Nanotechnological Strategies for Protein Delivery



Abstract
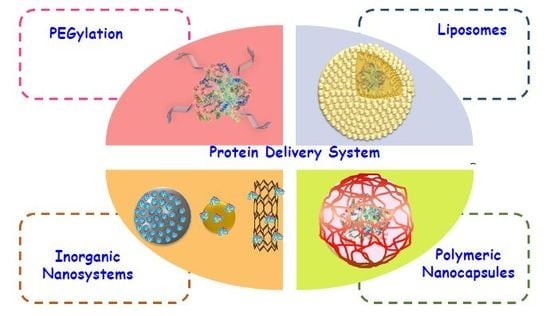
1. Introduction
2. PEGylation
3. Liposomes
4. Inorganic Nanoparticles
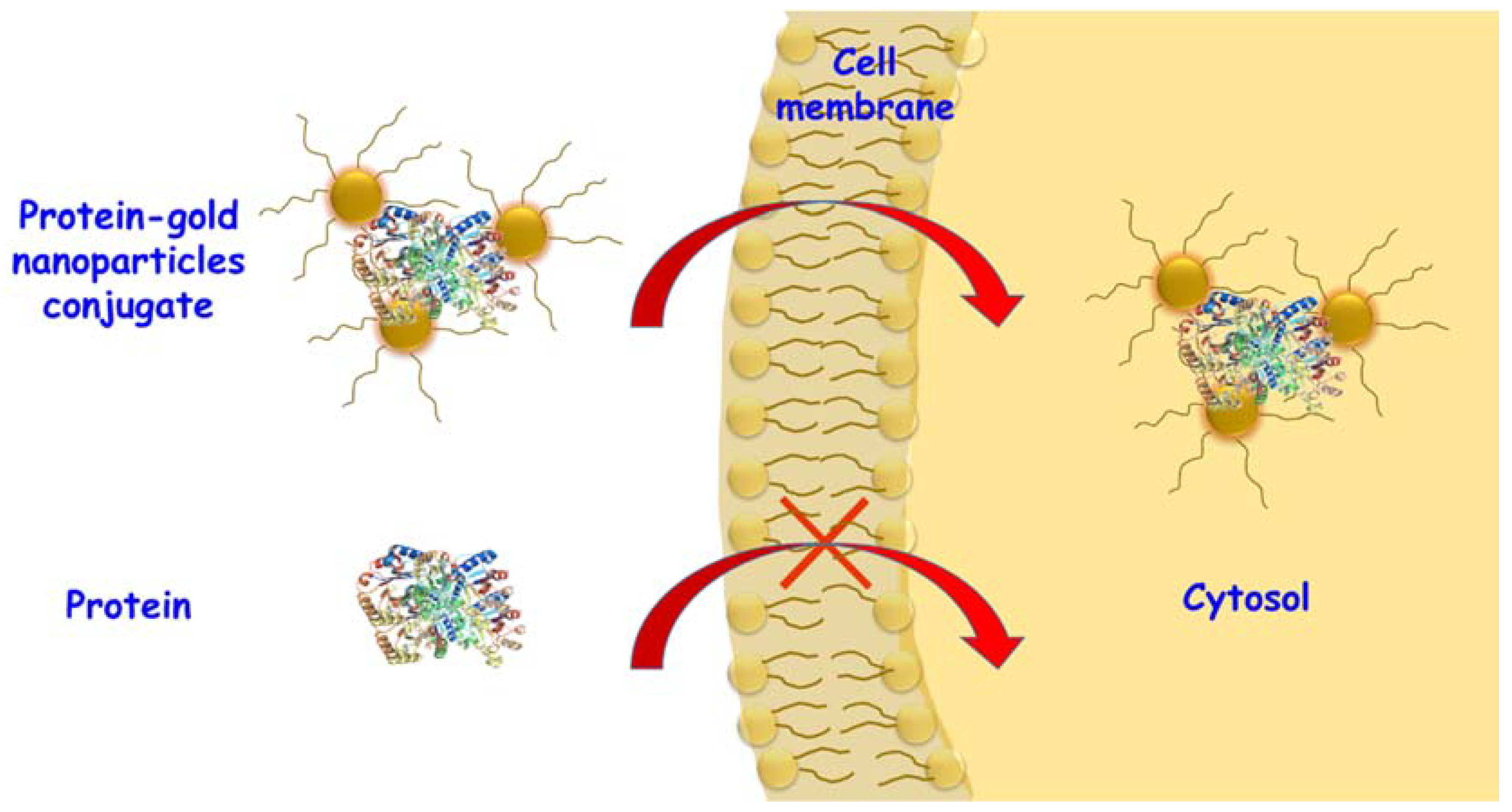
4.1. Mesoporous Silica Nanoparticles
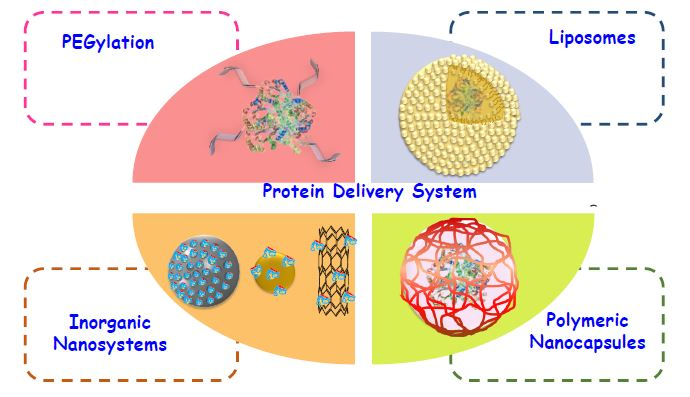
4.2. Gold Nanoparticles
4.3. Carbon Nanotubes
5. Polymeric Nanocapsules
5.1. Non-Degradable Nanocapsules
5.1.1. Acrylamide-Based Nanocapsules
5.1.2. Phosphorylcholine Nanocapsules
5.1.3. N-Vinylpyrrolidone Nanocapsules
5.2. Degradable Nanocapsules
5.2.1. pH-Respondive Nanocapsules
5.2.2. Enzyme Responsive Nanocapsules
5.2.3. Redox-Responsive Nanocapsules
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [PubMed]
- Childers, N.K.; Bruce, M.G.; McGhee, J.R. Molecular Mechanisms of Immunoglobulin a Defense. Annu. Rev. Microbiol. 1989, 43, 503–536. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1–PGC-1α interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Vassart, G.; Dumont, J.E. The Thyrotropin Receptor and the Regulation of Thyrocyte Function and Growth. Endocr. Rev. 1992, 13, 596–611. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Lukas, J.; Müller, H.; Lützhøt, D.; Strauss, M.; Bartek, J. Cyclin D1 protein expression and function in human breast cancer. Int. J. Cancer 1994, 57, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Gelse, K.; Pöschl, E.; Aigner, T. Collagens—Structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [PubMed]
- McKittrick, J.; Chen, P.-Y.; Bodde, S.G.; Yang, W.; Novitskaya, E.E.; Meyers, M.A. The Structure, Functions, and Mechanical Properties of Keratin. JOM 2012, 64, 449–468. [Google Scholar] [CrossRef]
- Aas, F.E.; Li, X.; Edwards, J.; Hongrø Solbakken, M.; Deeudom, M.; Vik, Å.; Moir, J.; Koomey, M.; Aspholm, M. Cytochrome c-based domain modularity governs genus-level diversification of electron transfer to dissimilatory nitrite reduction. Environ. Microbiol. 2015, 17, 2114–2132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hess, D.T.; Qian, Z.; Hausladen, A.; Fonseca, F.; Chaube, R.; Reynolds, J.D.; Stamler, J.S. Hemoglobin βCys93 is essential for cardiovascular function and integrated response to hypoxia. Proc. Natl. Acad. Sci. USA 2015, 112, 6425–6430. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.M.; Petering, D.H. Coboglobins: Oxygen-Carrying Cobalt-Reconstituted Hemoglobin and Myoglobin. Proc. Natl. Acad. Sci. USA 1970, 67, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Du, J.; Yan, M.; Dhaliwal, A.; Wen, J.; Liu, F.; Segura, T.; Lu, Y. Synthesis of protein nano-conjugates for cancer therapy. Nano Res. 2011, 4, 425–433. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Weisgraber, K.H.; Goedert, M.; Saunders, A.M.; Huang, D.; Corder, E.H.; Dong, L.-M.; Jakes, R.; Alberts, M.J.; Gilbert, J.R.; et al. Hypothesis: Microtubule Instability and Paired Helical Filament Formation in the Alzheimer Disease Brain Are Related to Apolipoprotein E Genotype. Exp. Neurol. 1994, 125, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 Cochaperone Aha1 Downregulation Rescues Misfolding of CFTR in Cystic Fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, Y.; Hsieh, R.S.; Wang, N.; Tai, W.; Joo, K.I.; Wang, P.; Gu, Z.; Tang, Y. Clickable protein nanocapsules for targeted delivery of recombinant p53 protein. J. Am. Chem. Soc. 2014, 136, 15319–15325. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.; Debs, R.J.; Desprez, P.-Y. Id genes and proteins as promising targets in cancer therapy. Trends Mol. Med. 2004, 10, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Patterson, H.; Nibbs, R.; McInnes, I.; Siebert, S. Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases. Clin. Exp. Immunol. 2014, 176, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nikles, D.; Bach, P.; Boller, K.; Merten, C.A.; Montrasio, F.; Heppner, F.L.; Aguzzi, A.; Cichutek, K.; Kalinke, U.; Buchholz, C.J. Circumventing tolerance to the prion protein (PrP): Vaccination with PrP-displaying retrovirus particles induces humoral immune responses against the native form of cellular PrP. J. Virol. 2005, 79, 4033–4042. [Google Scholar] [CrossRef] [PubMed]
- Lagassé, H.A.D.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Sauna, Z.E.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Research 2017, 6, 113. [Google Scholar] [CrossRef] [PubMed]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Chi, E.Y.; Krishnan, S.; Randolph, T.W.; Carpenter, J.F. Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 2003, 20, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A. Dominant forces in protein folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Shirley, B.A.; McNutt, M.; Gajiwala, K. Forces contributing to the conformational stability of proteins. FASEB J. 1996, 10, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Jaenicke, R. Protein folding: Local structures, domains, subunits, and assemblies. Biochemistry 1991, 30, 3147–3161. [Google Scholar] [CrossRef] [PubMed]
- Ruddy, S.; Gigli, I.; Austen, K.F. The Complement System of Man. N. Engl. J. Med. 1972, 287, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Müller-Eberhard, H.J. Molecular Organization and Function of the Complement System. Annu. Rev. Biochem. 1988, 57, 321–347. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Yang, X.; Arvizo, R.; Zhu, Z.-J.; Agasti, S.S.; Mo, Z.; Rotello, V.M. Intracellular Delivery of a Membrane-Impermeable Enzyme in Active Form Using Functionalized Gold Nanoparticles. J. Am. Chem. Soc. 2010, 132, 2642–2645. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Xu, D.; Wu, D.; Whittaker, J.W.; Terkeltaub, R.; Lu, Y. Nanocapsules of oxalate oxidase for hyperoxaluria treatment. Nano Res. 1898, 1, 8–11. [Google Scholar] [CrossRef]
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A.A. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can. Med. Assoc. J. 1922, 12, 141–146. [Google Scholar] [PubMed]
- Goeddel, D.V.; Kleid, D.G.; Bolivar, F.; Heyneker, H.L.; Yansura, D.G.; Crea, R.; Hirose, T.; Kraszewski, A.; Itakura, K.; Riggs, A.D. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc. Natl. Acad. Sci. USA 1979, 76, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Adeniyi-Jones, R.O.; Knight, G.; Leiper, J.M.; Wiles, P.G.; Jones, R.H.; Keen, H.; MacCuish, A.C.; Ward, J.D.; Watkins, P.J.; et al. Biosynthetic human insulin in the treatment of diabetes. A double-blind crossover trial in established diabetic patients. Lancet 1982, 2, 354–357. [Google Scholar] [CrossRef]
- Keen, H.; Glynne, A.; Pickup, J.C.; Viberti, G.C.; Bilous, R.W.; Jarrett, R.J.; Marsden, R. Human insulin produced by recombinant DNA technology: Safety and hypoglycaemic potency in healthy men. Lancet 1980, 2, 398–401. [Google Scholar] [CrossRef]
- Richter, B.; Neises, G. “Human” insulin versus animal insulin in people with diabetes mellitus. In The Cochrane Database of Systematic Reviews (Protocol); John Wiley & Sons, Ltd.: Chichester, UK, 2003; p. CD003816. [Google Scholar]
- Pensions, D.B.; Risk, U. The Purple Book; Pension Protection Fund: Wymondham, UK, 2014; pp. 1–2. [Google Scholar]
- Fee, C.J.; Van Alstine, J.M. PEG-proteins: Reaction engineering and separation issues. Chem. Eng. Sci. 2006, 61, 924–939. [Google Scholar] [CrossRef]
- Pasut, G.; Veronese, F.M. Polymer–drug conjugation, recent achievements and general strategies. Prog. Polym. Sci. 2007, 32, 933–961. [Google Scholar] [CrossRef]
- Knauf, M.J.; Bell, D.P.; Hirtzer, P.; Luo, Z.P.; Young, J.D.; Katre, N.V. Relationship of effective molecular size to systemic clearance in rats of recombinant interleukin-2 chemically modified with water-soluble polymers. J. Biol. Chem. 1988, 263, 15064–15070. [Google Scholar] [PubMed]
- Bhat, R.; Timasheff, S.N. Steric exclusion is the principal source of the preferential hydration of proteins in the presence of polyethylene glycols. Protein Sci. 1992, 1, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M. Peptide and protein PEGylation: A review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar] [CrossRef]
- Abuchowski, A.; van Es, T.; Palczuk, N.C.; Davis, F.F. Alteration of Immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 1977, 252, 3578–3581. [Google Scholar] [PubMed]
- Abuchowski, A.; Mccoy, J.R.; Palczuk, N.C.; Es, T.V.A.N.; Davis, F.F. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 1977, 252, 3582–3586. [Google Scholar] [PubMed]
- Levy, Y.; Hershfield, M.S.; Fernandez-Mejia, C.; Polmar, S.H.; Scudiery, D.; Berger, M.; Sorensen, R.U. Adenosine deaminase deficiency with late onset of recurrent infections: Response to treatment with polyethylene glycol-modified adenosine deaminase. J. Pediatr. 1988, 113, 312–317. [Google Scholar] [CrossRef]
- Alconcel, S.N.S.; Baas, A.S.; Maynard, H.D. FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. [Google Scholar] [CrossRef]
- Rajender Reddy, K.; Modi, M.W.; Pedder, S. Use of peginterferon alfa-2a (40 KD) (Pegasys®) for the treatment of hepatitis C. Adv. Drug Deliv. Rev. 2002, 54, 571–586. [Google Scholar] [CrossRef]
- Bennett, C.L.; Djulbegovic, B.; Norris, L.B.; Armitage, J.O. Colony-Stimulating Factors for Febrile Neutropenia during Cancer Therapy. N. Engl. J. Med. 2013, 368, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Romero-Weaver, A.L.; Wan, X.S.; Diffenderfer, E.S.; Lin, L.; Kennedy, A.R. Kinetics of Neutrophils in Mice Exposed to Radiation and/or Granulocyte Colony-Stimulating Factor Treatment. Radiat. Res. 2013, 180, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Villa, G.; de Francisco, A.L.M.; Albertazzi, A.; Adrogue, H.J.; Dougherty, F.C.; Beyer, U. Effect of a continuous erythropoietin receptor activator (CERA) on stable haemoglobin in patients with CKD on dialysis: Once monthly administration. Curr. Med. Res. Opin. 2007, 23, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.K.; Hempel, G.; Koling, S.; Chan, L.S.; Fisher, T.; Meiselman, H.J.; Garratty, G. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 2007, 110, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Takeda, M.; Sakamoto, H.; Kimoto, A.; Kobayashi, C.; Takasaki, N.; Yuki, K.; Tanaka, K.; Takenaga, M.; Igarashi, R.; et al. Accelerated Blood Clearance Phenomenon upon Repeated Injection of PEG-modified PLA-nanoparticles. Pharm. Res. 2009, 26, 2270–2279. [Google Scholar] [CrossRef] [PubMed]
- Ravin, H.A.; Seligman, A.M.; Fine, J. Polyvinyl Pyrrolidone as a Plasma Expander. N. Engl. J. Med. 1952, 247, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Haaf, F.; Sanner, A.; Straub, F. Polymers of N-Vinylpyrrolidone: Synthesis, Characterization and Uses. Polym. J. 1985, 17, 143–152. [Google Scholar] [CrossRef]
- Caliceti, P.; Schiavon, O.; Morpurgo, M.; Veronese, F.M.; Sartore, L.; Ranucci, E.; Ferruti, P. Physico-Chemical and Biological Properties of Monofunctional Hydroxy Teriminating Poly(N-Vinylpyrrolidone) Conjugated Superoxide Dismutase. J. Bioact. Compat. Polym. 1995, 10, 103–120. [Google Scholar] [CrossRef]
- Caliceti, P.; Schiavon, O.; Veronese, F.M. Immunological properties of uricase conjugated to neutral soluble polymers. Bioconjug. Chem. 2001, 12, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Tang, Y.; Brocchini, S.; Choi, J.; Godwin, A. Poly(2-methacryloyloxyethyl phosphorylcholine) for Protein Conjugation. Bioconjug. Chem. 2008, 19, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Wileman, T.E.; Foster, R.L.; Elliott, P.N.C. Soluble asparaginase-dextran conjugates show increased circulatory persistence and lowered antigen reactivity. J. Pharm. Pharmacol. 1986, 38, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Zinderman, C.E.; Landow, L.L.; Wise, R.P. Anaphylactoid Reactions to Dextran 40 and 70: Reports to the US Food and Drug Administration (FDA): 246. Pharmacoepidemiol. Drug Saf. 2006, 15, S115–S116. [Google Scholar]
- Zinderman, C.E.; Landow, L.; Wise, R.P. Anaphylactoid reactions to Dextran 40 and 70: Reports to the United States Food and Drug Administration, 1969 to 2004. J. Vasc. Surg. 2006, 43, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Jevševar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of therapeutic proteins. Biotechnol. J. 2010, 5, 113–128. [Google Scholar] [CrossRef] [PubMed]
- ChariotTM. Simple, Efficient Protein Delivery. Available online: https://www.activemotif.com/catalog/37/chariot-protein-delivery-reagent (accessed on 25 April 2018).
- Szoka, F.; Papahadjopoulos, D. Biochemistry Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation (drug delivery/encapsulation/lipid vesicles/encapsulated macromolecules). Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Geng, J.; Ah Yi, H.; Gogia, S.; Neelamegham, S.; Jacobs, A.; Lovell, J.F. Functionalization of cobalt porphyrin–phospholipid bilayers with his-tagged ligands and antigens. Nat. Chem. 2015, 7, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Oude Blenke, E.; Klaasse, G.; Merten, H.; Plückthun, A.; Mastrobattista, E.; Martin, N.I. Liposome functionalization with copper-free “click chemistry”. J. Control. Release 2015, 202, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Reto, A. Schwendener Liposomes as vaccine delivery systems: A review of the recent advances. Ther. Adv. Vaccines 2014, 2, 159–182. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Zelphati, O.; Wang, Y.; Kitada, S.; Reed, J.C.; Felgner, P.L.; Corbeil, J. Intracellular delivery of proteins with a new lipid-mediated delivery system. J. Biol. Chem. 2001, 276, 35103–35110. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Jin, J.; Yan, M.; Lu, Y. Synthetic Nanocarriers for Intracellular Protein Delivery. Curr. Drug Metab. 2012, 13, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Iwaoka, S.; Nakamura, T.; Takano, S.; Tsuchiya, S.; Aramaki, Y. Cationic liposomes induce apoptosis through p38 MAP kinase-caspase-8-Bid pathway in macrophage-like RAW264.7 cells. J. Leukoc. Biol. 2006, 79, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Scherphof, G.L.; Dijkstra, J.; Spanjer, H.H.; Derksen, J.T.P.; Roerdink, F.H. Uptake and Intracellular Processing of Targeted and Nontargeted Liposomes by Rat Kupffer Cells In Vivo and In Vitro. Ann. N. Y. Acad. Sci. 1985, 446, 368–384. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Lu, Y. Enzyme therapeutics for systemic detoxification. Adv. Drug Deliv. Rev. 2015, 90, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kiwada, H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 2008, 354, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Scherphof, G.; Roerdink, F.; Waite, M.; Parks, J. Disintegration of phosphatidylcholine liposomes in plasma as a result of interaction with high-density lipoproteins. Biochim. Biophys. Acta 1978, 542, 296–307. [Google Scholar] [CrossRef]
- Fanciullino, R.; Ciccolini, J. Liposome-Encapsulated Anticancer Drugs: Still Waiting for the Magic Bullet? Curr. Med. Chem. 2009, 16, 4361–4373. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Liang, M.; Wen, J.; Liu, Y.; Lu, Y.; Chen, I.S.Y. Single siRNA nanocapsules for enhanced RNAi delivery. J. Am. Chem. Soc. 2012, 134, 13542–13545. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.P.; Zeng, Q.H.; Lu, G.Q.; Yu, A.B. Inorganic nanoparticles as carriers for efficient cellular delivery. Chem. Eng. Sci. 2006, 61, 1027–1040. [Google Scholar] [CrossRef]
- Baeza, A.; Colilla, M.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin. Drug Deliv. 2015, 12, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Gu, Z.; Ottewell, T.; Yu, C. Silica-based nanoparticles for therapeutic protein delivery. J. Mater. Chem. B 2017, 5, 3241–3252. [Google Scholar] [CrossRef]
- Tu, J.; Boyle, A.L.; Friedrich, H.; Bomans, P.H.H.; Bussmann, J.; Sommerdijk, N.A.J.M.; Jiskoot, W.; Kros, A. Mesoporous Silica Nanoparticles with Large Pores for the Encapsulation and Release of Proteins. ACS Appl. Mater. Interfaces 2016, 8, 32211–32219. [Google Scholar] [CrossRef] [PubMed]
- Hoon Han, D.; Na, H.-K.; Hoon Choi, W.; Hoon Lee, J.; Kyung Kim, Y.; Won, C.; Lee, S.-H.; Pyo Kim, K.; Kuret, J.; Min, D.-H.; et al. Direct cellular delivery of human proteasomes to delay tau aggregation. Nat. Commun. 2014, 5, 5633. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.I.; Trewyn, B.G.; Lin, V.S.Y. Mesoporous silica nanoparticles for intracellular delivery of membrane-impermeable proteins. J. Am. Chem. Soc. 2007, 129, 8845–8849. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Davies, G.L.; Davis, J.J. Engineering cytochrome-modified silica nanoparticles to induce programmed cell death. Chem. Eur. J. 2013, 19, 17891–17898. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Mukherjee, P. Biological properties of “naked” metal nanoparticles. Adv. Drug Deliv. Rev. 2008, 17, 1289–1306. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Han, G.; De, M.; Kim, C.K.; Rotello, V.M. Gold nanoparticles in delivery applications. Adv. Drug Deliv. Rev. 2008, 60, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- De, M.; Ghosh, P.S.; Rotello, V.M. Applications of Nanoparticles in Biology. Adv. Mater. 2008, 20, 4225–4241. [Google Scholar] [CrossRef]
- Nativo, P.; Prior, I.A.; Brust, M. Uptake and Intracellular Fate of Surface-Modified Gold Nanoparticles. ACS Nano 2008, 2, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Bastús, N.G.; Pereiro, E.; López-Iglesias, C.; Puntes, V.F.; Kogan, M.J.; Giralt, E. Shuttling Gold Nanoparticles into Tumoral Cells with an Amphipathic Proline-Rich Peptide. ChemBioChem 2009, 10, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, A.G.; Xie, H.; Liu, Y.; Coleman, D.; Ryan, J.; Glomm, W.R.; Shipton, M.K.; Franzen, S.; Feldheim, D.L. Cellular Trajectories of Peptide-Modified Gold Particle Complexes: Comparison of Nuclear Localization Signals and Peptide Transduction Domains. Bioconjug. Chem. 2004, 15, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Simard, J.M.; Worrall, J.W.E.; Rotello, V.M. Tunable reactivation of nanoparticle-inhibited β-galactosidase by glutathione at intracellular concentrations. J. Am. Chem. Soc. 2004, 126, 13987–13991. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cao, L.; Shvartsman, D.; Silva, E.A.; Mooney, D.J. Targeted delivery of nanoparticles to ischemic muscle for imaging and therapeutic angiogenesis. Nano Lett. 2010, 11, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Kam, N.W.S.; Dai, H. Carbon nanotubes as intracellular protein transporters: Generality and biological functionality. J. Am. Chem. Soc. 2005, 127, 6021. [Google Scholar] [CrossRef] [PubMed]
- Kam, N.W.S.; Jessop, T.C.; Wender, P.A.; Dai, H. Nanotube molecular transporters: Internalization of carbon nanotube-protein conjugates into mammalian cells. J. Am. Chem. Soc. 2004, 126, 6850–6851. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Moore, J.M.; Huang, G.; Mount, A.S.; Rao, A.M.; Larcom, L.L.; Ke, P.C. RNA polymer translocation with single-walled carbon nanotubes. Nano Lett. 2004, 4, 2473–2477. [Google Scholar] [CrossRef]
- Bianco, A.; Hoebeke, J.; Godefroy, S.; Chaloin, O.; Pantarotto, D.; Briand, J.P.; Muller, S.; Prato, M.; Partidos, C.D. Cationic carbon nanotubes bind to CpG oligodeoxynucleotides and enhance their immunostimulatory properties. J. Am. Chem. Soc. 2005, 127, 58–59. [Google Scholar] [CrossRef] [PubMed]
- Pantarotto, D.; Briand, J.-P.; Prato, M.; Bianco, A. Translocation of bioactive peptides across cell membranes by carbon nanotubes. Chem. Commun. 2004, 1, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Pantarotto, D.; Singh, R.; McCarthy, D.; Erhardt, M.; Briand, J.-P.; Prato, M.; Kostarelos, K.; Bianco, A. Functionalized Carbon Nanotubes for Plasmid DNA Gene Delivery. Angew. Chem. 2004, 116, 5354–5358. [Google Scholar] [CrossRef]
- Shi Kam, N.W.; O’Connell, M.; Wisdom, J.A.; Dai, H. Carbon nanotubes as multifunctional biological transporters and near-infrared agents for selective cancer cell destruction. Proc. Natl. Acad. Sci. USA 2005, 102, 11600–11605. [Google Scholar] [CrossRef] [PubMed]
- Kam, N.W.S.; Liu, Z.; Dai, H. Functionalization of carbon nanotubes via cleavable disulfide bonds for efficient intracellular delivery of siRNA and potent gene silencing. J. Am. Chem. Soc. 2005, 127, 12492–12493. [Google Scholar] [CrossRef] [PubMed]
- Strachota, B.; Matějka, L.; Zhigunov, A.; Konefał, R.; Spěváček, J.; Dybal, J.; Puffr, R. Poly(N-isopropylacrylamide)–clay based hydrogels controlled by the initiating conditions: Evolution of structure and gel formation. Soft Matter 2015, 11, 9291–9306. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Du, J.; Li, J.; Yan, M.; Zhu, Q.; Jin, X.; Zhu, X.; Hu, Z.; Tang, Y.; Lu, Y. Construction of robust enzyme nanocapsules for effective organophosphate decontamination, detoxification, and protection. Adv. Mater. 2013, 25, 2212–2218. [Google Scholar] [CrossRef] [PubMed]
- Donarski, W.J.; Dumas, D.P.; Heitmeyer, D.P.; Lewis, V.E.; Raushel, F.M. Structure-Activity Relationships in the Hydrolysis of Substrates by the Phosphotriesterase from Pseudomonas diminutat. Biochemistry 1989, 28, 4650–4655. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jin, X.; Liu, Y.; Li, F.; Zhang, L.; Zhu, X.; Lu, Y. Robust enzyme–silica composites made from enzyme nanocapsules. Chem. Commun. 2015, 51, 9628–9631. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Liu, S.; Feng, J.; Kimura, S.; Wada, M.; Kuga, S.; Zhang, L. Cellulose-Silica Nanocomposite Aerogels by In Situ Formation of Silica in Cellulose Gel. Angew. Chem. 2012, 124, 2118–2121. [Google Scholar] [CrossRef]
- Ramanathan, M.; Luckarift, H.R.; Sarsenova, A.; Wild, J.R.; Ramanculov, E.K.; Olsen, E.V.; Simonian, A.L. Lysozyme-mediated formation of protein–silica nano-composites for biosensing applications. Colloids Surf. B Biointerfaces 2009, 73, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, V.A.; Paliwal, S.; Balasubramanian, S.; Nepal, D.; Davis, V.; Wild, J.; Ramanculov, E.; Simonian, A. Enhanced stability of enzyme organophosphate hydrolase interfaced on the carbon nanotubes. Colloids Surf. B Biointerfaces 2010, 77, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, J.; Yan, M.; Lau, M.Y.; Hu, J.; Han, H.; Yang, O.O.; Liang, S.; Wei, W.; Wang, H.; et al. Biomimetic enzyme nanocomplexes and their use as antidotes and preventive measures for alcohol intoxication. Nat. Nanotechnol. 2013, 8, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Duan, W.; Ahmed, S.; Mallouk, T.E.; Sen, A. Small power: Autonomous nano- and micromotors propelled by self-generated gradients. Nano Today 2013, 8, 531–554. [Google Scholar] [CrossRef]
- Simmchen, J.; Baeza, A.; Ruiz-Molina, D.; Vallet-Regí, M. Improving catalase-based propelled motor endurance by enzyme encapsulation. Nanoscale 2014, 6, 8907–8913. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Liu, Y.; Jin, X.; Liu, G.; Wen, J.; Zhang, L.; Li, J.; Yuan, X.; Chen, I.S.Y.; Chen, W.; et al. Phosphorylcholine polymer nanocapsules prolong the circulation time and reduce the immunogenicity of therapeutic proteins. Nano Res. 2016, 9, 1022–1031. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y.; Liu, G.; Xu, D.; Liang, S.; Zhu, X.; Lu, Y.; Wang, H. Prolonging the plasma circulation of proteins by nano-encapsulation with phosphorylcholine-based polymer. Nano Res. 2016, 9, 2424–2432. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, W.; Zhu, X.; Lu, Y. Encapsulating Therapeutic Proteins with Polyzwitterions for Lower Macrophage Nonspecific Uptake and Longer Circulation Time. ACS Appl. Mater. Interfaces 2017, 9, 7972–7978. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, D.; Jin, X.; Liu, G.; Liang, S.; Wang, H.; Chen, W.; Zhu, X.; Lu, Y. Nanocapsules of therapeutic proteins with enhanced stability and long blood circulation for hyperuricemia management. J. Control. Release 2017, 255, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of Extracellular Matrix Assembly in Interstitial Transport in Solid Tumors Role of Extracellular Matrix Assembly in Interstitial Transport in Solid Tumors 1. Cancer Res. 2000, 60, 2497–2503. [Google Scholar] [PubMed]
- Parodi, A.; Haddix, S.G.; Taghipour, N.; Scaria, S.; Taraballi, F.; Cevenini, A.; Yazdi, I.K.; Corbo, C.; Palomba, R.; Khaled, S.Z.; et al. Bromelain Surface Modification Increases the Diffusion of Silica Nanoparticles in the Tumor Extracellular Matrix. ACS Nano 2014, 8, 9874–9883. [Google Scholar] [CrossRef] [PubMed]
- Villegas, M.R.; Baeza, A.; Vallet Regí, M. Hybrid Collagenase Nanocapsules for Enhanced Nanocarrier Penetration in Tumoral Tissues. ACS Appl. Mater. Interfaces 2015, 7, 24075–24081. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Du, J.; Gu, Z.; Liang, M.; Hu, Y.; Zhang, W.; Priceman, S.; Wu, L.; Zhou, Z.H.; Liu, Z.; et al. A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat. Nanotechnol. 2010, 5, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wen, J.; Meng, Y.; Zhang, K.; Zhu, J.; Ren, Y.; Qian, X.; Yuan, X.; Lu, Y.; Kang, C. Efficient delivery of therapeutic miRNA nanocapsules for tumor suppression. Adv. Mater. 2015, 27, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Yan, M.; Hu, B.; Joo, K.L.; Biswas, A.; Huang, Y.; Lu, Y.; Wang, P.; Tang, Y. Protein nanocapsule weaved with enzymatically degradable polymerie network. Nano Lett. 2009, 9, 4533–4538. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Joo, K.-I.; Liu, J.; Zhao, M.; Fan, G.; Wang, P.; Gu, Z.; Tang, Y. Endoprotease-Mediated Intracellular Protein Delivery Using Nanocapsules. ACS Nano 2011, 5, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Anderson, S.M.; Du, J.; Yan, M.; Wang, J.; Shen, M.; Lu, Y.; Segura, T. Controlled protein delivery based on enzyme-responsive nanocapsules. Adv. Mater. 2011, 23, 4549–4553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Nih, L.; Carmichael, S.T.; Lu, Y.; Segura, T. Enzyme-Responsive Delivery of Multiple Proteins with Spatiotemporal Control. Adv. Mater. 2015, 27, 3620–3625. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Tate, S.S. Glutathione and Related γ-Glutamyl Compounds: Biosynthesis and Utilization. Annu. Rev. Biochem. 1976, 45, 559–604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Biswas, A.; Hu, B.; Joo, K.I.; Wang, P.; Gu, Z.; Tang, Y. Redox-responsive nanocapsules for intracellular protein delivery. Biomaterials 2011, 32, 5223–5230. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Hu, B.; Gu, Z.; Joo, K.; Wang, P.; Tang, Y. Degradable polymeric nanocapsul for efficient intracellular delivery of a high molecular weight tumor-selective protein complex. Nano Today 2013, 8, 11–20. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Klein, C.; Mü Ller, L.; Hansen, S.; Buchner, J. p53 contains large unstructured regions in its native state. J. Mol. Biol. 2002, 322, 917–927. [Google Scholar] [CrossRef]
- Dutta, K.; Hu, D.; Zhao, B.; Ribbe, A.E.; Zhuang, J.; Thayumanavan, S. Templated Self-Assembly of a Covalent Polymer Network for Intracellular Protein Delivery and Traceless Release. J. Am. Chem. Soc. 2017, 139, 5676–5679. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
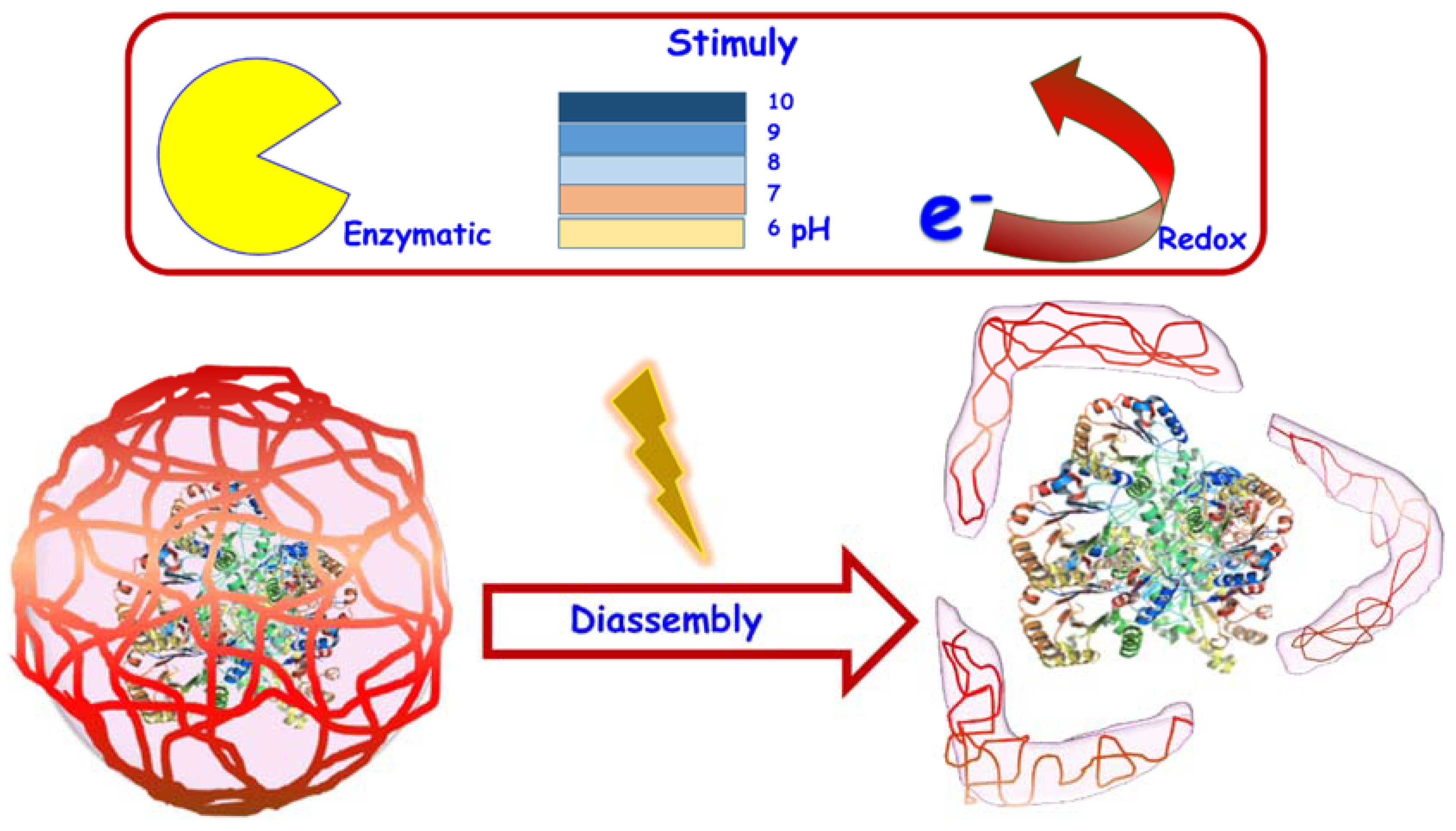{kind=link}
{kind=link}
| Strategies for Protein Delivery | |
|---|---|
| Pegylation | This strategy consists in the conjugation of polymeric chains on protein surface. This allow increase significally the times of circulation of proteins in bloodstream, however has a limited efficacy to protect against proteases attack. |
| Liposomes | Liposomes are biocompatible and cell-like nanodevices. Proteins can be delivered inside the aqueous core of liposomes or attached on their surface. Liposomes are characterized by high flexibility but their use are limited by small stability in human body. |
| Inorganic nanoparticles | Mesoporous silica nanoparticles, gold nanoparticles and carbon nanotubes allow the delivery of proteins on their surface or inside them. They are characterized by a high sturdiness but have a poor flexibility. |
| Polymeric nanocapsules | This strategy consist in a polymerization in situ around the protein making a polymeric coating. This strategy can be used to a large number of proteins and allow the design of nanocapsules both degradable and non-degradable. This class of systems combine the sturdiness of inorganic nanoparticles with flexibility of liposomes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villegas, M.R.; Baeza, A.; Vallet-Regí, M. Nanotechnological Strategies for Protein Delivery. Molecules 2018, 23, 1008. https://doi.org/10.3390/molecules23051008
Villegas MR, Baeza A, Vallet-Regí M. Nanotechnological Strategies for Protein Delivery. Molecules. 2018; 23(5):1008. https://doi.org/10.3390/molecules23051008
Chicago/Turabian StyleVillegas, María Rocío, Alejandro Baeza, and María Vallet-Regí. 2018. "Nanotechnological Strategies for Protein Delivery" Molecules 23, no. 5: 1008. https://doi.org/10.3390/molecules23051008
APA StyleVillegas, M. R., Baeza, A., & Vallet-Regí, M. (2018). Nanotechnological Strategies for Protein Delivery. Molecules, 23(5), 1008. https://doi.org/10.3390/molecules23051008