polymerase Reveal How Promoters Direct Initiation and Pausing. Science 2013, 339, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.; Wang, S.; Soares, F.; Fischer, M.; Meng, F.; Du, Z.; Lin, C.; Meyer, C.; DeCaprio, J.A.; et al. Transcriptional landscape of the human cell cycle. Proc. Natl. Acad. Sci. USA 2017, 114, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Hah, N.; Murakami, S.; Nagari, A.; Danko, C.G.; Kraus, W.L. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013, 23, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Chae, M.; Krishnakumar, R.; Danko, C.G.; Kraus, W.L. Dynamic reorganization of the AC16 cardiomyocyte transcriptome in response to TNFα signaling revealed by integrated genomic analyses. BMC Genom. 2014, 15, 155. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.A.; Mellert, H.; Dengler, V.; Andryzik, Z.; Guarnieri, A.; Freeman, J.A.; Luo, X.; Kraus, W.L.; Dowell, R.D.; Espinosa, J.M. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife 2014, 3, e02200. [Google Scholar] [CrossRef] [PubMed]
- Puc, J.; Kozbial, P.; Li, W.; Tan, Y.; Liu, Z.; Suter, T.; Ohgi, K.A.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Ligand-Dependent Enhancer Activation Regulated by Topoisomerase-I Activity. Cell 2015, 160, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Danko, C.G.; Hyland, S.L.; Core, L.J.; Martins, A.L.; Waters, C.T.; Lee, H.W.; Cheung, V.G.; Kraus, W.L.; Lis, J.T.; Siepel, A. Identification of active transcriptional regulatory elements from GRO-seq data. Nat. Meth. 2015, 12, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Azofeifa, J.G.; Allen, M.A.; Hendrix, J.R.; Read, T.; Rubin, J.D.; Dowell, R.D. Enhancer RNA profiling predicts transcription factor activity. Genome Res. 2018, 28, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Azofeifa, J.G.; Dowell, R.D. A generative model for the behavior of RNA polymerase. Bioinformatics 2017, 33, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Azofeifa, J.G.; Allen, M.A.; Lladser, M.E.; Dowell, R.D. An Annotation Agnostic Algorithm for Detecting Nascent RNA Transcripts in GRO-Seq. IEEE/ACM Trans. Comput. Biol. Bioinf. 2017, 14, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- Sammons, M.A.; Zhu, J.; Drake, A.M.; Berger, S.L. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 2015, 25, 179–188. [Google Scholar] [CrossRef] [PubMed]
- King, H.W.; Klose, R.J. The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. eLife 2017, 6, e22631. [Google Scholar] [CrossRef] [PubMed]
- Vrljicak, P.; Lucas, E.S.; Lansdowne, L.; Lucciola, R.; Muter, J.; Dyer, N.P.; Brosens, J.J.; Ott, S. Analysis of chromatin accessibility in decidualizing human endometrial stromal cells. FASEB J. 2018, 32, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Daftary, G.S.; Lomberk, G.A.; Buttar, N.S.; Allen, T.W.; Grzenda, A.; Zhang, J.; Zheng, Y.; Mathison, A.J.; Gada, R.P.; Calvo, E.; et al. Detailed Structural-Functional Analysis of the Krüppel-like Factor 16 (KLF16) Transcription Factor Reveals Novel Mechanisms for Silencing Sp/KLF Sites Involved in Metabolism and Endocrinology. J. Biol. Chem. 2012, 287, 7010–7025. [Google Scholar] [CrossRef] [PubMed]
- Tamm, K.; Rõõm, M.; Salumets, A.; Metsis, M. Genes targeted by the estrogen and progesterone receptors in the human endometrial cell lines HEC1A and RL95-2. Reprod. Biol. Endocrinol. 2009, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Azofeifa, J.; Allen, M.A.; Lladser, M.E.; Dowell, R. FStitch: A Fast and Simple Algorithm for Detecting Nascent RNA Transcripts. In Proceedings of the 5th BCB ’14, ACM Conference on Bioinformatics, Computational Biology, and Health Informatics, Newport Beach, CA, USA, 20–23 September 2014; ACM: New York, NY, USA, 2014; pp. 174–183. [Google Scholar]
Sample Availability: Not available.. |




{kind=link}
{kind=link}
{kind=link}
| Mutated TF | Disease/Symptoms |
|---|---|
| RUNX1 | familial platelet disorder with associated myeloid malignancy [4] |
| GRHL3 | cleft Palate [5] |
| MITF | deafness [6]; Waardenburg syndrome (hearing loss) [7] |
| LMX1B | nail–patella syndrome [8] (poorly developed nails and kneecaps) |
| TFAM | mitochondrial DNA depletion syndrome [9] |
| NKX2-5 | congenital heart disease [10] |
| TBX5 | Holt–Oram syndrome [11] (impared development of the heart and upper limbs) |
| MAF | congenital cataract [12] (severe visual impairment in infants) |
| TCF4 | Pitt–Hopkins syndrome [13] (intellectual disability and developmental delay, breathing problems, recurrent seizures) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripodi, I.J.; Allen, M.A.; Dowell, R.D. Detecting Differential Transcription Factor Activity from ATAC-Seq Data. Molecules 2018, 23, 1136. https://doi.org/10.3390/molecules23051136
Tripodi IJ, Allen MA, Dowell RD. Detecting Differential Transcription Factor Activity from ATAC-Seq Data. Molecules. 2018; 23(5):1136. https://doi.org/10.3390/molecules23051136
Chicago/Turabian StyleTripodi, Ignacio J., Mary A. Allen, and Robin D. Dowell. 2018. "Detecting Differential Transcription Factor Activity from ATAC-Seq Data" Molecules 23, no. 5: 1136. https://doi.org/10.3390/molecules23051136
APA StyleTripodi, I. J., Allen, M. A., & Dowell, R. D. (2018). Detecting Differential Transcription Factor Activity from ATAC-Seq Data. Molecules, 23(5), 1136. https://doi.org/10.3390/molecules23051136