
Transcription Factors as Therapeutic Targets in Chronic Kidney Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Nuclear Factor Erythroid-2 Related Factor 2 (Nrf2)
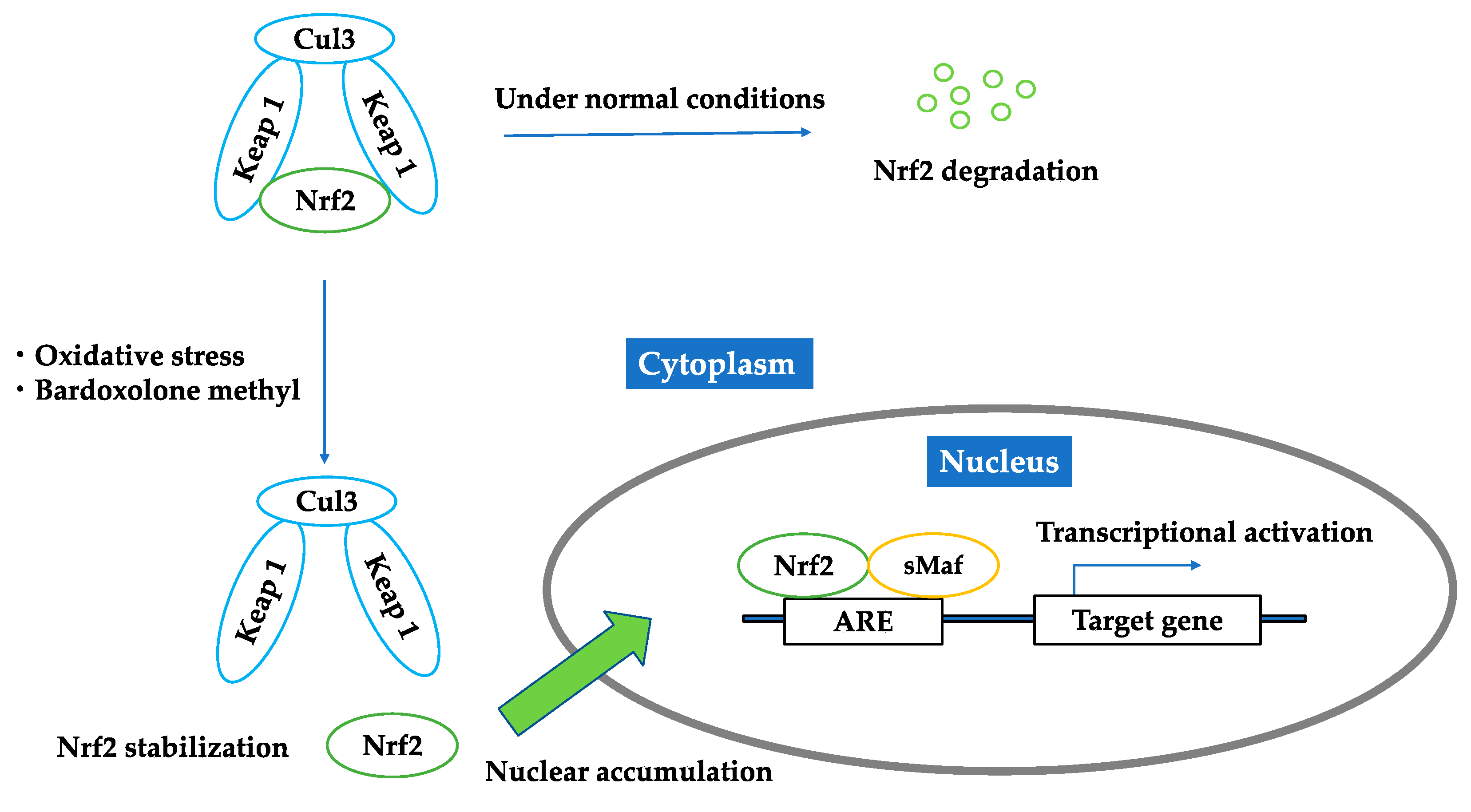
2.1. Nrf2–Keap1 Antioxidant Pathway
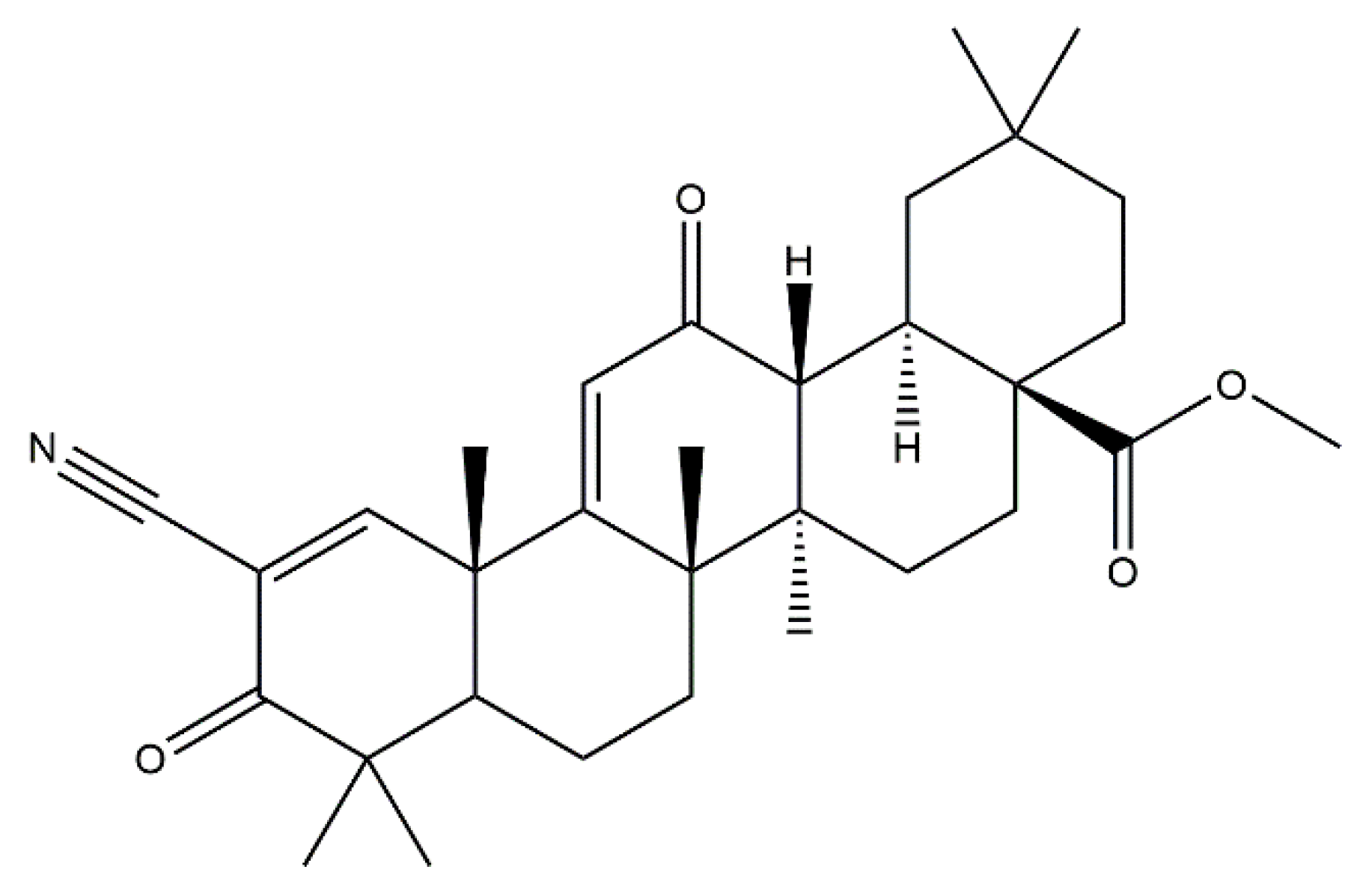
2.2. Bardoxolone Methyl as a Nrf2 Activator
2.3. Brief Summary of Targeting Nrf2
3. Hypoxia-Inducible Factor (HIF)
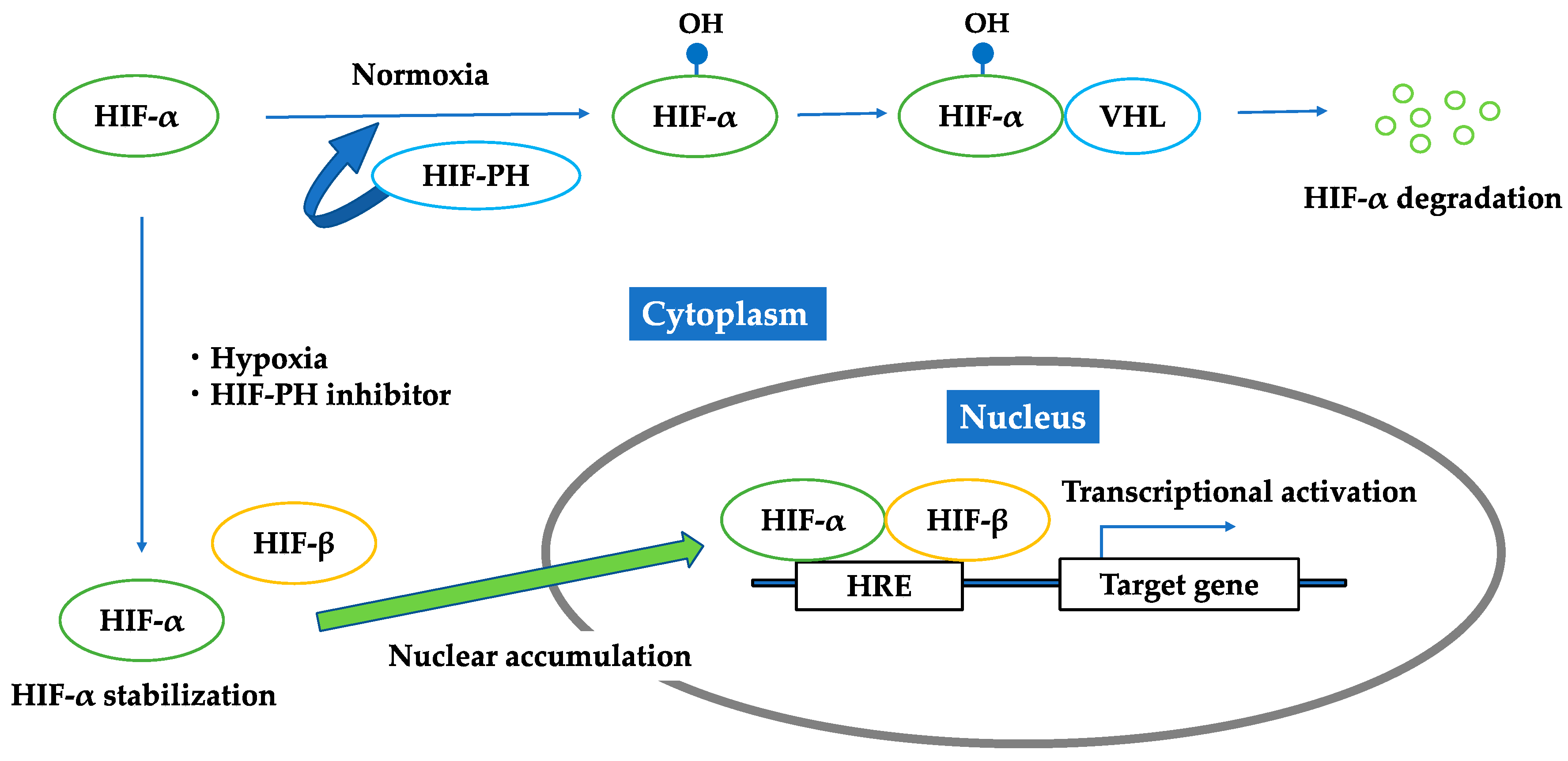
3.1. HIF–HRE Pathway
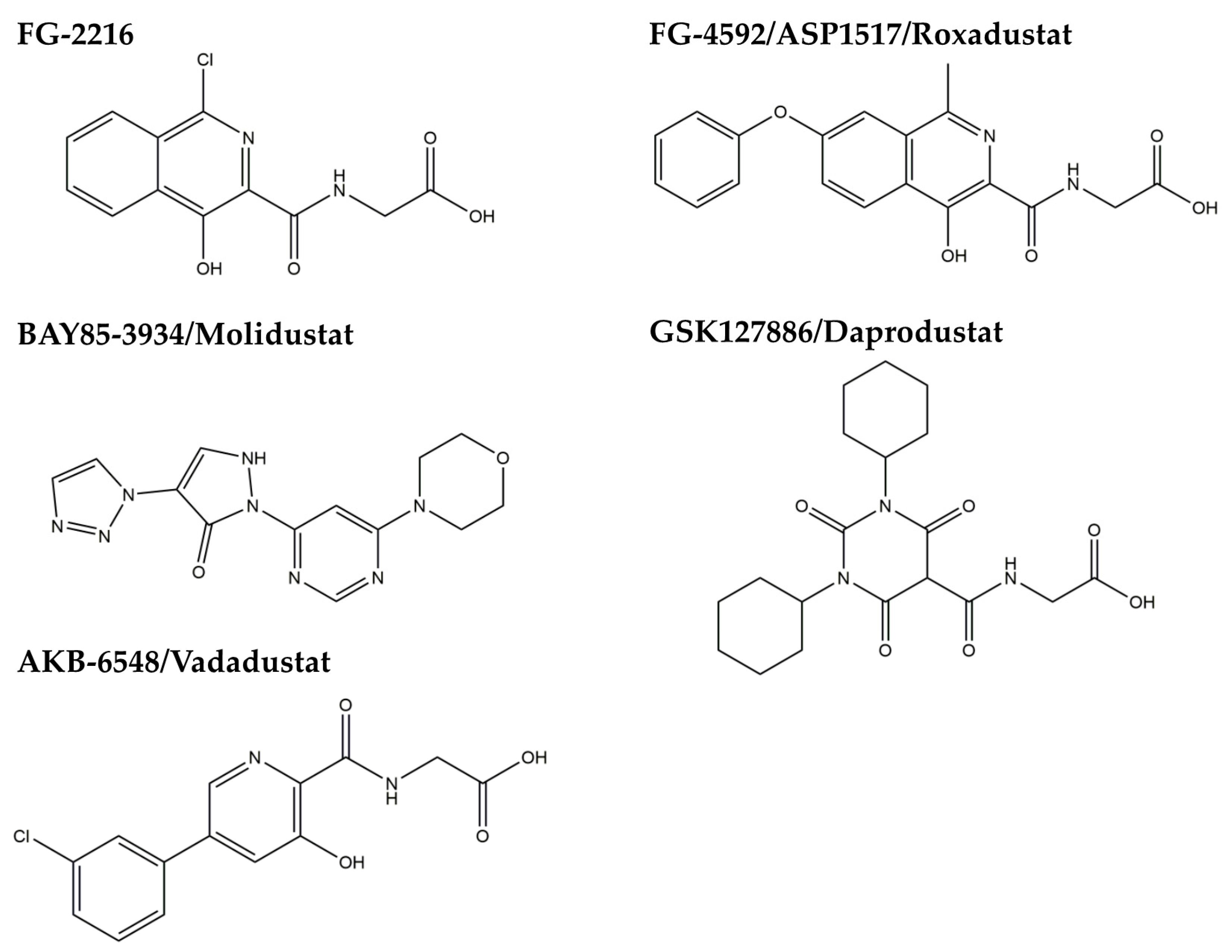
3.2. HIF-PH Inhibitor as an HIF Stabilizer
3.3. Brief Summary of Targeting HIF
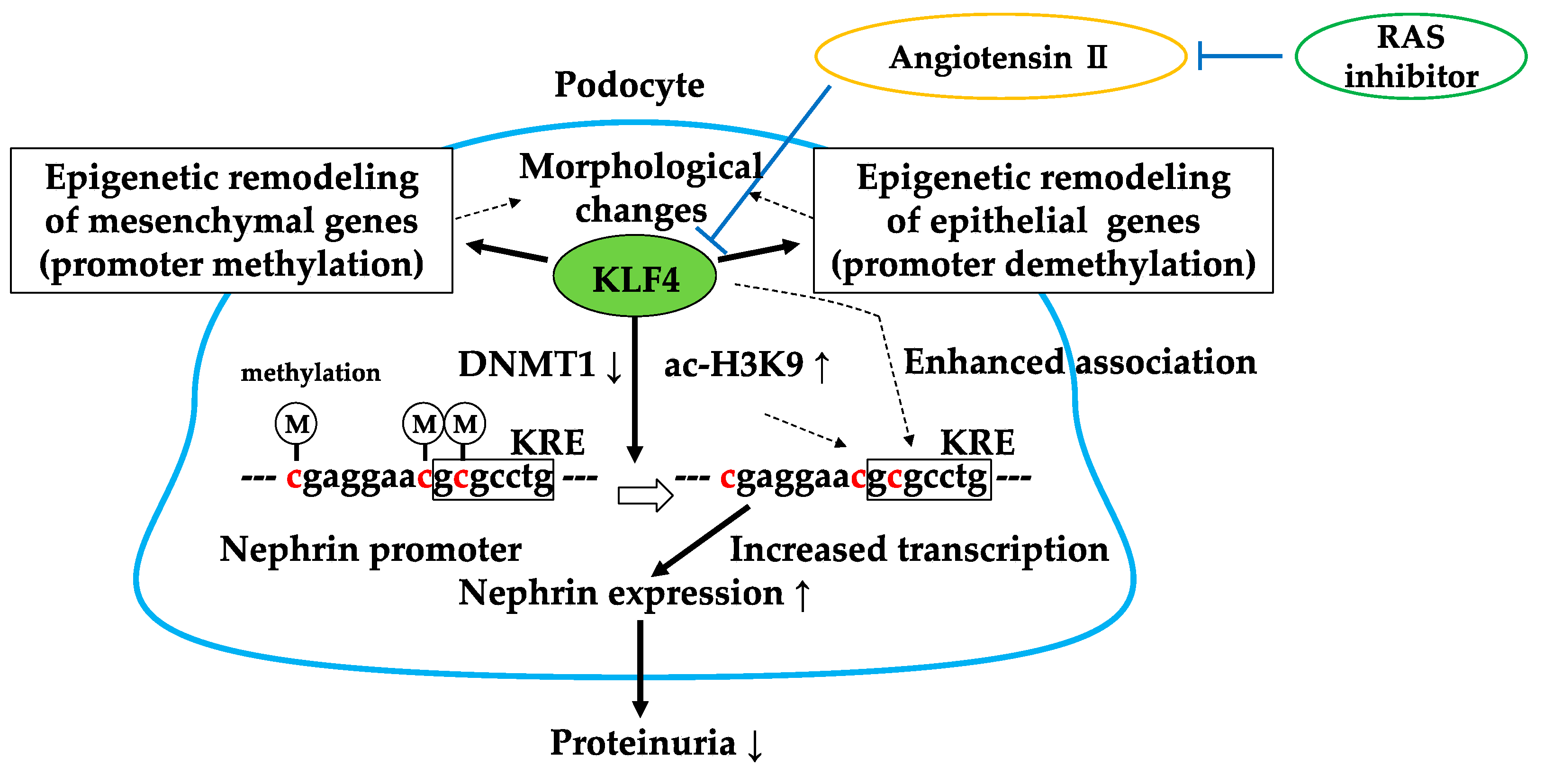
4. Kruppel-Like Factor 4 (KLF4)
4.1. KLF4 Modulates Podocyte Phenotype and Attenuates Proteinuria
4.2. RAS Inhibitor Attenuates Proteinuria in Part via KLF4
4.3. Brief Summary of Targeting KLF4
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar]
- Couser, W.G.; Remuzzi, G.; Mendis, S.; Tonelli, M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011, 80, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; de Jong, P.E.; Coresh, J.; El Nahas, M.; Astor, B.C.; Matsushita, K.; Gansevoort, R.T.; Kasiske, B.L.; Eckardt, K.U. The definition, classification, and prognosis of chronic kidney disease: A KDIGO Controversies Conference report. Kidney Int. 2011, 80, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O‘Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef] [PubMed]
- Cottone, S.; Lorito, M.C.; Riccobene, R.; Nardi, E.; Mule, G.; Buscemi, S.; Geraci, C.; Guarneri, M.; Arsena, R.; Cerasola, G. Oxidative stress, inflammation and cardiovascular disease in chronic renal failure. J. Nephrol. 2008, 21, 175–179. [Google Scholar] [PubMed]
- Mimura, I.; Nangaku, M. The suffocating kidney: Tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 2010, 6, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The Role of Nrf2 in Cardiovascular Function and Disease. Oxidative Med. Cell. Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Johnson, J.A. Nrf2—A therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88 Pt B, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Sato, T.; Rodriguez-Iturbe, B.; Vaziri, N.D. Role of intrarenal angiotensin system activation, oxidative stress, inflammation, and impaired nuclear factor-erythroid-2-related factor 2 activity in the progression of focal glomerulosclerosis. J. Pharmacol. Exp. Ther. 2011, 337, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, M.A.; Nicholas, S.B.; Norris, K.C.; Vaziri, N.D. Role of impaired Nrf2 activation in the pathogenesis of oxidative stress and inflammation in chronic tubulo-interstitial nephropathy. Nephrol. Dial. Transplant. 2013, 28, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Bolati, D.; Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Indoxyl sulfate, a uremic toxin, downregulates renal expression of Nrf2 through activation of NF-kappaB. BMC Nephrol. 2013, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Hector, A.; Kuijpers, T.; Roos, D.; Hartl, D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin. Dev. Immunol. 2012, 2012, 252460. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Itoh, K.; Enomoto, A.; Hirayama, A.; Yamaguchi, N.; Kobayashi, M.; Morito, N.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001, 60, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Hirayama, A.; Ishizaki, K.; Yamada, A.; Takeuchi, M.; Yamagishi, S.; Morito, N.; Nakano, T.; Ojima, M.; Shimohata, H.; et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells 2008, 13, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T.; Yore, M.M.; Fu, L.; Lopchuk, J.M.; Gribble, G.W. New synthetic triterpenoids: Potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J. Nat. Prod. 2011, 74, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Wang, Y.; Senitko, M.; Meyer, C.; Wigley, W.C.; Ferguson, D.A.; Grossman, E.; Chen, J.; Zhou, X.J.; Hartono, J.; et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARgamma, and HO-1. Am. J. Physiol. Ren. Physiol. 2011, 300, F1180–F1192. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Sharma, A.; Stefanovic, N.; Yuen, D.Y.; Karagiannis, T.C.; Meyer, C.; Ward, K.W.; Cooper, M.E.; de Haan, J.B. Derivative of bardoxolone methyl, dh404, in an inverse dose-dependent manner lessens diabetes-associated atherosclerosis and improves diabetic kidney disease. Diabetes 2014, 63, 3091–3103. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Krauth, M.; Huff, J.W.; Ferguson, D.A.; Ruiz, S.; Meyer, C.J.; Warnock, D.G. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am. J. Nephrol. 2011, 33, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J.; Dominguez, J.H. Rapid progression of diabetic nephropathy is linked to inflammation and episodes of acute renal failure. Am. J. Nephrol. 2010, 32, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Carrero, J.J.; Stenvinkel, P. Inflammation as a risk factor and target for therapy in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 662–668. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Agarwal, R.; Audhya, P.; Bakris, G.L.; Chin, M.; Krauth, M. Rationale and trial design of Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes: The Occurrence of Renal Events (BEACON). Am. J. Nephrol. 2013, 37, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Reisman, S.A.; Bakris, G.L.; O’Grady, M.; Linde, P.G.; McCullough, P.A.; Packham, D.; Vaziri, N.D.; Ward, K.W.; Warnock, D.G.; et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 2014, 39, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Bakris, G.L.; Block, G.A.; Chertow, G.M.; Goldsberry, A.; Inker, L.A.; Heerspink, H.J.L.; O’Grady, M.; Pergola, P.E.; Wanner, C.; et al. Bardoxolone Methyl Improves Kidney Function in Patients with Chronic Kidney Disease Stage 4 and Type 2 Diabetes: Post-Hoc Analyses from Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes Study. Am. J. Nephrol. 2018, 47, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Sarnak, M.J.; Levey, A.S. Bardoxolone methyl, chronic kidney disease, and type 2 diabetes. N. Engl. J. Med. 2011, 365, 1746–1747. [Google Scholar] [PubMed]
- Tayek, J.A.; Kalantar-Zadeh, K. The extinguished BEACON of bardoxolone: Not a Monday morning quarterback story. Am. J. Nephrol. 2013, 37, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Porrini, E.L.; Gaspari, F.; Motterlini, N.; Cannata, A.; Carrara, F.; Cella, C.; Ferrari, S.; Stucchi, N.; Parvanova, A.; et al. Glomerular hyperfiltration and renal disease progression in type 2 diabetes. Diabetes Care 2012, 35, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Fine, L.G.; Orphanides, C.; Norman, J.T. Progressive renal disease: The chronic hypoxia hypothesis. Kidney Int. Suppl. 1998, 65, 74S–78S. [Google Scholar]
- Nangaku, M.; Inagi, R.; Miyata, T.; Fujita, T. Hypoxia and hypoxia-inducible factor in renal disease. Nephron Exp. Nephrol. 2008, 110, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Eckardt, K.U. Hypoxia and the HIF system in kidney disease. J. Mol. Med. (Berl) 2007, 85, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Gunaratnam, L.; Bonventre, J.V. HIF in kidney disease and development. J. Am. Soc. Nephrol. 2009, 20, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nangaku, M. Recent advances and clinical application of erythropoietin and erythropoiesis-stimulating agents. Exp. Cell Res. 2012, 318, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Hirsila, M.; Koivunen, P.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Wish, J.B. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients with CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Katavetin, P.; Miyata, T.; Inagi, R.; Tanaka, T.; Sassa, R.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. High glucose blunts vascular endothelial growth factor response to hypoxia via the oxidative stress-regulated hypoxia-inducible factor/hypoxia-responsible element pathway. J. Am. Soc. Nephrol. 2006, 17, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Khamaisi, M.; Abassi, Z.; Shilo, V.; Weksler-Zangen, S.; Goldfarb, M.; Shina, A.; Zibertrest, F.; Eckardt, K.U.; Rosen, S.; et al. Adaptation to hypoxia in the diabetic rat kidney. Kidney Int. 2008, 73, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yamaguchi, J.; Higashijima, Y.; Nangaku, M. Indoxyl sulfate signals for rapid mRNA stabilization of Cbp/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 2 (CITED2) and suppresses the expression of hypoxia-inducible genes in experimental CKD and uremia. FASEB J. 2013, 27, 4059–4075. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nangaku, M. Drug discovery for overcoming chronic kidney disease (CKD): Prolyl-hydroxylase inhibitors to activate hypoxia-inducible factor (HIF) as a novel therapeutic approach in CKD. J. Pharmacol. Sci. 2009, 109, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Matsumoto, M.; Inagi, R.; Miyata, T.; Kojima, I.; Ohse, T.; Fujita, T.; Nangaku, M. Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int. 2005, 68, 2714–2725. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kojima, I.; Ohse, T.; Inagi, R.; Miyata, T.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. Hypoxia-inducible factor modulates tubular cell survival in cisplatin nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2005, 289, F1123–F1133. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Eckardt, K.U. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat. Rev. Nephrol. 2016, 12, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Theilig, F.; Enke, A.K.; Scolari, B.; Polzin, D.; Bachmann, S.; Koesters, R. Tubular deficiency of von Hippel-Lindau attenuates renal disease progression in anti-GBM glomerulonephritis. Am. J. Pathol. 2011, 179, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Wang, G.; Shevchuk, M.M.; Nanus, D.M.; Gudas, L.J. Generation of a mouse model of Von Hippel-Lindau kidney disease leading to renal cancers by expression of a constitutively active mutant of HIF1alpha. Cancer Res. 2011, 71, 6848–6856. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J.; Fu, L.; Minton, D.R.; Mongan, N.P.; Nanus, D.M. The role of HIF1alpha in renal cell carcinoma tumorigenesis. J. Mol. Med. (Berlin) 2014, 92, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Mallipattu, S.K.; Estrada, C.C.; He, J.C. The critical role of Kruppel-like factors in kidney disease. Am. J. Physiol. Ren. Physiol. 2017, 312, F259–F265. [Google Scholar] [CrossRef] [PubMed]
- Mallipattu, S.K.; Guo, Y.; Revelo, M.P.; Roa-Pena, L.; Miller, T.; Ling, J.; Shankland, S.J.; Bialkowska, A.B.; Ly, V.; Estrada, C.; et al. Kruppel-Like Factor 15 Mediates Glucocorticoid-Induced Restoration of Podocyte Differentiation Markers. J. Am. Soc. Nephrol. 2017, 28, 166–184. [Google Scholar] [CrossRef] [PubMed]
- Mallipattu, S.K.; Liu, R.; Zheng, F.; Narla, G.; Ma‘ayan, A.; Dikman, S.; Jain, M.K.; Saleem, M.; D‘Agati, V.; Klotman, P.; et al. Kruppel-like factor 15 (KLF15) is a key regulator of podocyte differentiation. J. Biol. Chem. 2012, 287, 19122–19135. [Google Scholar] [CrossRef] [PubMed]
- Mallipattu, S.K.; Horne, S.J.; D‘Agati, V.; Narla, G.; Liu, R.; Frohman, M.A.; Dickman, K.; Chen, E.Y.; Ma‘ayan, A.; Bialkowska, A.B.; et al. Kruppel-like factor 6 regulates mitochondrial function in the kidney. J. Clin. Investig. 2015, 125, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Sasamura, H.; Nakamura, M.; Azegami, T.; Oguchi, H.; Sakamaki, Y.; Itoh, H. KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J. Clin. Investig. 2014, 124, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.T.; Pevsner, J.; Yang, V.W. The biology of the mammalian Kruppel-like family of transcription factors. Int. J. Biochem. Cell Biol. 2000, 32, 1103–1121. [Google Scholar] [CrossRef]
- Katz, J.P.; Perreault, N.; Goldstein, B.G.; Lee, C.S.; Labosky, P.A.; Yang, V.W.; Kaestner, K.H. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development 2002, 129, 2619–2628. [Google Scholar] [PubMed]
- Segre, J.A.; Bauer, C.; Fuchs, E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 1999, 22, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Maherali, N.; Ahfeldt, T.; Rigamonti, A.; Utikal, J.; Cowan, C.; Hochedlinger, K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 2008, 3, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Aoi, T.; Yae, K.; Nakagawa, M.; Ichisaka, T.; Okita, K.; Takahashi, K.; Chiba, T.; Yamanaka, S. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 2008, 321, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Mori, H.; Masaki, T. Angiotensin II blockade and renal protection. Curr. Pharm. Des. 2013, 19, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Sasamura, H.; Nakamura, M.; Sakamaki, Y.; Azegami, T.; Oguchi, H.; Tokuyama, H.; Wakino, S.; Hayashi, K.; Itoh, H. Renin-angiotensin blockade resets podocyte epigenome through Kruppel-like Factor 4 and attenuates proteinuria. Kidney Int. 2015, 88, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Lin, H.H.; Tang, M.J. Matrix-Stiffness-Regulated Inverse Expression of Kruppel-Like Factor 5 and Kruppel-Like Factor 4 in the Pathogenesis of Renal Fibrosis. Am. J. Pathol. 2015, 185, 2468–2481. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Tang, W.; Yuan, Q.; Peng, L.; Yu, P. Epigenetic repression of Kruppel-like factor 4 through Dnmt1 contributes to EMT in renal fibrosis. Int. J. Mol. Med. 2015, 35, 1596–1602. [Google Scholar] [CrossRef] [PubMed]
- Mreich, E.; Chen, X.M.; Zaky, A.; Pollock, C.A.; Saad, S. The role of Kruppel-like factor 4 in transforming growth factor-beta-induced inflammatory and fibrotic responses in human proximal tubule cells. Clin. Exp. Pharmacol. Physiol. 2015, 42, 680–686. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hishikawa, A.; Hayashi, K.; Itoh, H. Transcription Factors as Therapeutic Targets in Chronic Kidney Disease. Molecules 2018, 23, 1123. https://doi.org/10.3390/molecules23051123
Hishikawa A, Hayashi K, Itoh H. Transcription Factors as Therapeutic Targets in Chronic Kidney Disease. Molecules. 2018; 23(5):1123. https://doi.org/10.3390/molecules23051123
Chicago/Turabian StyleHishikawa, Akihito, Kaori Hayashi, and Hiroshi Itoh. 2018. "Transcription Factors as Therapeutic Targets in Chronic Kidney Disease" Molecules 23, no. 5: 1123. https://doi.org/10.3390/molecules23051123
APA StyleHishikawa, A., Hayashi, K., & Itoh, H. (2018). Transcription Factors as Therapeutic Targets in Chronic Kidney Disease. Molecules, 23(5), 1123. https://doi.org/10.3390/molecules23051123