
Carotenoid Lutein Selectively Inhibits Breast Cancer Cell Growth and Potentiates the Effect of Chemotherapeutic Agents through ROS-Mediated Mechanisms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Lutein Selectively Inhibits the Growth of Human Breast Cancer Cells
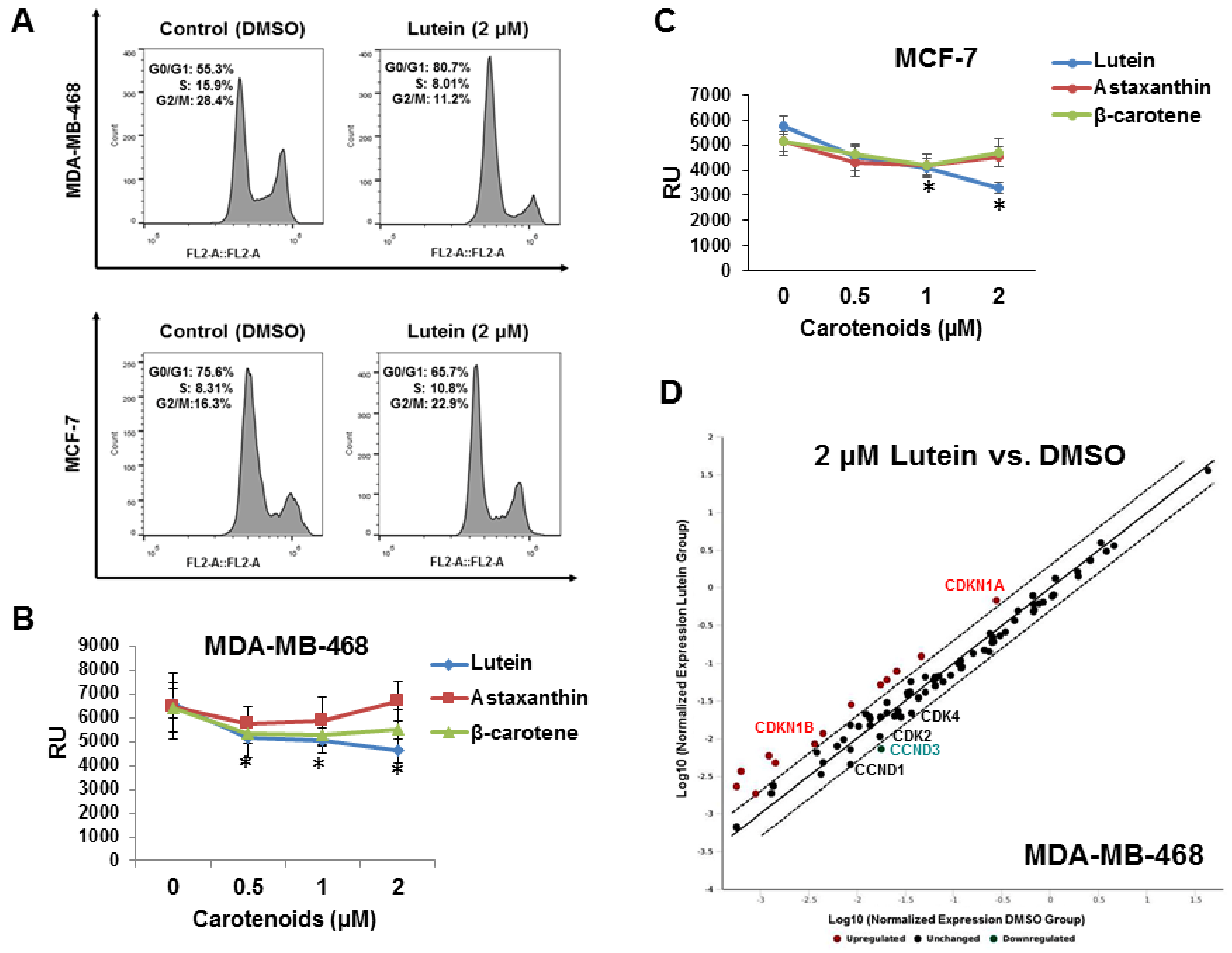
2.2. Lutein Induces Cell Cycle Arrest in Human Breast Cancer Cells
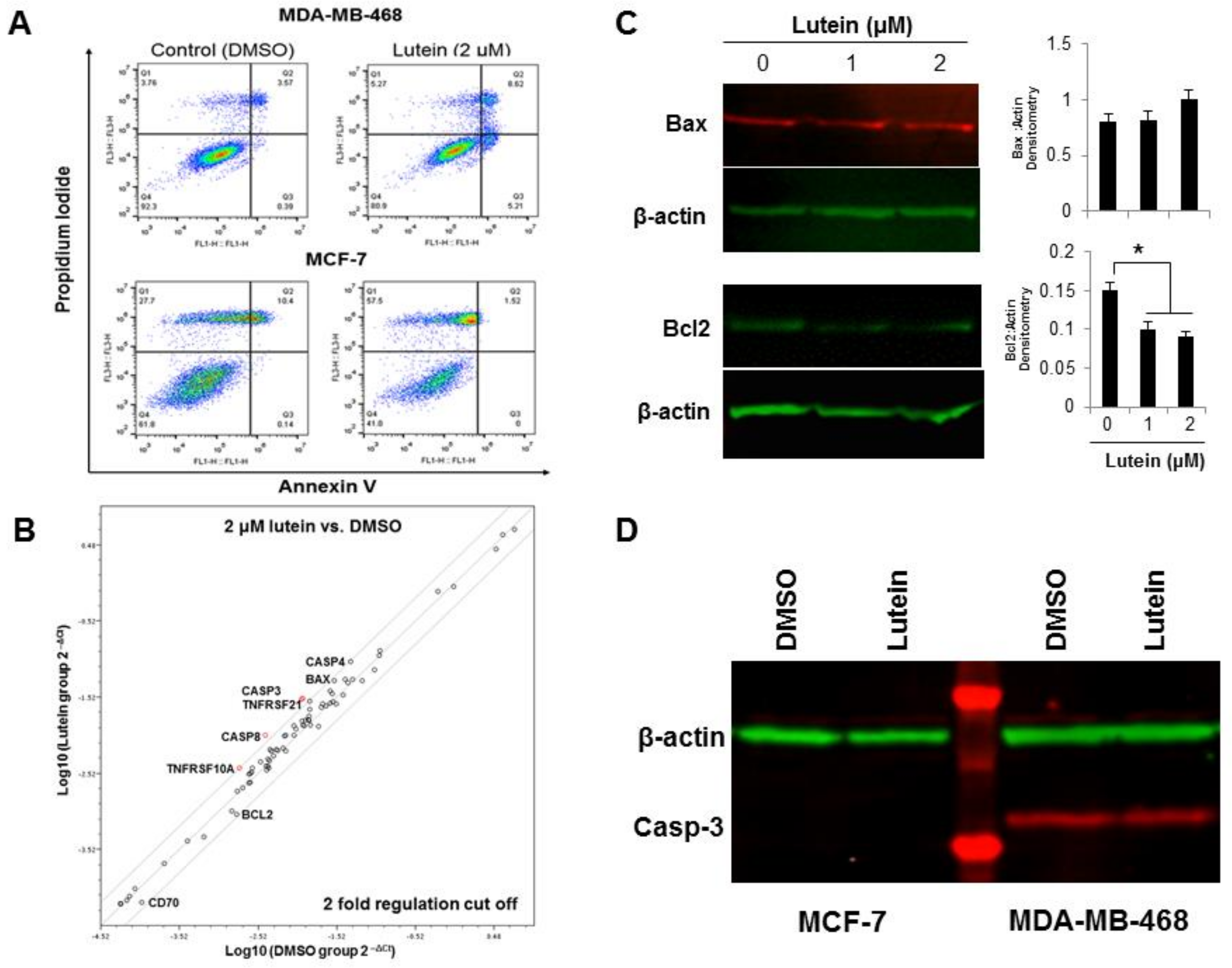
2.3. Lutein Induces Minimal Apoptotic Cell Death in Breast Cancer Cells
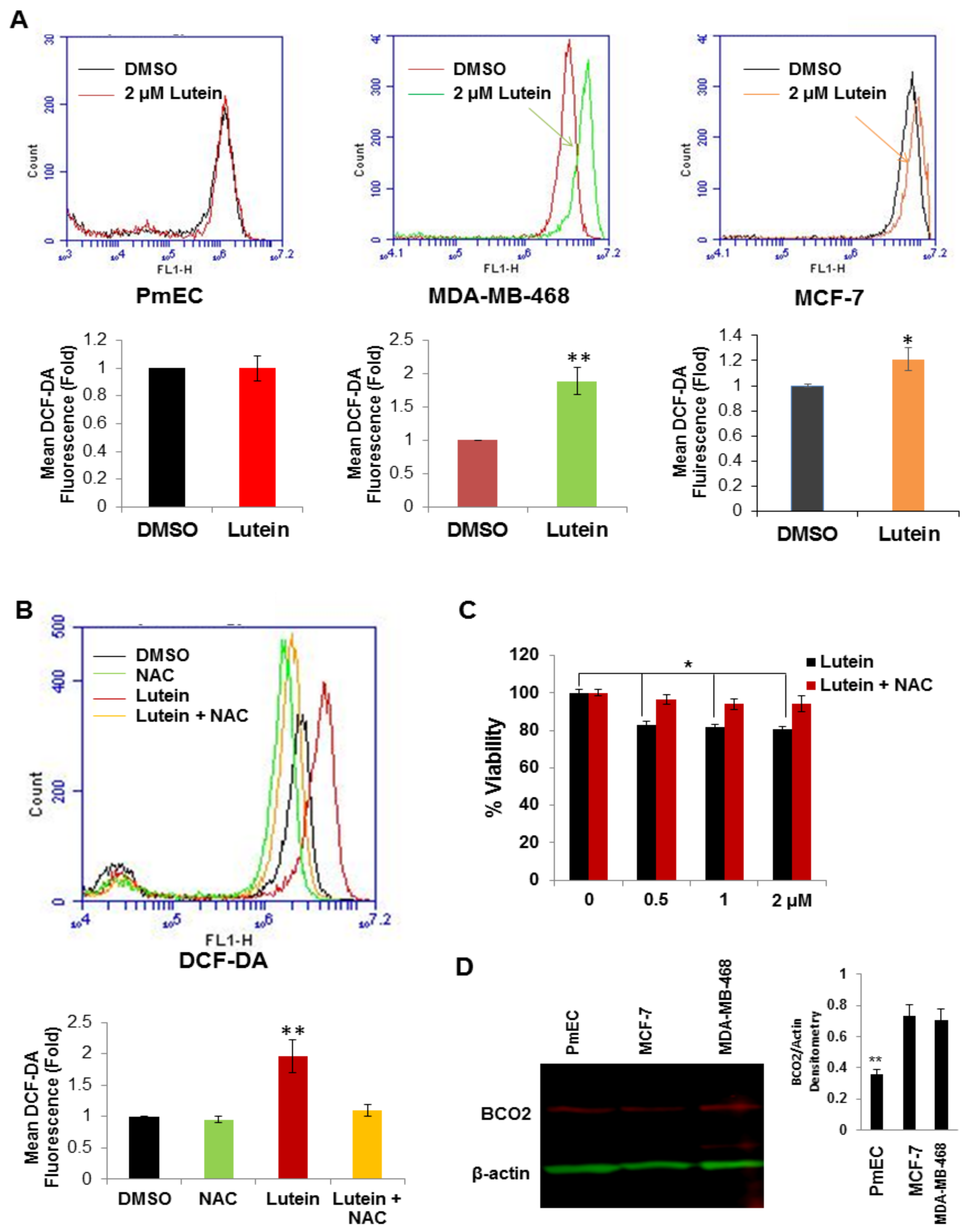
2.4. Lutein Selectively Increases Intracellular ROS Production in Human Breast Cancer Cells
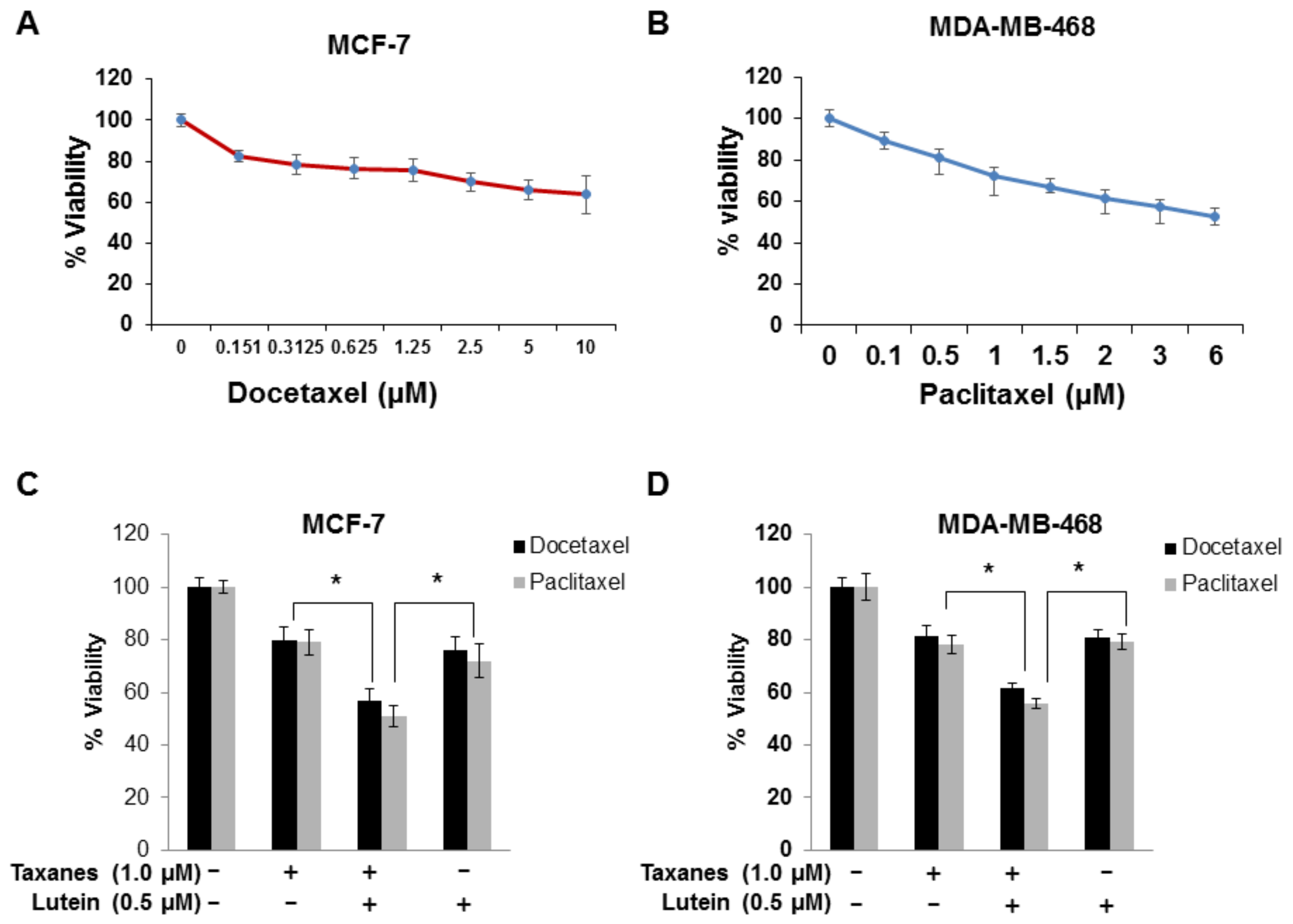
2.5. Lutein Enhances the Cytotoxic Effects of Chemotherapeutic Taxanes in Human Breast Cancer Cells
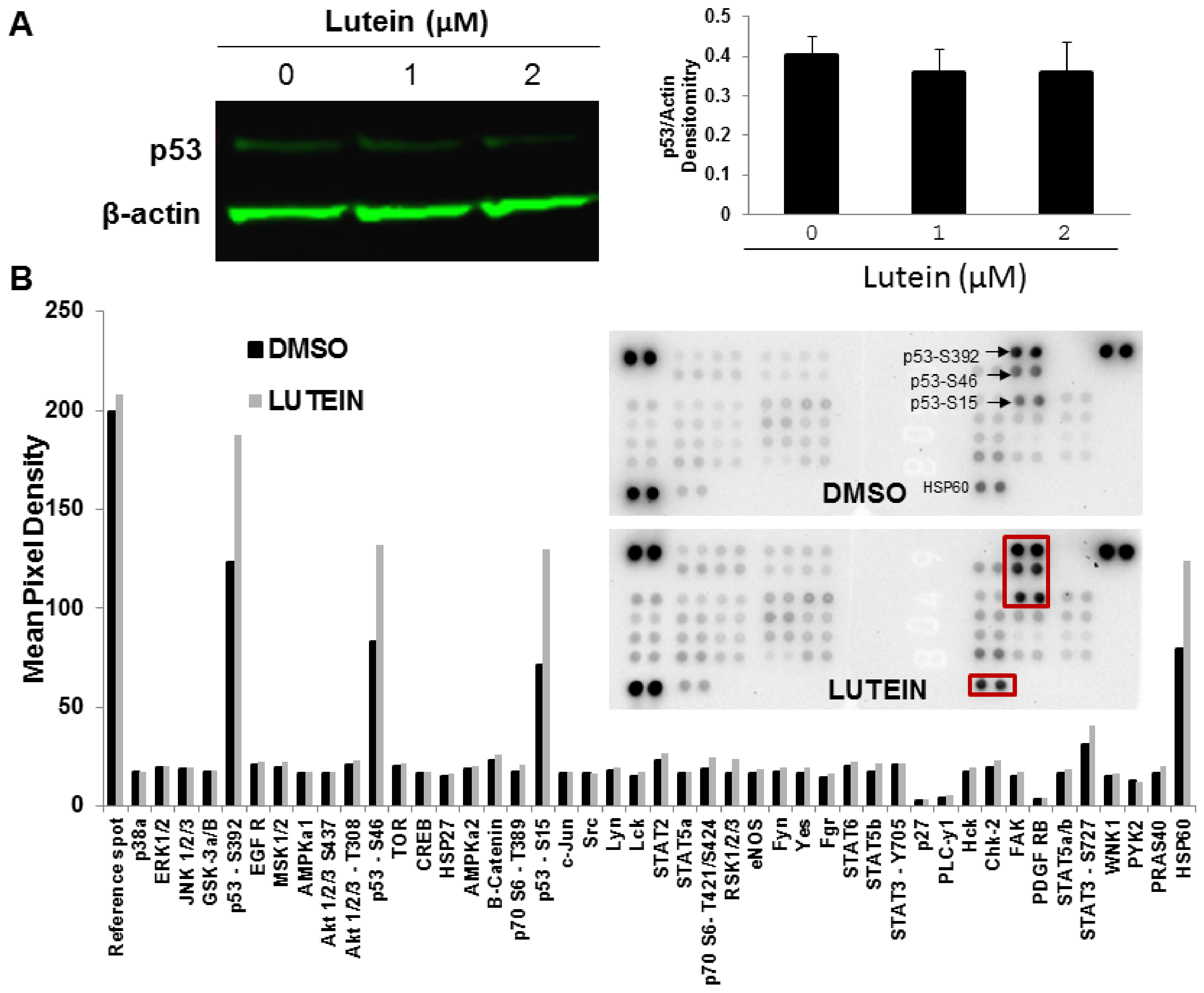
2.6. Lutein Activates the p53 Signaling Pathway and Upregulates HSP60 in MDA-MB-468 Cells
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell and Cell Culture
4.3. Cell Proliferation/Viability Assays
4.4. Colony Formation Assay
4.5. Cell Cycle Analysis
4.6. EdU Proliferation Assays
4.7. Flow Cytometric Assay of Apoptosis
4.8. Quantitative RT–PCR Apoptosis and Cell Cycle Focused Gene Arrays
4.9. Determination of Intracellular ROS Production
4.10. Quantitative Fluorescent Western Blot Analysis
4.11. Phospho-Antibody Array Analysis
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. New Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Eliassen, A.H.; Hendrickson, S.J.; Brinton, L.A.; Buring, J.E.; Campos, H.; Dai, Q.; Dorgan, J.F.; Franke, A.A.; Gao, Y.T.; Goodman, M.T.; et al. Circulating carotenoids and risk of breast cancer: Pooled analysis of eight prospective studies. J. Natl. Cancer Inst. 2012, 104, 1905–1916. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Lu, M.S.; Wang, L.; Mo, X.F.; Luo, W.P.; Du, Y.F.; Zhang, C.X. Specific serum carotenoids are inversely associated with breast cancer risk among Chinese women: A case-control study. Br. J. Nutr. 2016, 115, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Spiegelman, D.; Baglietto, L.; Bernstein, L.; Boggs, D.A.; van den Brandt, P.A.; Buring, J.E.; Gapstur, S.M.; Giles, G.G.; Giovannucci, E.; et al. Carotenoid intakes and risk of breast cancer defined by estrogen receptor and progesterone receptor status: A pooled analysis of 18 prospective cohort studies. Am. J. Clin. Nutr. 2012, 95, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Chan, D.S.; Vieira, A.R.; Navarro Rosenblatt, D.A.; Vieira, R.; Greenwood, D.C.; Norat, T. Dietary compared with blood concentrations of carotenoids and breast cancer risk: A systematic review and meta-analysis of prospective studies. Am. J. Clin. Nutr. 2012, 96, 356–373. [Google Scholar] [CrossRef] [PubMed]
- Bakker, M.F.; Peeters, P.H.; Klaasen, V.M.; Bueno-de-Mesquita, H.B.; Jansen, E.H.; Ros, M.M.; Travier, N.; Olsen, A.; Tjonneland, A.; Overvad, K.; et al. Plasma carotenoids, vitamin C, tocopherols, and retinol and the risk of breast cancer in the European Prospective Investigation into Cancer and Nutrition cohort. Am. J. Clin. Nutr. 2016, 103, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Baumgartner, R.N.; Yang, D.; Slattery, M.L.; Murtaugh, M.A.; Byers, T.; Hines, L.M.; Giuliano, A.R.; Baumgartner, K.B. No evidence of association between breast cancer risk and dietary carotenoids, retinols, vitamin C and tocopherols in Southwestern Hispanic and non-Hispanic White women. Breast Cancer Res. Treat. 2009, 114, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Demmig-Adams, B.; Adams, W.W. Antioxidants in photosynthesis and human nutrition. Science 2002, 298, 2149–2153. [Google Scholar] [CrossRef] [PubMed]
- Erdman, J.W., Jr.; Ford, N.A.; Lindshield, B.L. Are the health attributes of lycopene related to its antioxidant function? Arch. Biochem. Biophy. 2009, 483, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L., Jr.; Valanis, B.; Williams, J.H., Jr.; et al. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J. Natl. Cancer Inst. 1996, 88, 1550–1559. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 29, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.J. Role of lutein and zeaxanthin in visual and cognitive function throughout the lifespan. Nutr. Rev. 2014, 72, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Ribaya-Mercado, J.D.; Blumberg, J.B. Lutein and zeaxanthin and their potential roles in disease prevention. J. Am. Coll. Nutr. 2004, 23, 567S–587S. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, E.R.; Firdous, A.P.; Ramnath, V.; Kuttan, R. Effect of carotenoid lutein on N-nitrosodiethylamine-induced hepatocellular carcinoma and its mechanism of action. Eur. J. Cancer Prev. 2013, 22, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Rafi, M.M.; Kanakasabai, S.; Gokarn, S.V.; Krueger, E.G.; Bright, J.J. Dietary lutein modulates growth and survival genes in prostate cancer cells. J. Med. Food 2015, 18, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Chew, B.P.; Brown, C.M.; Park, J.S.; Mixter, P.F. Dietary lutein inhibits mouse mammary tumor growth by regulating angiogenesis and apoptosis. Anticancer Res. 2003, 23, 3333–3339. [Google Scholar] [PubMed]
- Sumantran, V.N.; Zhang, R.; Lee, D.S.; Wicha, M.S. Differential regulation of apoptosis in normal versus transformed mammary epithelium by lutein and retinoic acid. Cancer Epidemiol. Prev. Biomark. 2000, 9, 257–263. [Google Scholar]
- Tanaka, T.; Shnimizu, M.; Moriwaki, H. Cancer chemoprevention by carotenoids. Molecules 2012, 17, 3202–3242. [Google Scholar] [CrossRef] [PubMed]
- Young, A.J.; Lowe, G.M. Antioxidant and prooxidant properties of carotenoids. Arch. Biochem. Biophy. 2001, 385, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.; Lobo, G.P.; Golczak, M.; Li, H.N.; Klimova, T.; Hoppel, C.L.; Wyss, A.; Palczewski, K.; von Lintig, J. A mitochondrial enzyme degrades carotenoids and protects against oxidative stress. FASEB J. 2011, 25, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. CA A Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef]
- Lavrik, I.; Golks, A.; Krammer, P.H. Death receptor signaling. J. Cell Sci. 2005, 118, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-mediated apoptosis in mammals. Protein Cell 2014, 5, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.; Esposti, M.D.; Gilmore, A.P. Bcl-2 proteins and mitochondria—Specificity in membrane targeting for death. Biochim. Biophys. Acta 2011, 181, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Palozza, P. Prooxidant actions of carotenoids in biologic systems. Nutr. Rev. 1998, 56, 257–265. [Google Scholar] [CrossRef] [PubMed]
- El-Agamey, A.; Lowe, G.M.; McGarvey, D.J.; Mortensen, A.; Phillip, D.M.; Truscott, T.G.; Young, A.J. Carotenoid radical chemistry and antioxidant/pro-oxidant properties. Arch. Biochem. Biophy. 2004, 430, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Lin, J.F.; Chang, H.H.; Lee, G.A.; Hung, C.F. Lutein protects against methotrexate-induced and reactive oxygen species-mediated apoptotic cell injury of IEC-6 cells. PLoS ONE 2013, 8, e72553. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.M.; Chen, C.L.; Yang, C.M.; Wu, P.H.; Tsou, C.J.; Chiang, K.W.; Wu, W.B. The carotenoid lutein enhances matrix metalloproteinase-9 production and phagocytosis through intracellular ROS generation and ERK1/2, p38 MAPK, and RARbeta activation in murine macrophages. J. Leukocyte Biol. 2013, 93, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Lobo, G.P.; Isken, A.; Hoff, S.; Babino, D.; von Lintig, J. BCDO2 acts as a carotenoid scavenger and gatekeeper for the mitochondrial apoptotic pathway. Development 2012, 139, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, R.; Devaraj, S.N.; Padma, V.V. Lutein protects HT-29 cells against Deoxynivalenol-induced oxidative stress and apoptosis: Prevention of NF-kappaB nuclear localization and down regulation of NF-kappaB and Cyclo-Oxygenase-2 expression. Free Radic. Biol. Med. 2010, 49, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; He, Y.G.; Andersson, S. Cell type-specific expression of beta-carotene 9’,10’-monooxygenase in human tissues. J. Histochem. Cytochem. 2005, 53, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.M.; Marisiddaiah, R.; Zaripheh, S.; Wiener, D.; Rubin, L.P. Mitochondrial beta-Carotene 9,10 Oxygenase Modulates Prostate Cancer Growth via NF-kappa B Inhibition: A Lycopene-Independent Function. Mol. Cancer Res. 2016, 14, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, G.; Amengual, J.; Hoppel, C.L.; von Lintig, J. Evidence for compartmentalization of mammalian carotenoid metabolism. FASEB J 2014, 28, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Mathiasen, I.S.; Jaattela, M. Triggering caspase-independent cell death to combat cancer. Trends Mol. Med. 2002, 8, 212–220. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell. Biol. 2003, 2, 8576–8585. [Google Scholar] [CrossRef]
- Hao, M.; Lowy, A.M.; Kapoor, M.; Deffie, A.; Liu, G.; Lozano, G. Mutation of phosphoserine 389 affects p53 function in vivo. J. Biol. Chem. 1996, 271, 29380–29385. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.B.; Hsieh, J.K.; Zhong, S.; Heath, V.; Gusterson, B.; Crook, T.; Lu, X. Ser392 phosphorylation regulates the oncogenic function of mutant p53. Cancer Res. 2004, 64, 4749–4754. [Google Scholar] [CrossRef] [PubMed]
- Urushibara, M.; Kageyama, Y.; Akashi, T.; Otsuka, Y.; Takizawa, T.; Koike, M.; Kihara, K. HSP60 may predict good pathological response to neoadjuvant chemoradiotherapy in bladder cancer. Jpn. J. Clin. Oncol. 2007, 37, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Choy, G.; Tang, D.G. Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: Evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J. Biol. Chem. 2007, 282, 31289–31301. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.K.; Singh, A.; Yadav, N.K.; Cheruvu, S.H.; Hossain, Z.; Meena, S.; Maheshwari, S.; Singh, A.K.; Shahab, U.; Sharma, C.; et al. Anti-breast tumor activity of Eclipta extract in-vitro and in-vivo: Novel evidence of endoplasmic reticulum specific localization of Hsp60 during apoptosis. Sci. Rep. 2015, 5, 18457. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, X.; Smith, J.R.; Swanson, H.M.; Rubin, L.P. Carotenoid Lutein Selectively Inhibits Breast Cancer Cell Growth and Potentiates the Effect of Chemotherapeutic Agents through ROS-Mediated Mechanisms. Molecules 2018, 23, 905. https://doi.org/10.3390/molecules23040905
Gong X, Smith JR, Swanson HM, Rubin LP. Carotenoid Lutein Selectively Inhibits Breast Cancer Cell Growth and Potentiates the Effect of Chemotherapeutic Agents through ROS-Mediated Mechanisms. Molecules. 2018; 23(4):905. https://doi.org/10.3390/molecules23040905
Chicago/Turabian StyleGong, Xiaoming, Joshua R. Smith, Haley M. Swanson, and Lewis P. Rubin. 2018. "Carotenoid Lutein Selectively Inhibits Breast Cancer Cell Growth and Potentiates the Effect of Chemotherapeutic Agents through ROS-Mediated Mechanisms" Molecules 23, no. 4: 905. https://doi.org/10.3390/molecules23040905
APA StyleGong, X., Smith, J. R., Swanson, H. M., & Rubin, L. P. (2018). Carotenoid Lutein Selectively Inhibits Breast Cancer Cell Growth and Potentiates the Effect of Chemotherapeutic Agents through ROS-Mediated Mechanisms. Molecules, 23(4), 905. https://doi.org/10.3390/molecules23040905