
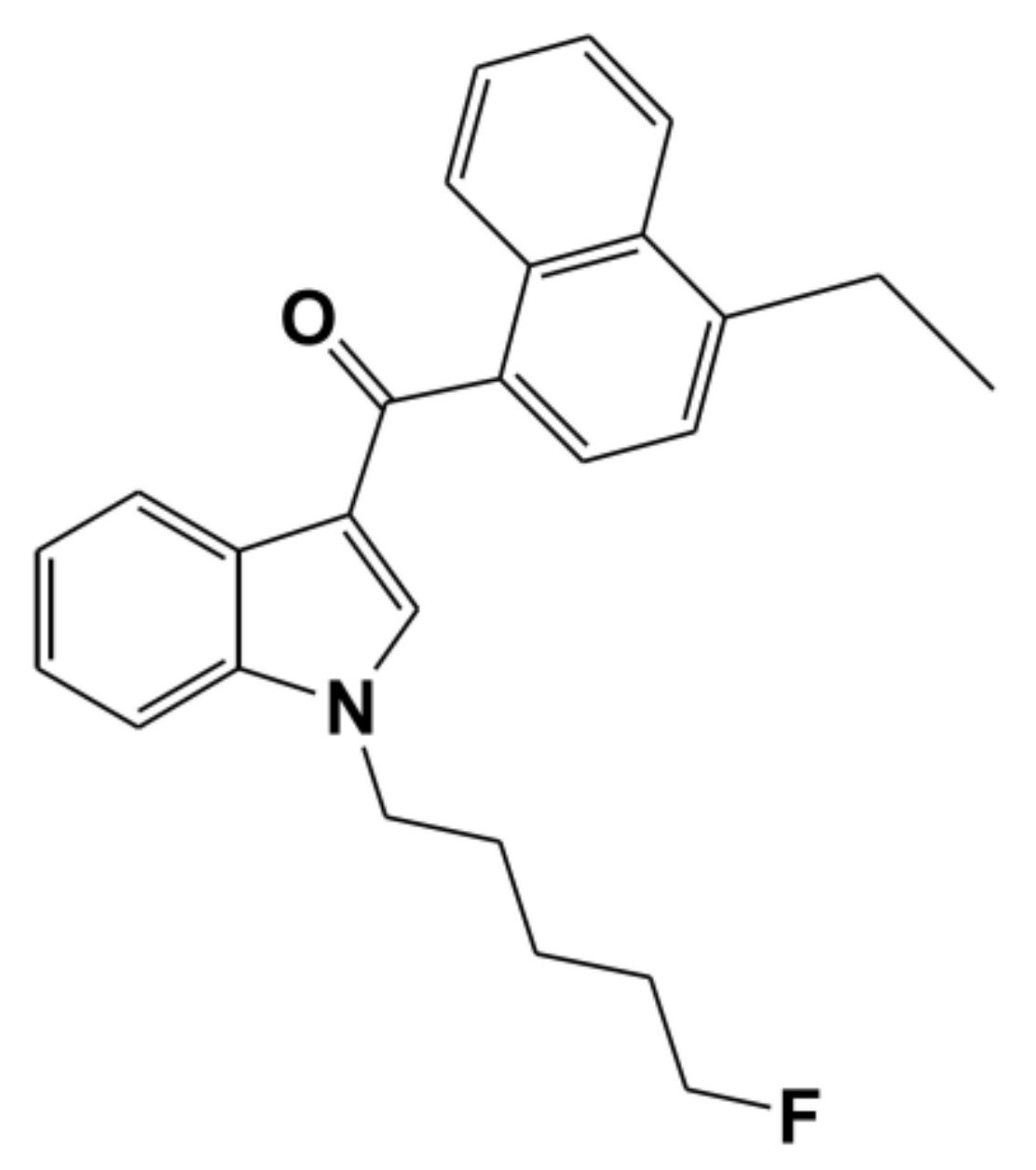
In Vitro Inhibitory Effects of Synthetic Cannabinoid EAM-2201 on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes



Abstract
:1. Introduction
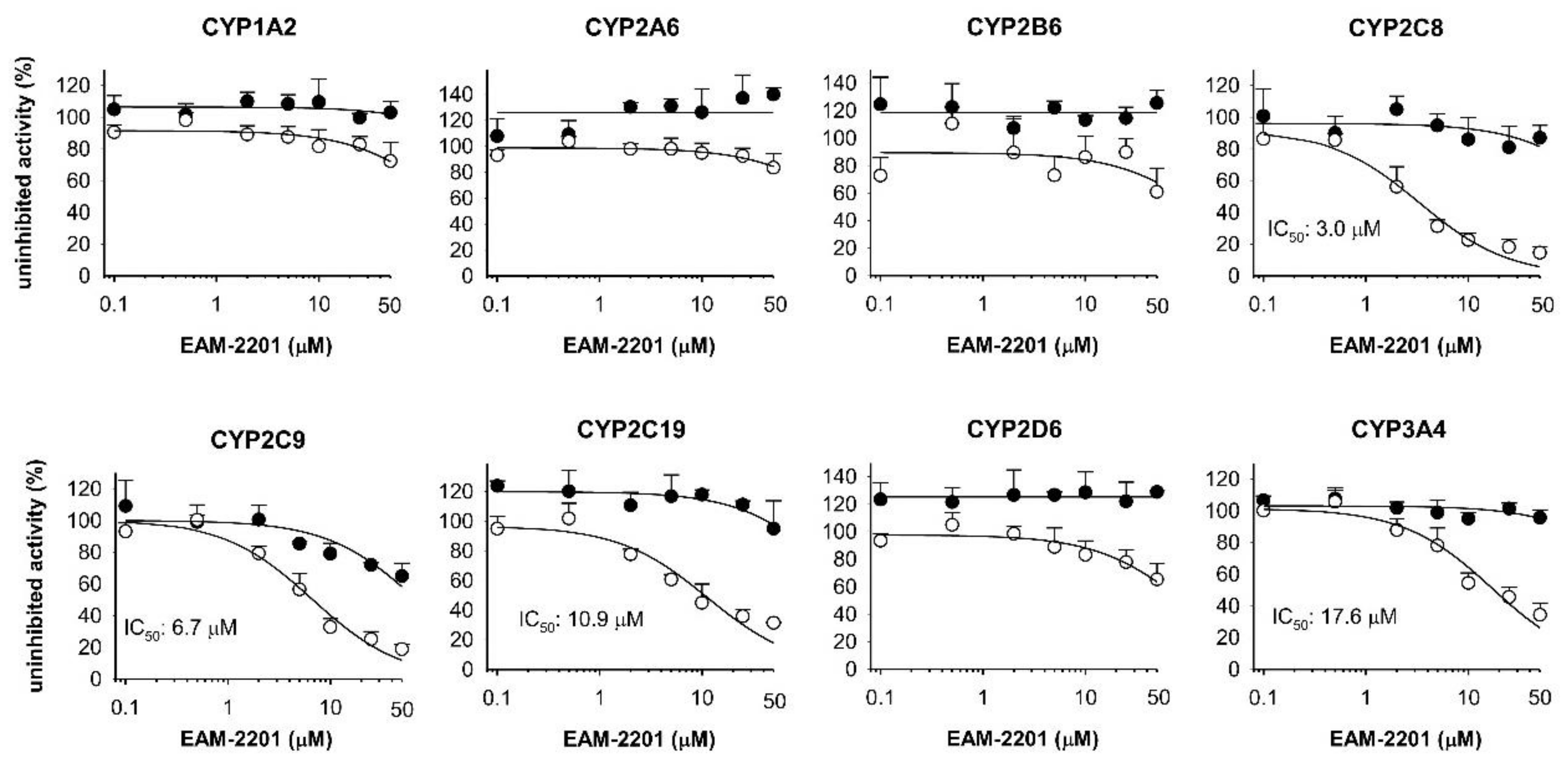
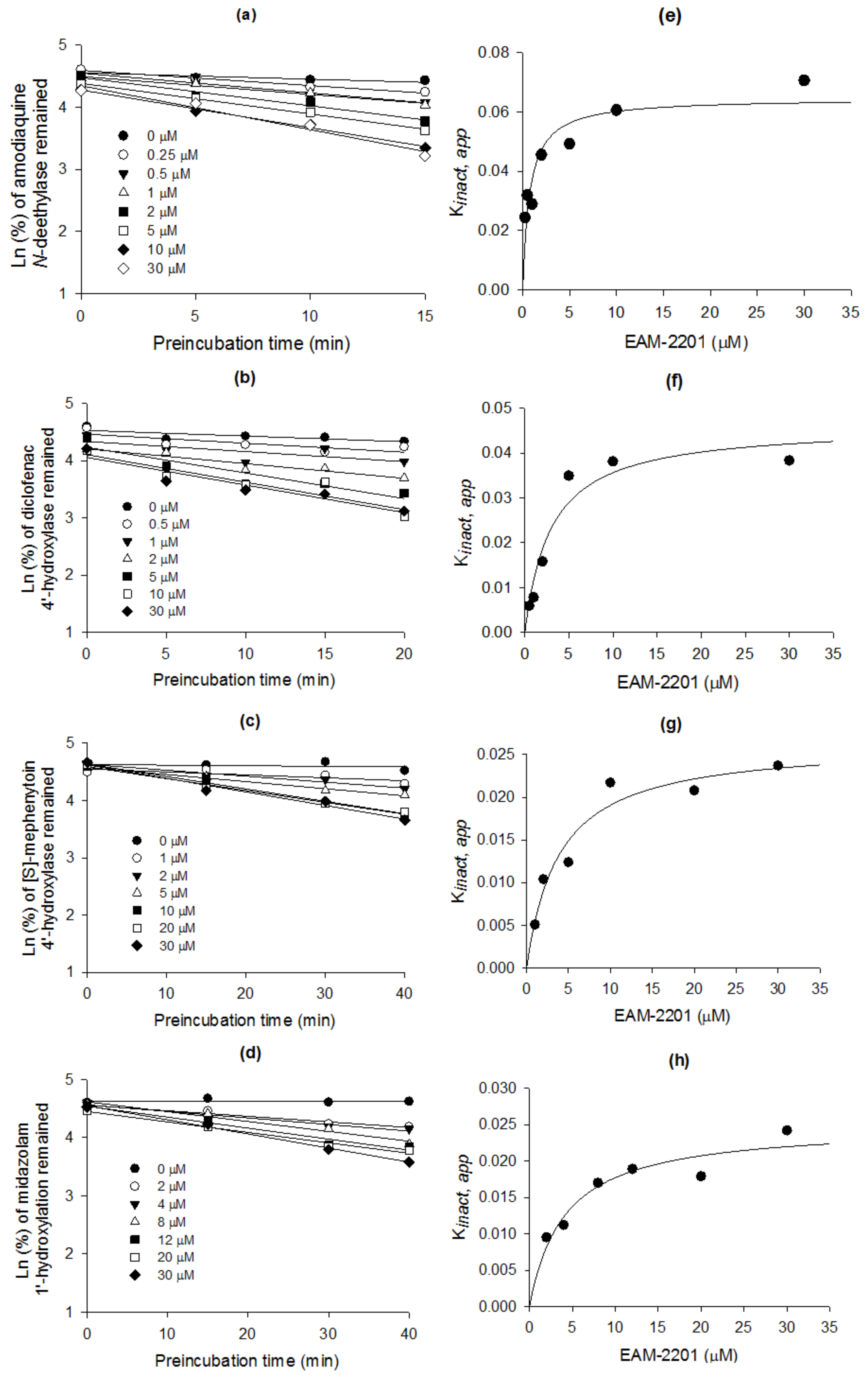
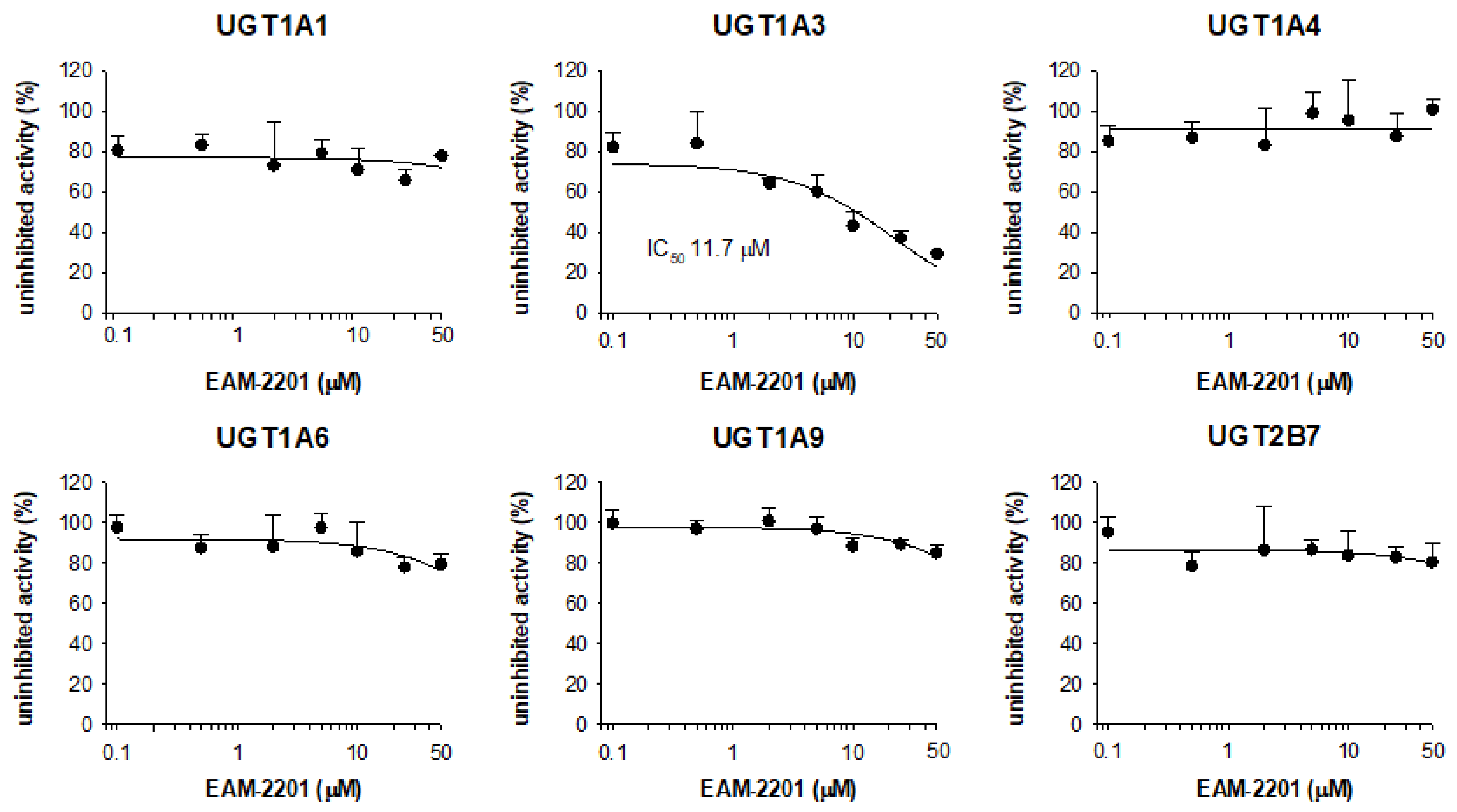
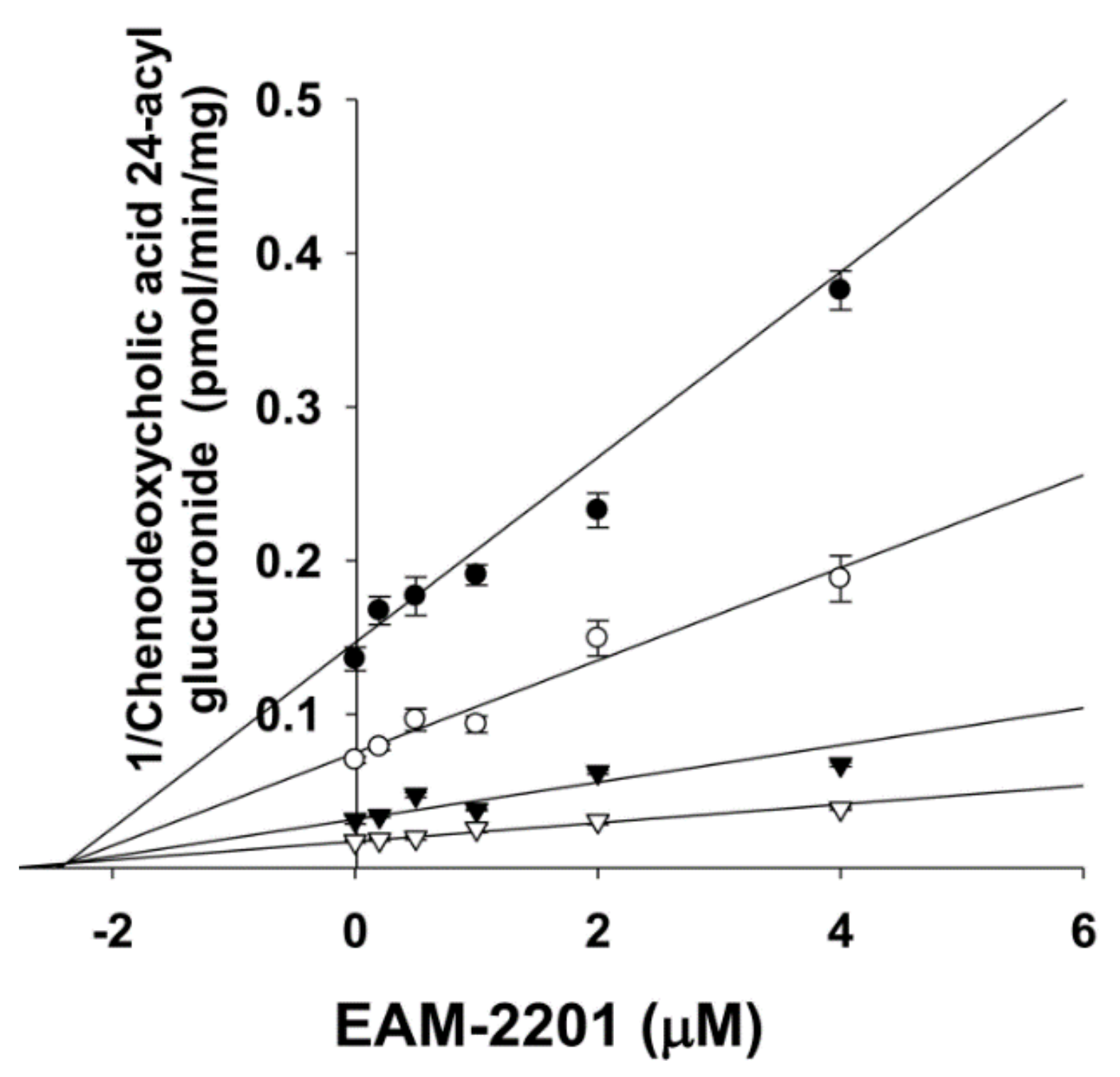
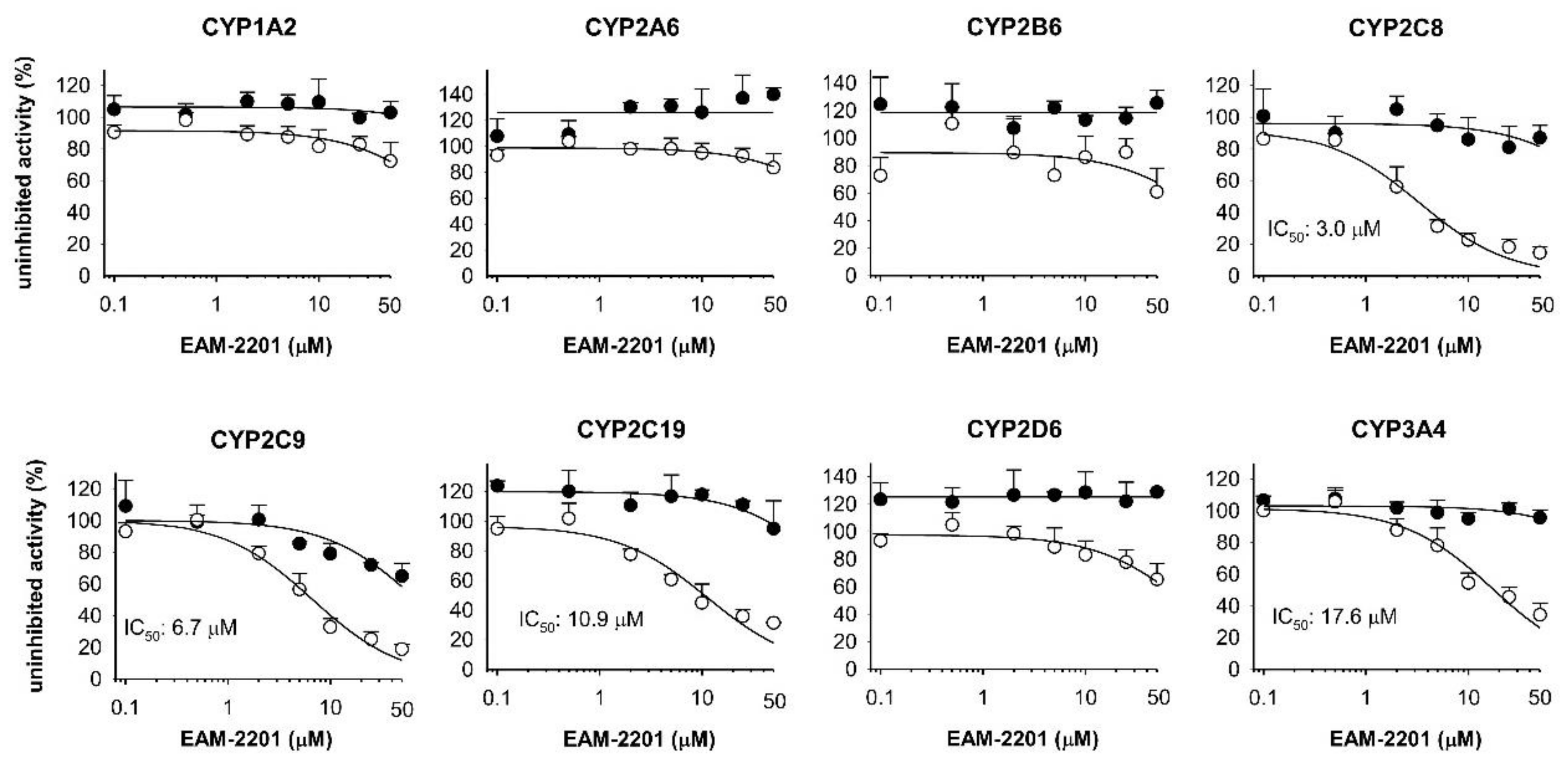
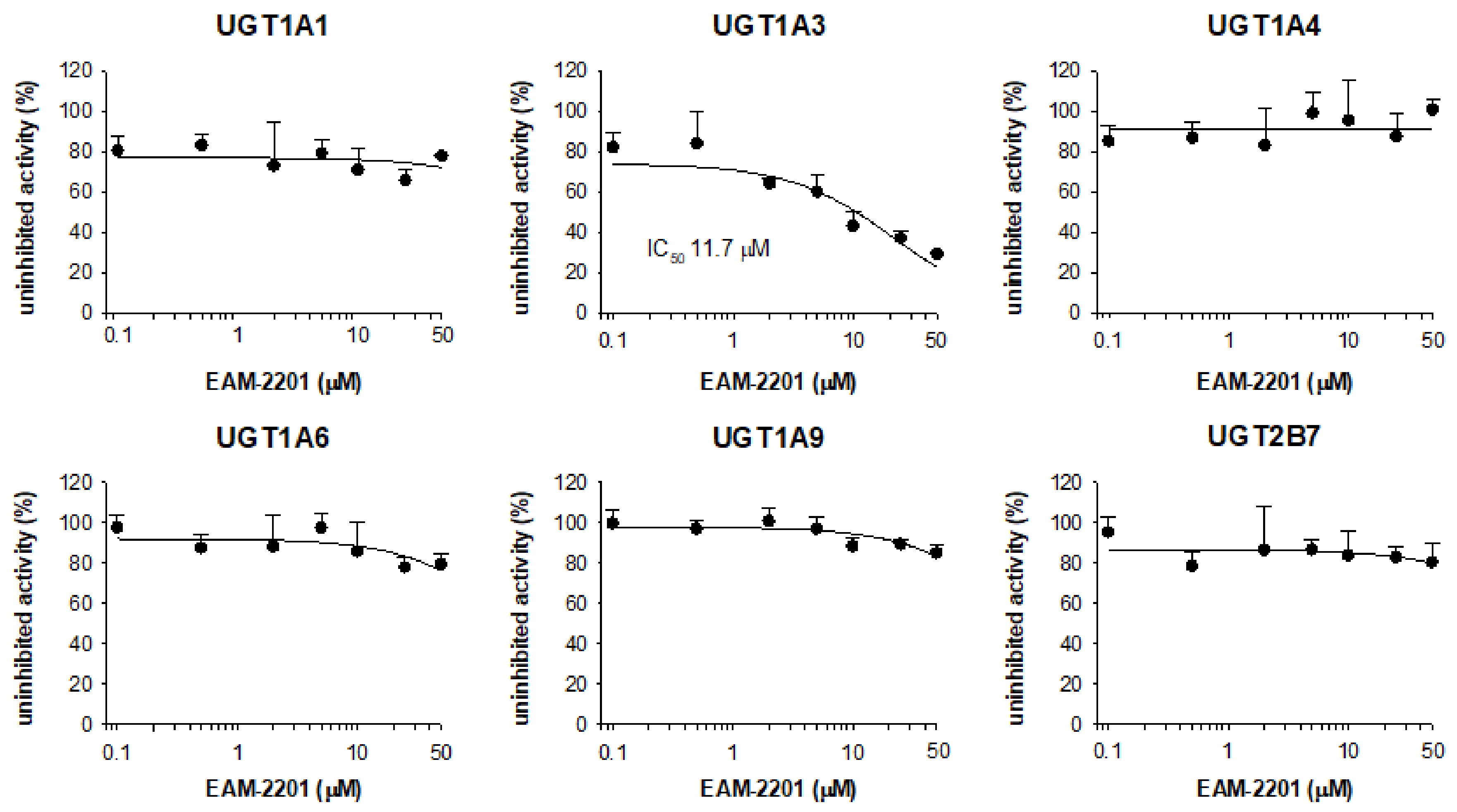
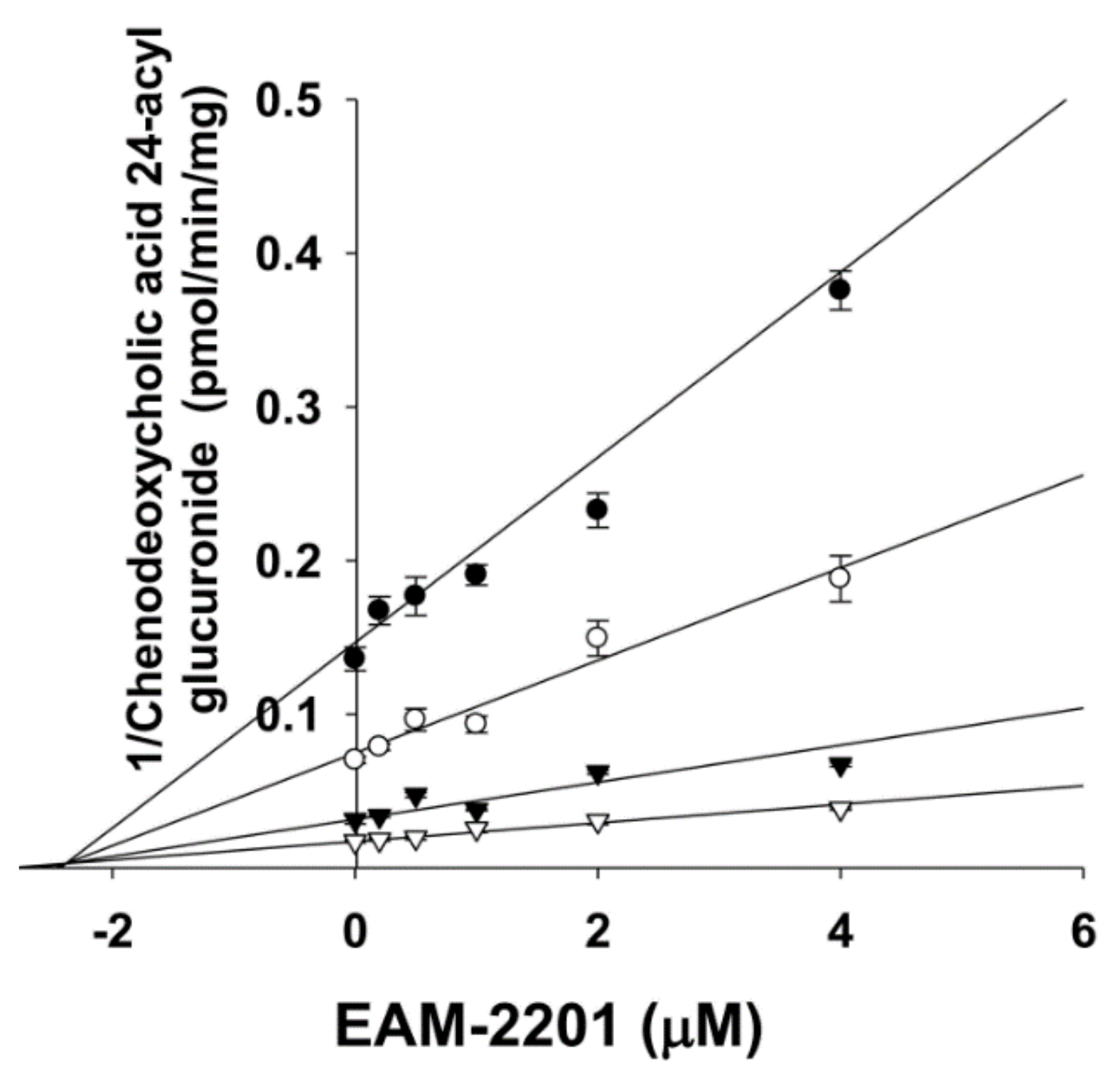
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Inhibitory Effects of EAM-2201 on Eight Major CYP Activities in Human Liver Microsomes
4.3. Inhibitory Effects of EAM-2201 on Six Major UGTs in Human Liver Microsomes
4.4. Time-Dependent Inhibition of CYP2C8, CYP2C9, CYP2C19 and CYP3A4 Activities by EAM-2201
4.5. Enzyme Kinetic Analysis
4.6. LC-MS/MS Analysis
4.7. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marusich, J.A.; Wiley, J.L.; Lefever, T.W.; Patel, P.R.; Thomas, B.F. Finding order in chemical chaos—Continuing characterization of synthetic cannabinoid receptor agonists. Neuropharmacology 2017. [Google Scholar] [CrossRef] [PubMed]
- Costain, W.J.; Tauskela, J.S.; Rasquinha, I.; Comas, T.; Hewitt, M.; Marleau, V.; Soo, E.C. Pharmacological characterization of emerging synthetic cannabinoids in HEK293T cells and hippocampal neurons. Eur. J. Pharmacol. 2016, 786, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, N.; Kawamura, M.; Kikura-Hanajiri, R.; Goda, Y. URB-754: A new class of designer drug and 12 synthetic cannabinoids detected in illegal products. Forensic Sci. Int. 2013, 227, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Gol, E.; Cok, I. Assessment of types of synthetic cannabinoids in narcotic cases assessed by the council of forensic medicine between 2011–2015, Ankara, Turkey. Forensic Sci. Int. 2017, 280, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Cannaert, A.; Storme, J.; Franz, F.; Auwarter, V.; Stove, C.P. Detection and activity profiling of synthetic cannabinoids and their metabolites with a newly developed bioassay. Anal. Chem. 2016, 88, 11476–11485. [Google Scholar] [CrossRef] [PubMed]
- Bilici, R. Synthetic cannabinoids. North. Clin. Istanb. 2014, 1, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Karila, L.; Benyamina, A.; Blecha, L.; Cottencin, O.; Billieux, J. The synthetic cannabinoids phenomenon. Curr. Pharm. Des. 2016, 22, 6420–6425. [Google Scholar] [CrossRef] [PubMed]
- Hess, C.; Stockhausen, S.; Kernbach-Wighton, G.; Madea, B. Death due to diabetic ketoacidosis: Induction by the consumption of synthetic cannabinoids? Forensic Sci. Int. 2015, 257, e6–e11. [Google Scholar] [CrossRef] [PubMed]
- Hess, C.; Murach, J.; Krueger, L.; Scharrenbroch, L.; Unger, M.; Madea, B.; Sydow, K. Simultaneous detection of 93 synthetic cannabinoids by liquid chromatography-tandem mass spectrometry and retrospective application to real forensic samples. Drug Test. Anal. 2017, 9, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.S.; Kong, T.Y.; Lee, J.Y.; Kim, J.Y.; In, M.K.; Lee, H.S. In vitro metabolism of a novel synthetic cannabinoid, EAM-2201, in human liver microsomes and human recombinant cytochrome P450s. J. Pharm. Biomed. Anal. 2016, 119, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Cerny, M.A. Prevalence of non-cytochrome P450-mediated metabolism in food and drug administration-approved oral and intravenous drugs: 2006–2015. Drug Metab. Dispos. 2016, 44, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.S.; Dalvie, D.K. Cytochrome P450 and non-cytochrome P450 oxidative metabolism: Contributions to the pharmacokinetics, safety and efficacy of xenobiotics. Drug Metab. Dispos. 2016, 44, 1229–1245. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kwon, S.S.; Kong, T.Y.; Cheong, J.C.; Kim, H.S.; In, M.K.; Lee, H.S. AM-2201 inhibits multiple cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2017, 22, 443. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.Y.; Kim, J.H.; Kwon, S.S.; Cheong, J.C.; Kim, H.S.; In, M.K.; Lee, H.S. Inhibition of cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferases by MAM-2201 in human liver microsomes. Arch. Pharm. Res. 2017, 40, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.D.; Marques-Magallanes, J.A.; Yuan, M.; Sun, W.; Tashkin, D.P.; Hankinson, O. Induction and regulation of the carcinogen-metabolizing enzyme CYP1A1 by marijuana smoke and delta (9)-tetrahydrocannabinol. Am. J. Respir. Cell Mol. Biol. 2001, 24, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Kushihara, M.; Yamamoto, I.; Watanabe, K. Characterization of major phytocannabinoids, cannabidiol and cannabinol, as isoform-selective and potent inhibitors of human CYP1 enzymes. Biochem. Pharmacol. 2010, 79, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Ebisawa, J.; Okushima, Y.; Yamamoto, I.; Watanabe, K. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: Role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci. 2011, 88, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Maeda, C.; Yamamoto, I.; Watanabe, K. Differential inhibition of human cytochrome P450 2A6 and 2B6 by major phytocannabinoids. Forensic Toxicol. 2011, 29, 117–124. [Google Scholar] [CrossRef]
- Yamaori, S.; Okamoto, Y.; Yamamoto, I.; Watanabe, K. Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab. Dispos. 2011, 39, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Koeda, K.; Kushihara, M.; Hada, Y.; Yamamoto, I.; Watanabe, K. Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metab. Pharmacokinet. 2012, 27, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Al Saabi, A.; Allorge, D.; Sauvage, F.L.; Tournel, G.; Gaulier, J.M.; Marquet, P.; Picard, N. Involvement of udp-glucuronosyltransferases UGT1A9 and UGT2B7 in ethanol glucuronidation and interactions with common drugs of abuse. Drug Metab. Dispos. 2013, 41, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Stout, S.M.; Cimino, N.M. Exogenous cannabinoids as substrates, inhibitors and inducers of human drug metabolizing enzymes: A systematic review. Drug Metab. Rev. 2014, 46, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Zendulka, O.; Dovrtelova, G.; Noskova, K.; Turjap, M.; Sulcova, A.; Hanus, L.; Jurica, J. Cannabinoids and cytochrome P450 interactions. Curr. Drug Metab. 2016, 17, 206–226. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Yamaori, S.; Okamoto, Y.; Yamamoto, I.; Watanabe, K. Cannabidiol is a potent inhibitor of the catalytic activity of cytochrome P450 2C19. Drug Metab. Pharmacokinet. 2013, 28, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.Y.; Kim, J.H.; Choi, W.G.; Lee, J.Y.; Kim, H.S.; Kim, J.Y.; In, M.K.; Lee, H.S. Metabolic characterization of (1-(5-fluoropentyl)-1H-indol-3-yl)(4-methyl-1-naphthalenyl)-methanone (MAM-2201) using human liver microsomes and cDNA-overexpressed cytochrome P450 enzymes. Anal. Bioanal. Chem. 2017, 409, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Polasek, T.M.; Elliot, D.J.; Lewis, B.C.; Miners, J.O. Mechanism-based inactivation of human cytochrome P4502C8 by drugs in vitro. J. Pharmacol. Exp. Ther. 2004, 311, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.S.; Yang, L.P.; Li, X.T.; Liu, J.P.; Zhou, Z.W.; Zhou, S.F. Human CYP2C8: Structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr. Drug Metab. 2009, 10, 1009–1047. [Google Scholar] [CrossRef] [PubMed]
- Hutzler, J.M.; Balogh, L.M.; Zientek, M.; Kumar, V.; Tracy, T.S. Mechanism-based inactivation of cytochrome P450 2C9 by tienilic acid and (+/−)-suprofen: A comparison of kinetics and probe substrate selection. Drug Metab. Dispos. 2009, 37, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Zhou, Z.W.; Yang, L.P.; Cai, J.P. Substrates, inducers, inhibitors and structure-activity relationships of human cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009, 16, 3480–3675. [Google Scholar] [CrossRef] [PubMed]
- Chimalakonda, K.C.; Seely, K.A.; Bratton, S.M.; Brents, L.K.; Moran, C.L.; Endres, G.W.; James, L.P.; Hollenberg, P.F.; Prather, P.L.; Radominska-Pandya, A.; et al. Cytochrome P450-mediated oxidative metabolism of abused synthetic cannabinoids found in K2/spice: Identification of novel cannabinoid receptor ligands. Drug Metab. Dispos. 2012, 40, 2174–2184. [Google Scholar] [CrossRef] [PubMed]
- Thanh, H.D.; Dijols, S.; Macherey, A.-C.; Goldstein, J.; Dansette, P.; Mansuy, D. Ticlopidine as a Selective Mechanism-Based Inhibitor of Human Cytochrome P450 2C19. Biochemistry 2001, 40, 12112–12122. [Google Scholar]
- Nishiya, Y.; Hagihara, K.; Kurihara, A.; Okudaira, N.; Farid, N.A.; Okazaki, O.; Ikeda, T. Comparison of mechanism-based inhibition of human cytochrome P450 2C19 by ticlopidine, clopidogrel and prasugrel. Xenobiotica 2009, 39, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Yamazaki, H. Comparison of cytochrome P450 2C subfamily members in terms of drug oxidation rates and substrate inhibition. Curr. Drug Metab. 2012, 13, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Albaugh, D.R.; Fullenwider, C.L.; Fisher, M.B.; Hutzler, J.M. Time-dependent inhibition and estimation of CYP3A clinical pharmacokinetic drug-drug interactions using plated human cell systems. Drug Metab. Dispos. 2012, 40, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Alonen, A.; Finel, M.; Kostiainen, R. The human UDP-glucuronosyltransferase UGT1A3 is highly selective towards N2 in the tetrazole ring of losartan, candesartan and zolarsartan. Biochem. Pharmacol. 2008, 76, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, T.J.; Aehlen, A.; Ehmer, U.; Kalthoff, S.; Manns, M.P.; Strassburg, C.P. Regulation of the human bile acid UDP-glucuronosyltransferase 1A3 by the farnesoid X receptor and bile acids. J. Hepatol. 2010, 52, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.S.; Kim, Y.W.; Kim, H.J.; Shin, H.J.; Shin, J.G.; Kim, K.H.; Chi, Y.H.; Paik, S.H.; Kim, D.H. Glucuronidation of fimasartan, a new angiotensin receptor antagonist, is mainly mediated by UGT1A3. Xenobiotica 2015, 45, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Su, M.K.; Seely, K.A.; Moran, J.H.; Hoffman, R.S. Metabolism of classical cannabinoids and the synthetic cannabinoid JWH-018. Clin. Pharmacol. Ther. 2015, 97, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.S.; Kim, J.H.; Jeong, H.U.; Cho, Y.Y.; Oh, S.R.; Lee, H.S. Inhibitory effects of aschantin on cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2016, 21, 554. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Activity | CYP | Ki (µM) | kinact (min−1) |
|---|---|---|---|
| Amodiaquine N-deethylase | 2C8 | 0.54 | 0.0633 |
| Diclofenac 4′-hydroxylase | 2C9 | 3.0 | 0.0462 |
| [S]-Mephenytoin 4′-hydroxylase | 2C19 | 3.8 | 0.0264 |
| Midazolam 1′-hydroxylase | 3A4 | 4.1 | 0.0250 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, T.Y.; Kwon, S.-S.; Cheong, J.C.; Kim, H.S.; Kim, J.Y.; Lee, H.S. In Vitro Inhibitory Effects of Synthetic Cannabinoid EAM-2201 on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules 2018, 23, 920. https://doi.org/10.3390/molecules23040920
Kong TY, Kwon S-S, Cheong JC, Kim HS, Kim JY, Lee HS. In Vitro Inhibitory Effects of Synthetic Cannabinoid EAM-2201 on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules. 2018; 23(4):920. https://doi.org/10.3390/molecules23040920
Chicago/Turabian StyleKong, Tae Yeon, Soon-Sang Kwon, Jae Chul Cheong, Hee Seung Kim, Jin Young Kim, and Hye Suk Lee. 2018. "In Vitro Inhibitory Effects of Synthetic Cannabinoid EAM-2201 on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes" Molecules 23, no. 4: 920. https://doi.org/10.3390/molecules23040920
APA StyleKong, T. Y., Kwon, S.-S., Cheong, J. C., Kim, H. S., Kim, J. Y., & Lee, H. S. (2018). In Vitro Inhibitory Effects of Synthetic Cannabinoid EAM-2201 on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules, 23(4), 920. https://doi.org/10.3390/molecules23040920