Monitoring of the Apple Fruit Moth: Detection of Genetic Variation and Structure Applying a Novel Multiplex Set of 19 STR Markers

 ,
, 
 ,
,
Abstract
:1. Introduction
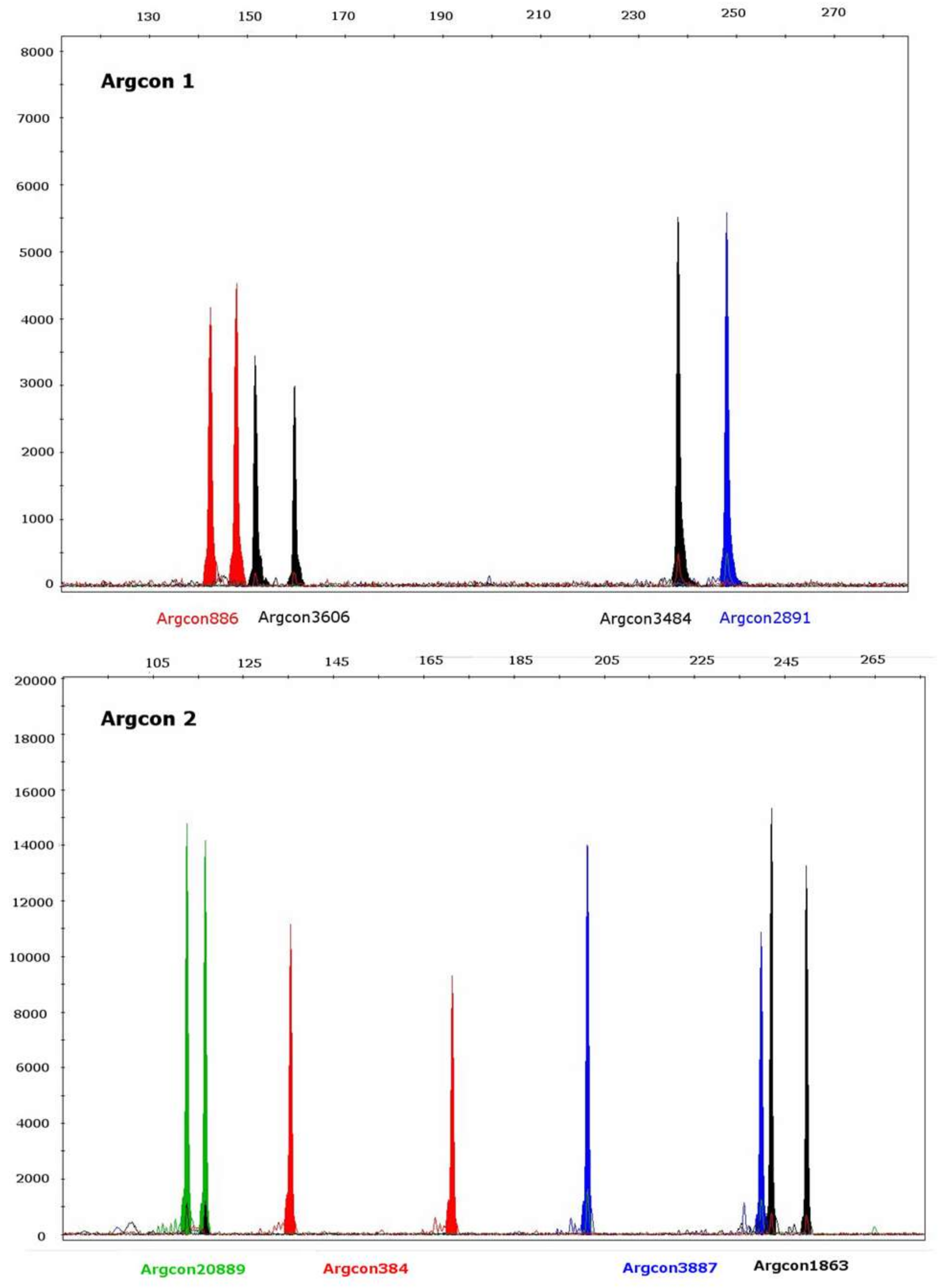
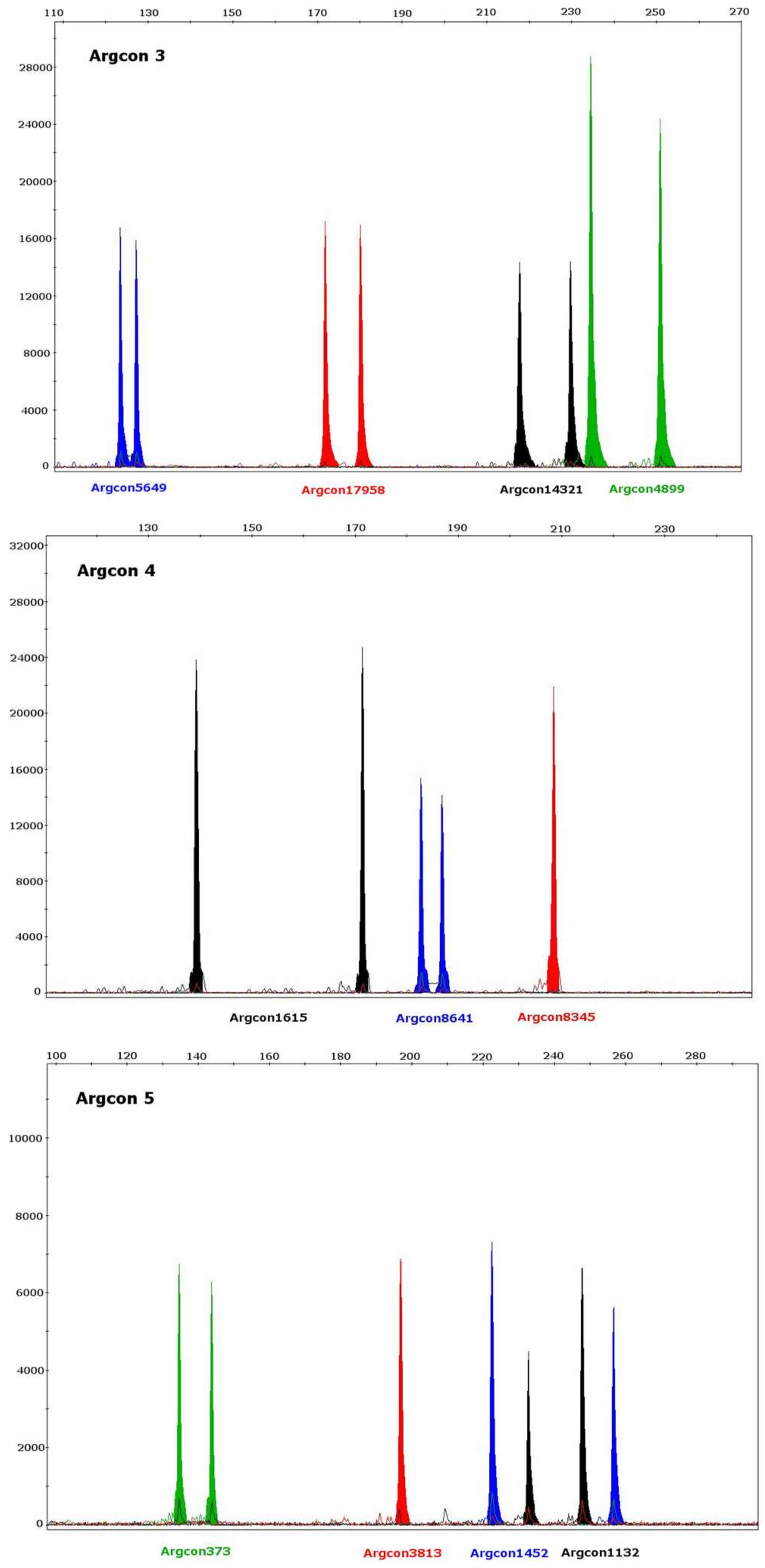
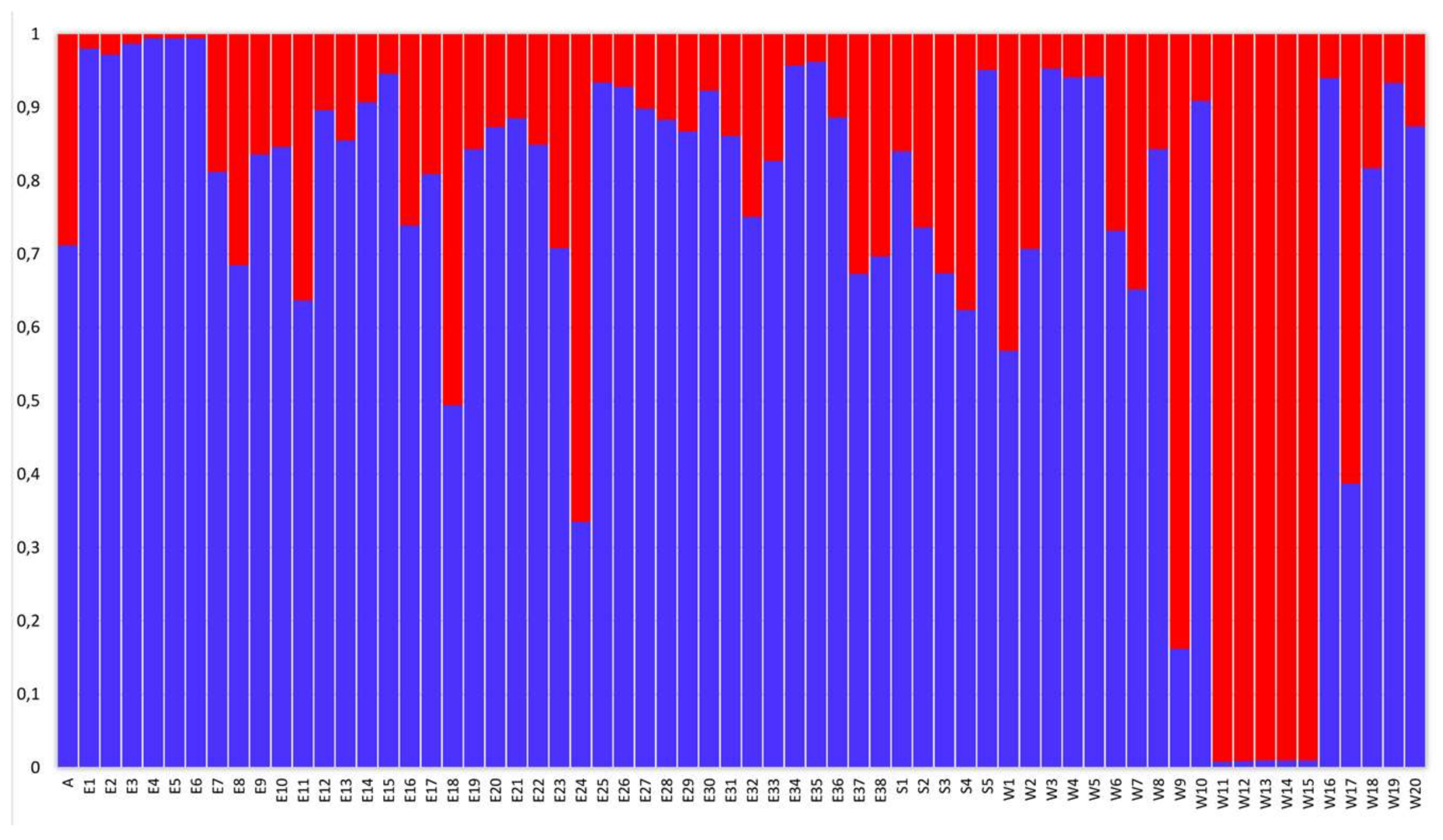
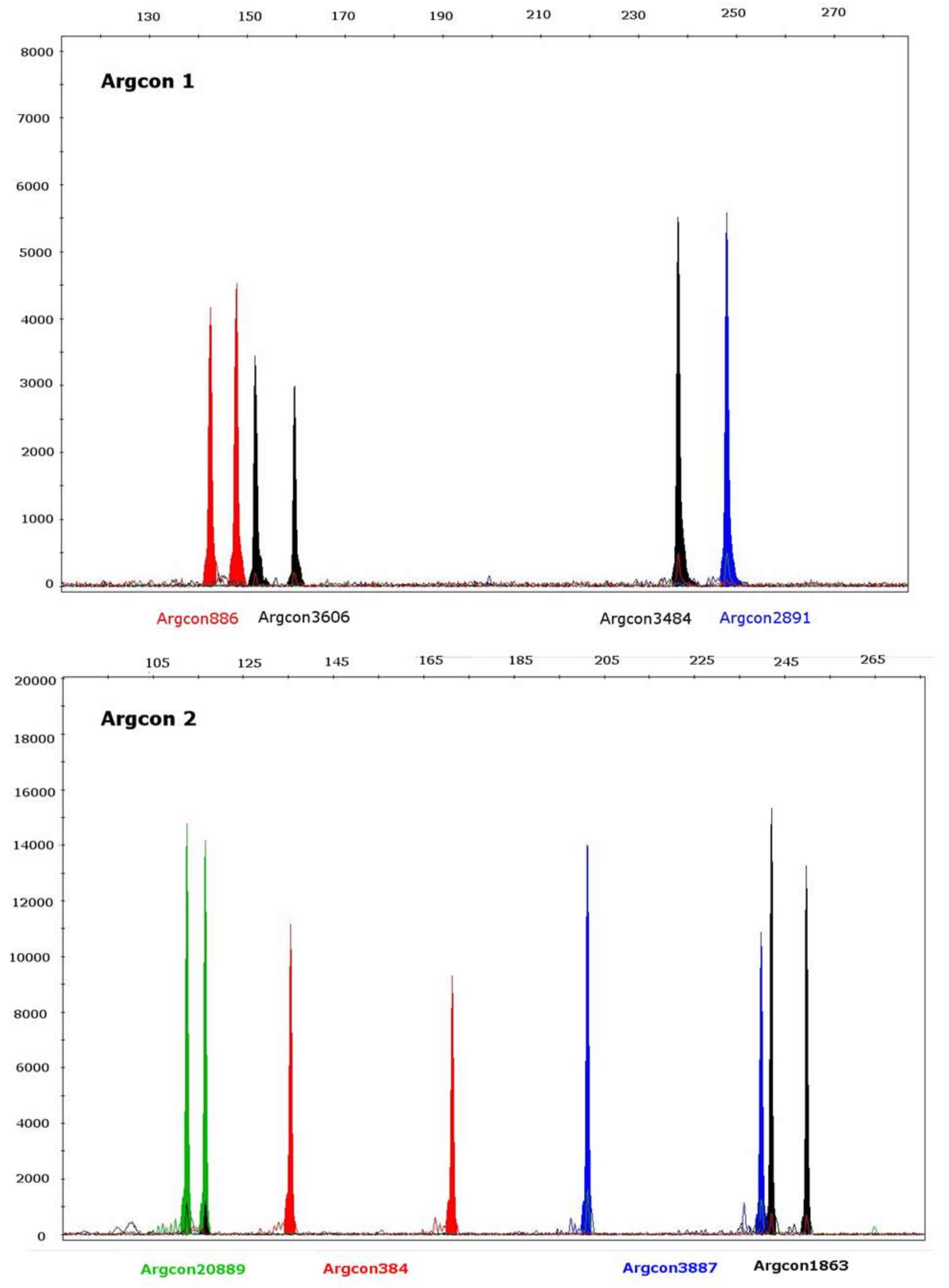
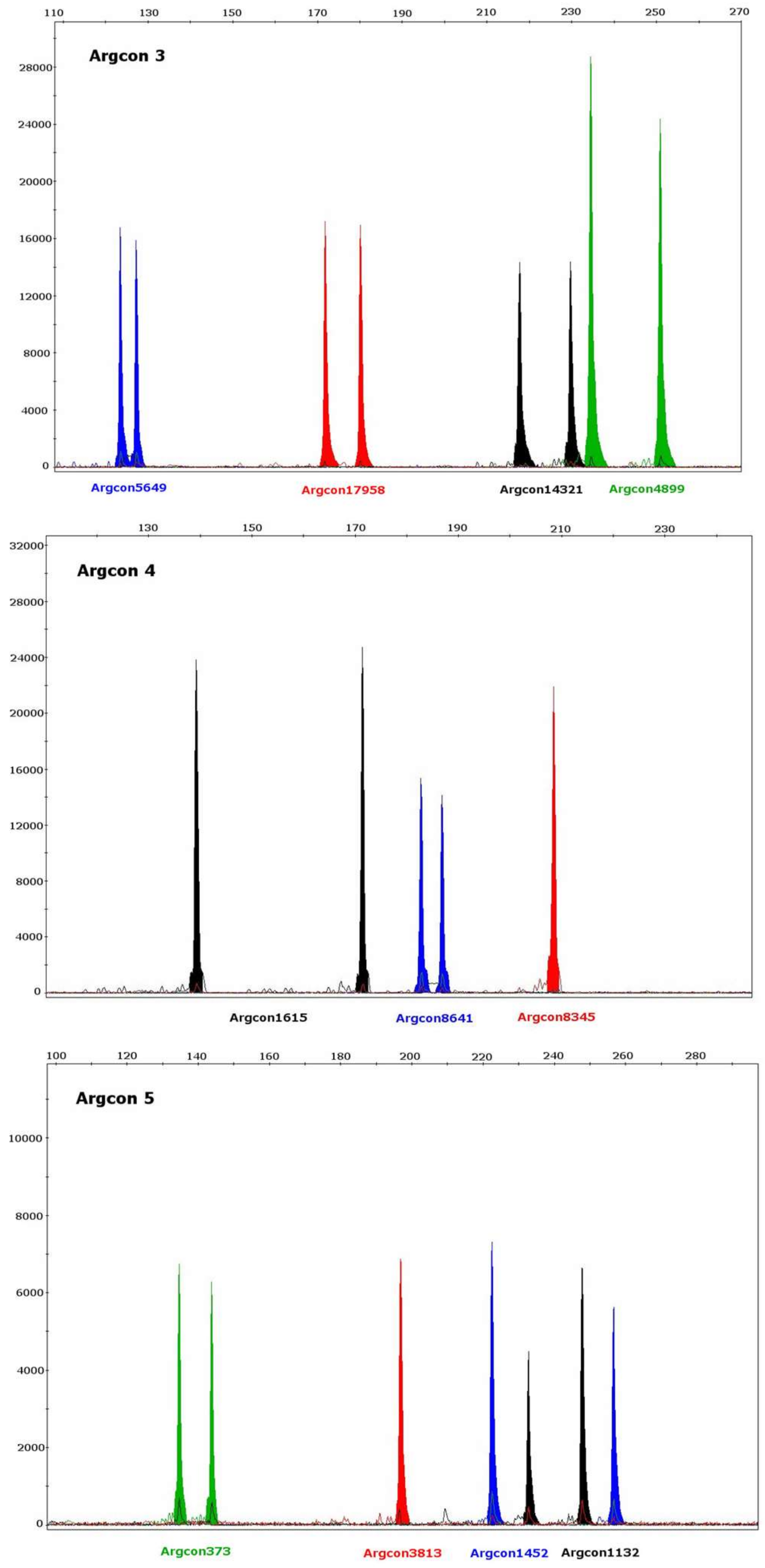
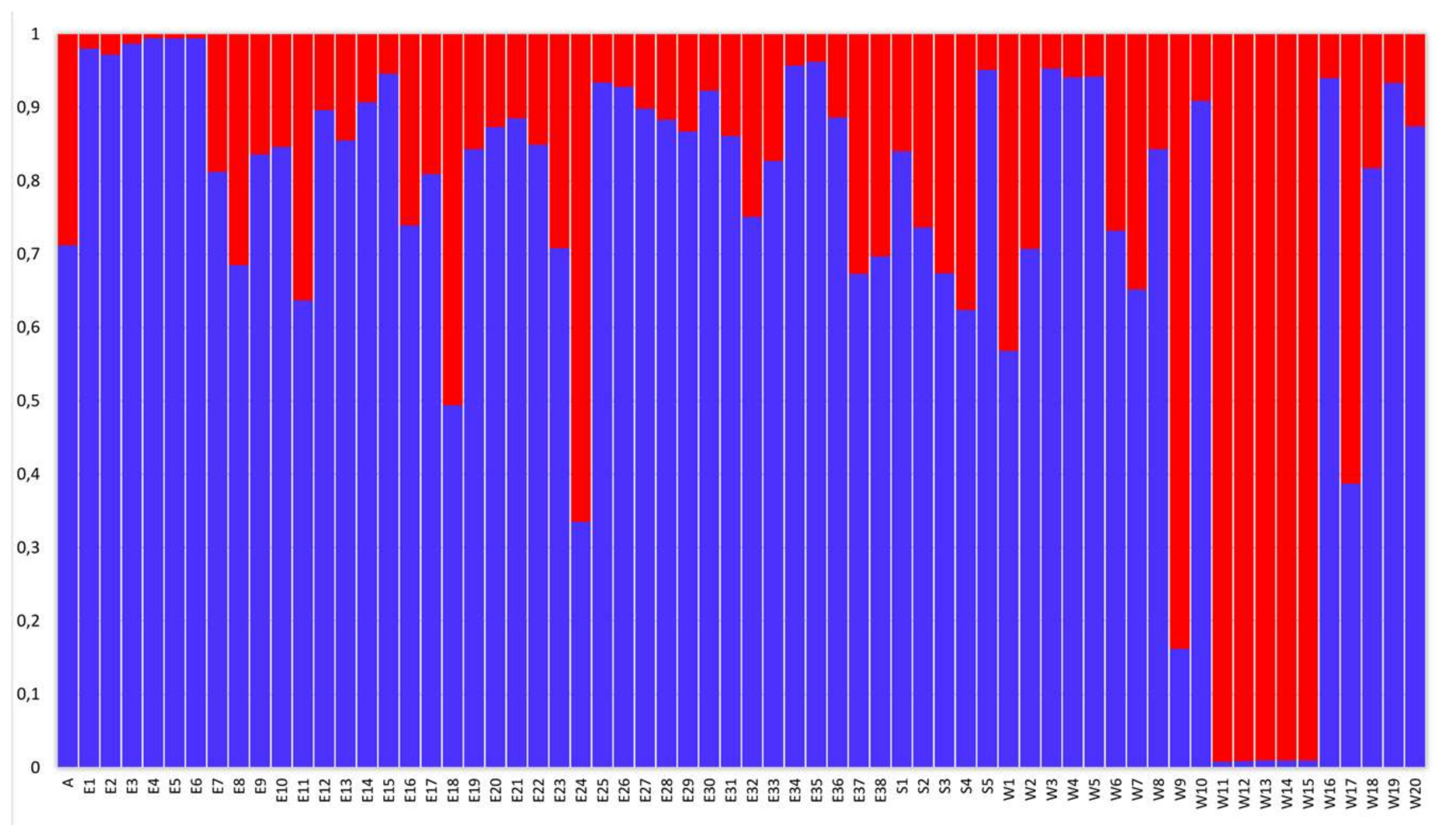
2. Results
3. Discussion
4. Materials and Methods
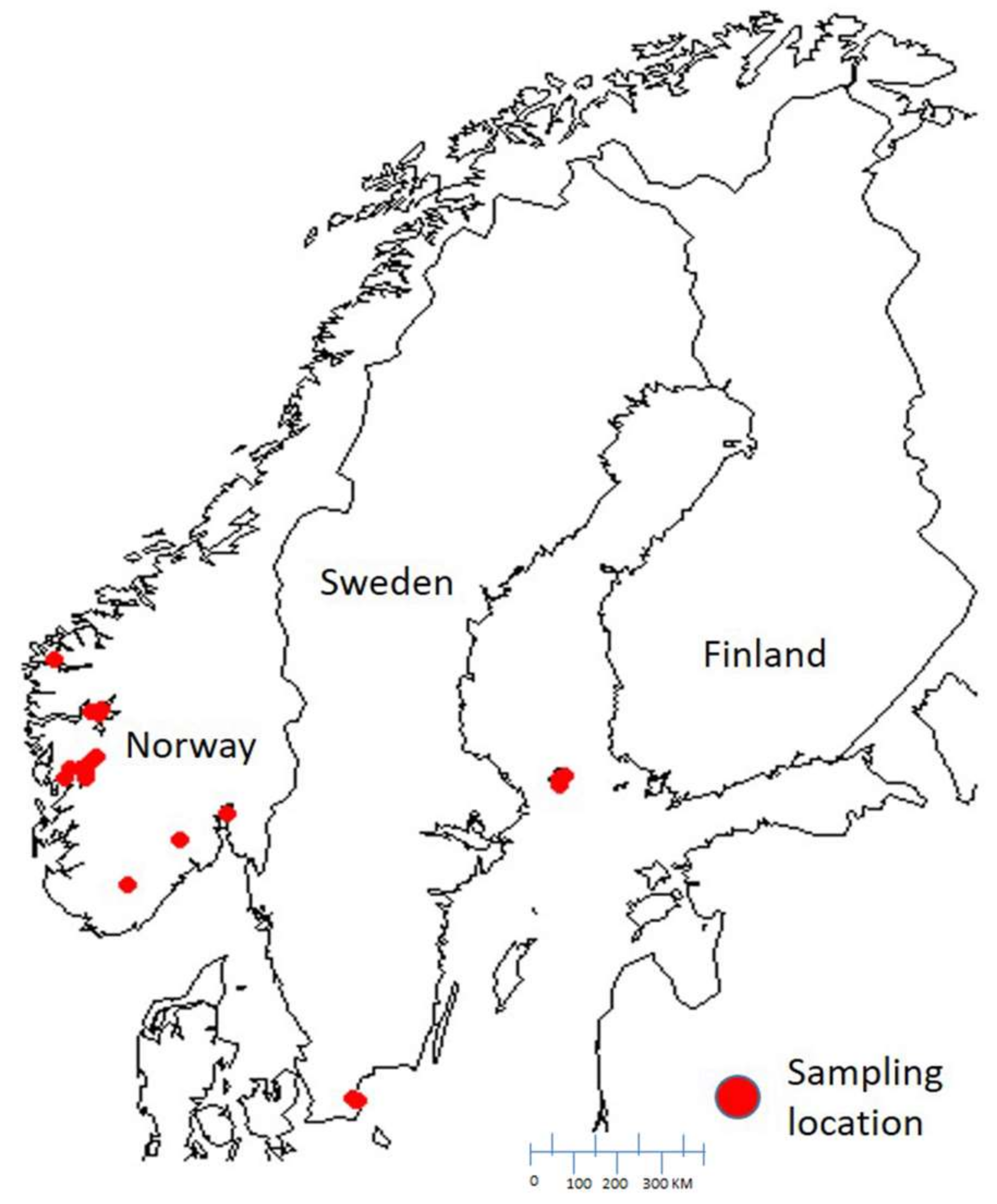
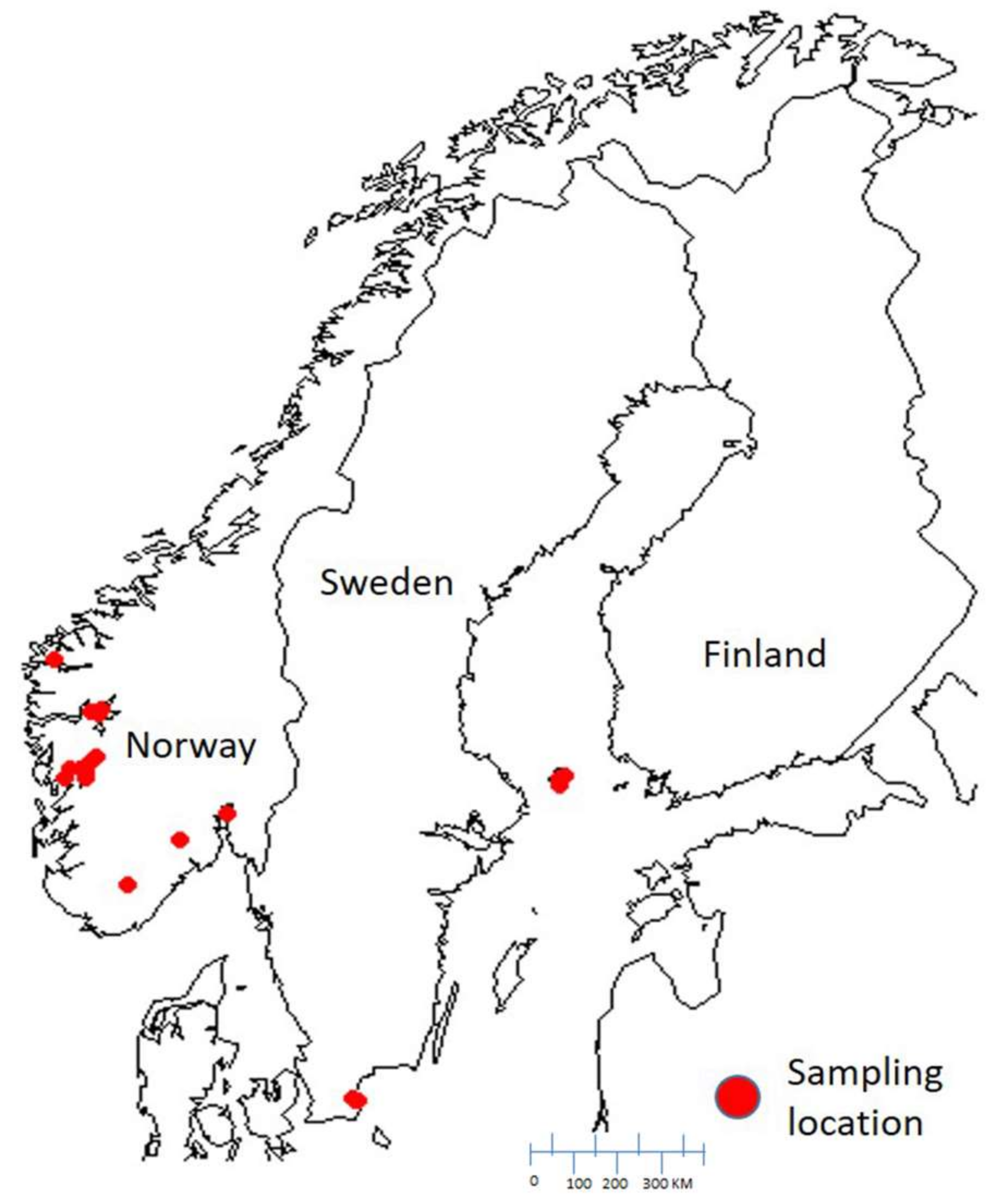
4.1. Sampling
4.2. Identification of STRs
4.3. PCR Amplification
4.4. Species Specificity
4.5. Data Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Okamoto, H. On the life-history of the apple fruit-miner, Argyresthia conjugella Zeller. Trans. Sappore Nat. Hist. Soc. 1917, 6, 213–219. [Google Scholar]
- Sharma, J.; Khajuria, D.; Dogra, G. Studies on the apple fruit moth, Argyresthia conjugella Zeller (Yponomeutidae: Lepidoptera): Identification, distribution and extent of damage in India. Int. J. Pest Manag. 1988, 34, 189–192. [Google Scholar]
- Fletcher, J. Report of the Entomologist and Botanist, 1896; Department of Agriculture, Central Experimental Farm: Ottawa, ON, Canada, 1897.
- Schöyen, W. Beretning om skadeinsekter og plantesygdomme i 1897. Kristiania 1898, 45, 8. [Google Scholar]
- Kobro, S.; Søreide, L.; Djønne, E.; Rafoss, T.; Jaastad, G.; Witzgall, P. Masting of rowan Sorbus aucuparia L. and consequences for the apple fruit moth Argyresthia conjugella Zeller. Popul. Ecol. 2003, 45, 25–30. [Google Scholar]
- Bengtsson, M.; Jaastad, G.; Knudsen, G.; Kobro, S.; Bäckman, A.C.; Pettersson, E.; Witzgall, P. Plant volatiles mediate attraction to host and non-host plant in apple fruit moth, Argyresthia conjugella. Entomol. Exp. Appl. 2006, 118, 77–85. [Google Scholar] [CrossRef]
- Knudsen, G.K.; Tasin, M. Spotting the invaders: A monitoring system based on plant volatiles to forecast apple fruit moth attacks in apple orchards. Basic Appl. Ecol. 2015, 16, 354–364. [Google Scholar] [CrossRef]
- Edland, A.; Meyer, B. Prognosar om angrep av rognebaermoll (Argyresthia conjugella Zeller) pa eple. Gartneryrket 1978, 2013, 563–564. [Google Scholar]
- Elameen, A.; Eiken, H.G.; Knudsen, G.K. Genetic diversity in apple fruit moth indicate different clusters in the two most important apple growing regions of Norway. Diversity 2016, 8, 10. [Google Scholar] [CrossRef]
- Diéguez-Uribeondo, J.; Fregeneda-Grandes, J.M.; Cerenius, L.; Pérez-Iniesta, E.; Aller-Gancedo, J.M.; Tellería, M.T.; Söderhäll, K.; Martín, M.P. Re-evaluation of the enigmatic species complex Saprolegnia diclina and Saprolegnia parasitica based on morphological, physiological and molecular data. Fungal Genet. Biol. 2007, 44, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Roux, O.; Gevrey, M.; Arvanitakis, L.; Gers, C.; Bordat, D.; Legal, L. ISSR-PCR: Tool for discrimination and genetic structure analysis of Plutella xylostella populations native to different geographical areas. Mol. Phylogenet. Evol. 2007, 43, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.B.; Batista, R.; Prado, P.I.; Brown, K.S.; Freitas, A.V. The importance of small scales to the fruit-feeding butterfly assemblages in a fragmented landscape. Biodivers. Conserv. 2012, 21, 811–827. [Google Scholar] [CrossRef]
- Tufto, J.; Lande, R.; Ringsby, T.H.; Engen, S.; Sæther, B.E.; Walla, T.R.; DeVries, P.J. Estimating brownian motion dispersal rate, longevity and population density from spatially explicit mark–recapture data on tropical butterflies. J. Anim. Ecol. 2012, 81, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Legal, L.; Dorado, O.; Machkour-M’Rabet, S.; Leberger, R.; Albre, J.; Mariano, N.A.; Gers, C. Ecological constraints and distribution of the primitive and enigmatic endemic Mexican butterfly Baronia brevicornis (Lepidoptera: Papilionidae). Can. Entomol. 2015, 147, 71–88. [Google Scholar] [CrossRef]
- Laffin, R.; Langor, D.; Sperling, F. Population structure and gene flow in the white pine weevil, Pissodes strobi (Coleoptera: Curculionidae). Ann. Entomol. Soc. Am. 2004, 97, 949–956. [Google Scholar] [CrossRef]
- Gómez, A. Molecular ecology of rotifers: From population differentiation to speciation. Hydrobiologia 2005, 546, 83–99. [Google Scholar] [CrossRef]
- Margaritopoulos, J.T.; Voudouris, C.C.; Olivares, J.; Sauphanor, B.; Mamuris, Z.; Tsitsipis, J.A.; Franck, P. Dispersal ability in codling moth: Mark–release–recapture experiments and kinship analysis. Agric. For. Entomol. 2012, 14, 399–407. [Google Scholar] [CrossRef]
- Voudouris, C.C.; Franck, P.; Olivares, J.; Sauphanor, B.; Mamuris, Z.; Tsitsipis, J.; Margaritopoulos, J. Comparing the genetic structure of codling moth Cydia pomonella (L.) from Greece and France: Long distance gene-flow in a sedentary pest species. Bull. Entomol. Res. 2012, 102, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Jiang, G.-F.; Liu, Y.-X.; He, Q.-X.; Blanchard, B. Population explosion in the yellow-spined bamboo locust Ceracris kiangsu and inferences for the impact of human activity. PLoS ONE 2014, 9, e89873. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, F.; Coutinho, R.; Kraenkel, R. Spatial dynamics of a population with stage-dependent diffusion. Commun. Nonlinear Sci. Numer. Simul. 2015, 22, 605–610. [Google Scholar] [CrossRef]
- Kaňuch, P.; Berggren, Å.; Cassel-Lundhagen, A. Genetic diversity of a successful colonizer: Isolated populations of Metrioptera roeselii regain variation at an unusually rapid rate. Ecol. Evol. 2014, 4, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-R.; Szmidt, A.E. Molecular markers in population genetics of forest trees. Scand. J. For. Res. 2001, 16, 199–220. [Google Scholar] [CrossRef]
- Squirrell, J.; Hollingsworth, P.; Woodhead, M.; Russell, J.; Lowe, A.; Gibby, M.; Powell, W. How much effort is required to isolate nuclear microsatellites from plants? Mol. Ecol. 2003, 12, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Willems, T.; Gymrek, M.; Highnam, G.; Mittelman, D.; Erlich, Y.; Consortium, G.P. The landscape of human STR variation. Genome Res. 2014, 24, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Linacre, A.; Gusmao, L.; Hecht, W.; Hellmann, A.; Mayr, W.; Parson, W.; Prinz, M.; Schneider, P.; Morling, N. ISFG: Recommendations regarding the use of non-human (animal) DNA in forensic genetic investigations. Forensic Sci. Int. Genet. 2011, 5, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Meglecz, E.; Petenian, F.; Danchin, E.; D’Acier, A.C.; Rasplus, J.Y.; Faure, E. High similarity between flanking regions of different microsatellites detected within each of two species of Lepidoptera: Parnassius apollo and Euphydryas aurinia. Mol. Ecol. 2004, 13, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-X. Lepidopteran microsatellite DNA: Redundant but promising. Trends Ecol. Evol. 2004, 19, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.H.; de Bruijn, I.; Haas, B.J.; Belmonte, R.; Löbach, L.; Christie, J.; van den Ackerveken, G.; Bottin, A.; Bulone, V.; Díaz-Moreno, S.M. Distinctive expansion of potential virulence genes in the genome of the Oomycete fish pathogen Saprolegnia parasitica. PLoS Genet. 2013, 9, e1003272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Reineke, A.; Assaf, H.; Kulanek, D.; Mori, N.; Pozzebon, A.; Duso, C. A novel set of microsatellite markers for the European grapevine moth Lobesia botrana isolated using next-generation sequencing and their utility for genetic characterization of populations from Europe and the Middle East. Bull. Entomol. Res. 2015, 105, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, M.J.; Carpten, J.D.; Smith, J.R. Modulation of non-templated nucleotide addition by Taq DNA polymerase: Primer modifications that facilitate genotyping. Biotechniques 1996, 20, 1004–1010. [Google Scholar] [PubMed]
- Li, Y.; Cooke, D.E.; Jacobsen, E.; van der Lee, T. Efficient multiplex simple sequence repeat genotyping of the Oomycete plant pathogen Phytophthora infestans. J. Microbiol. Methods 2013, 92, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.-K.; Sun, E.-T.; Jia, C.-N.; Kong, D.-D.; Jiang, Y.-X. Genetic diversity and differentiation of Lepidoglyphus destructor (Acari: Glycyphagidae) inferred from Inter-Simple Sequence Repeat (ISSR) fingerprinting. Syst. Appl. Acarol. 2014, 19, 491–498. [Google Scholar] [CrossRef]
- Sauné, L.; Abella, F.; Kerdelhué, C. Isolation, characterization and PCR multiplexing of 17 microsatellite loci in the pine processionary moth Thaumetopoea pityocampa (Lepidoptera, Notodontidae). Conserv. Genet. Resour. 2015, 7, 755–757. [Google Scholar] [CrossRef]
- Aarnes, S.G.; Fløystad, I.; Schregel, J.; Vindstad, O.P.L.; Jepsen, J.U.; Eiken, H.G.; Ims, R.A.; Hagen, S.B. Identification and evaluation of 21 novel microsatellite markers from the autumnal moth (Epirrita autumnata) (Lepidoptera: Geometridae). Int. J. Mol. Sci. 2015, 16, 22541–22554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, N.; Corrêa, A.; Alves-Pereira, A.; Campos, J.; Zucchi, M.; Omoto, C. Cross-species amplification and polymorphism of microsatellite loci in Helicoverpa armigera and Helicoverpa zea (Lepidoptera: Noctuidae) in Brazilian cropping systems. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wen, J.; Wei, S.; Liu, J.; Yang, F.; Chen, M. Characterization of novel microsatellite markers for Hyphantria cunea and implications for other Lepidoptera. Bull. Entomol. Res. 2015, 105, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Z.; Cao, L.-J.; Zhu, J.-Y.; Wei, S.-J. Development and characterization of novel microsatellite markers for the peach fruit moth Carposina sasakii (Lepidoptera: Carposinidae) using next-generation sequencing. Int. J. Mol. Sci. 2016, 17, 362. [Google Scholar] [CrossRef] [PubMed]
- Endersby, N.; McKechnie, S.; Vogel, H.; Gahan, L.; Baxter, S.; Ridland, P.; Weeks, A. Microsatellites isolated from diamondback moth, Plutella xylostella (L.), for studies of dispersal in Australian populations. Mol. Ecol. Resour. 2005, 5, 51–53. [Google Scholar] [CrossRef]
- Ishiguro, N.; Tsuchida, K. Polymorphic microsatellite loci for the rice stem borer, Chilo suppressalis (walker) (Lepidoptera: Crambidae). Appl. Entomol. Zool. 2006, 41, 565–568. [Google Scholar] [CrossRef]
- Delamaire, S.; Esselink, G.D.; Samiei, L.; Courtin, C.; Magnoux, E.; Rousselet, J.; Smulders, M.J. Isolation and characterization of six microsatellite loci in the larch budmoth Zeiraphera diniana (Lepidoptera: Tortricidae). Eur. J. Entomol. 2010, 107, 267. [Google Scholar] [CrossRef]
- Petenian, F.; Meglécz, E.; Genson, G.; Rasplus, J.Y.; Faure, E. Isolation and characterization of polymorphic microsatellites in Parnassius apollo and Euphydryas aurinia (Lepidoptera). Mol. Ecol. Resour. 2005, 5, 243–245. [Google Scholar] [CrossRef]
- Berman, M.; Austin, C.M.; Miller, A.D. Characterization of the complete mitochondrial genome and 13 microsatellite loci through next-generation sequencing for the new Caledonian spider-ant Leptomyrmex pallens. Mol. Biol. Rep. 2014, 41, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Drag, L.; Kosnar, J.; Cizek, L. Development and characterization of ten polymorphic microsatellite loci for the great Capricorn beetle (Cerambyx cerdo) (Coleoptera: Cerambycidae). Conserv. Genet. Resour. 2013, 5, 907–909. [Google Scholar] [CrossRef]
- Elisa Posso, C.; González, R.; Cárdenas, H.; Gallego, G.; Duque, M.C.; Suarez, M.F. Random amplified polymorphic DNA analysis of Anopheles nuneztovari (Diptera: Culicidae) from western and Northeastern Colombia. Memórias do Instituto Oswaldo Cruz 2003, 95, 469–476. [Google Scholar] [CrossRef]
- Ciosi, M.; Masiga, D.K.; Turner, C.M.; Valenzuela, J.G. Laboratory colonisation and genetic bottlenecks in the tsetse fly Glossina pallidipes. PLoS Negl. Trop. Dis. 2014, 8, e2697. [Google Scholar] [CrossRef] [PubMed]
- Charman, T.G.; Sears, J.; Green, R.E.; Bourke, A.F. Conservation genetics, foraging distance and nest densityof the scarce great yellow bumblebee (Bombus distinguendus). Mol. Ecol. 2010, 19, 2661–2674. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, A. Isolation and characterization of five microsatellite loci in the Glanville fritillary butterfly (Melitaea cinxia). Mol. Ecol. Resour. 2006, 6, 163–164. [Google Scholar] [CrossRef]
- Franck, P.; Reyes, M.; Olivares, J.; Sauphanor, B. Genetic architecture in codling moth populations: Comparison between microsatellite and insecticide resistance markers. Mol. Ecol. 2007, 16, 3554–3564. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, S.A.; Thistlewood, H.; Keyghobadi, N. Population genetic structure of the western cherry fruit fly Rhagoletis indifferens (Diptera: Tephritidae) in British Columbia, Canada. Agric. For. Entomol. 2014, 16, 33–44. [Google Scholar] [CrossRef]
- Fuentes-Contreras, E.; Espinoza, J.L.; Lavandero, B.; Ramírez, C.C. Population genetic structure of codling moth (Lepidoptera: Tortricidae) from apple orchards in central Chile. J. Econ. Entomol. 2008, 101, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Pichon, A.; Arvanitakis, L.; Roux, O.; Kirk, A.A.; Alauzet, C.; Bordat, D.; Legal, L. Genetic differentiation among various populations of the diamondback moth, Plutella xylostella Lepidoptera Yponomeutidae. Bull. Entomol. Res. 2006, 96, 137–144. [Google Scholar] [CrossRef] [PubMed]
- De La Poza, M.; Farinós, G.P.; Beroiz, B.; Ortego, F.; Hernández-Crespo, P.; Castañera, P. Genetic structure of Sesamia nonagrioides (lefebvre) populations in the Mediterranean area. Environ. Entomol. 2008, 37, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Timm, A.E.; Geertsema, H.; Warnich, L. Gene flow among Cydia pomonella (Lepidoptera: Tortricidae) geographic and host populations in South Africa. J. Econ. Entomol. 2006, 99, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Thaler, R.; Brandstätter, A.; Meraner, A.; Chabicovski, M.; Parson, W.; Zelger, R.; Dalla Via, J.; Dallinger, R. Molecular phylogeny and population structure of the codling moth (Cydia pomonella) in Central Europe: Li. AFLP analysis reflects human-aided local adaptation of a global pest species. Mol. Phylogenet. Evol. 2008, 48, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, H.; Siegfried, B.D. Genetic differentiation of western corn rootworm populations (Coleoptera: Chrysomelidae) relative to insecticide resistance. Ann. Entomol. Soc. Am. 2012, 105, 232–240. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatic 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. In Computer Science and Biology, Proceedings of the German Conference on Bioinformatics, Hannover, Germany, 4–6 October 1999; Department of Bioinformatics: Braunschweig, Germany, 1999; Volume 99, pp. 45–56. [Google Scholar]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. Genepop (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.; Yang, R.; Boyle, T.; Ye, Z.; Xiyan, J. Popgene32, Microsoft Windows-Based Freeware for Population Genetic Analysis, Version 1.32; Molecular Biology and Biotechnology Centre, University of Alberta: Edmonton, AB, Canada, 2000. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [PubMed]
- Schneider, S.; Roessli, D.; Excoffier, L. Arlequin: A software for population genetics data analysis. Genetic and Biometry Laboratory. Department of Anthropology, University of Geneva, Switzerland. User Manu. Ver 2000, 2, 2496–2497. [Google Scholar]
- Whitlock, M.C.; McCauley, D.E. Indirect measures of gene flow and migration: Fst ≠ 1/(4nm + 1). Heredity 1999, 82, 117–125. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Limited sample material may be available from the authors upon request. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Size Range (bp) Illumina MiSeq n = 5 | Size Range (bp) ABI3730 Capillary Electrophoresis n = 64 | Repeat Motif (Illumina MiSeq) * |
|---|---|---|---|
| Argcon373 | 97–153 | 98–246 | (TTGT)22 |
| Argcon384 | 121–196 | 129–207 | (AACA)10 |
| Argcon886 | 119–137 | 129–201 | (TTTC)7 |
| Argcon1132 | 205–247 | 210–268 | (AGAA)7 |
| Argcon1452 | 214–247 | 208–292 | (TCTT)9 |
| Argcon1615 | 120–164 | 129–169 | (TTTG)10 |
| Argcon1863 | 231–246 | 235–249 | (ACAT)7 |
| Argcon2891 | 239–251 | 237–258 | (TATC)7 |
| Argcon3484 | 230–251 | 230–258 | (TTGT)8 |
| Argcon3606 | 143–160 | 151–183 | (ACAT)9 |
| Argcon3813 | 179–199 | 188–216 | (TTCT)7 |
| Argcon3887 | 185–261 | 188–340 | (AAGA)9 |
| Argcon4899 | 217–273 | 214–386 | (TTCT)9 |
| Argcon5649 | 105–140 | 111–151 | (AAAC)7 |
| Argcon8345 | 196–256 | 204–260 | (TTTC)8 |
| Argcon8461 | 120–242 | 122–306 | (TTTC)16 |
| Argcon14321 | 199–228 | 205–233 | (TATC)8 |
| Argcon17958 | 154–192 | 164–284 | (GTTT)9 |
| Argcon20889 | 97–143 | 104–156 | (TTTC)12 |
| Multiplex Panel | Locus | Primer Sequences (5′-3′) a | PCR Concentration, Dye | Gene-Bank Accession No. |
|---|---|---|---|---|
| I | Argcon_3606 | F: AGTGAACCTACTGAGCGTCC | 0.05 µM, NED | MF156559 |
| R: GTTTCTTCCCTTATGGGAAAAGGCGTG | ||||
| Argcon_3484 | F: GGGCAGCTGTTTCCCAATTC | 0.10 µM, NED | MF156558 | |
| R: GTTTCTTCTCCTCGTGCATTTTTGGG | ||||
| Argcon_2891 | F: AGAACTGGGCCTCACGATAC | 0.10 µM, FAM | MF156557 | |
| R: GTTTCTTGTTATCGGCATTCCACAAGGG | ||||
| Argcon_886 | F: ACCCGACCTGAACATATCCG | 0.10 µM, PET | MF156552 | |
| R: GTTTCTTCCATCGTTGGCACTTACGAG | ||||
| II | Argcon_20889 | F: TGTGTCTAGTTTCTTGTATTTGTTGC | 0.10 µM, VIC | MF156568 |
| R: GTTTCTTTAGTGTGGGCTAAGGGATGC | ||||
| Argcon_384 | F: CATGTCTCCTCTTTGCAGCG | 0.15 µM, PET | MF156551 | |
| R: GTTTCTTGTAAGGGAGTGTCGTGTTGC | ||||
| Argcon_1863 | F: CGCCCCGGATTCTCAACTAC | 0.20 µM, NED | MF156556 | |
| R: GTTTCTTTCACCCCTCTCTGTATTCGTC | ||||
| Argcon_3887 | F: CATTGTTGACAGCTCGGCAC | 0.25 µM, FAM | MF156561 | |
| R: GTTTCTTGTGAGCCTTTCGGATTTGGG | ||||
| III | Argcon_14321 | F: TGTTTTGTTCAATCTGTATTACTTGTC | 0.15 µM, NED | MF156566 |
| R: GTTTCTTACAGGGGGACAATCCAATCTAC | ||||
| Argcon_5649 | F: AGCCCTACGACTCCATCAAC | 0.10 µM, FAM | MF156563 | |
| R: GTTTCTTGCTAAACTATCCGTCGGCAC | ||||
| Argcon_17958 | F: GCTCAGTGTATCAGGTACGAG | 0.20 µM, PET | MF156567 | |
| R: GTTTCTTCGCTGTTCTACATGGAGCTG | ||||
| Argcon_4899 | F: TCATGGTTTGGCATGTCGAG | 0.10 µM, VIC | MF156562 | |
| R: GTTTCTTAGTTCAAATCCGTCCTAAAAGC | ||||
| IV | Argcon_1615 | F: CCCCTTATTTGAGCAGTTGAGC | 0.05 µM, NED | MF156555 |
| R: GTTTCTTGCAACATTATTTCGTCCGCAG | ||||
| Argcon_8345 | F: GCTCAAACGGTTGTCCCTAC | 0.20 µM, PET | MF156564 | |
| R: GTTTCTTGTATGCTACGGTTACAGGGC | ||||
| Argcon_8461 | F: ACTTTACTGGCCTAGGTGCG | 0.15 µM, FAM | MF156565 | |
| R: GTTTCTTCCAGGTGAAACATCGTGAGG | ||||
| V | Argcon_373 | F: AGTACCTCGTCGATACGCAC | 0.05 µM, VIC | MF156550 |
| R: GTTTCTTAGGGGTGTCAGGATGTGATG | ||||
| Argcon_3813 | F: GCTGTCGTAAACCCTTCCAC | 0.10 µM, PET | MF156560 | |
| R: GTTTCTTGGCCCCATTGGTTCCATAAC | ||||
| Argcon_1452 | F: AATAACAGTGGCACACCACG | 0.15 µM, FAM | MF156554 | |
| R: GTTTCTTTATGCGGATTCCAAACGCAG | ||||
| Argcon_1132 | F: TGTCTATGGAAGCCCCGATG | 0.15 µM, NED | MF156553 | |
| R: GTTTCTTGTTCCAAGATTTGCCGCTCC |
| Locus | NA | NG | HO | HE | Nei | FIS | HWE p Values a |
|---|---|---|---|---|---|---|---|
| Argcon2891 | 10 | 63 | 0.3333 | 0.6060 | 0.6012 | 0.4455 | 0.0087 ** |
| Argcon3606 | 7 | 64 | 0.6000 | 0.6136 | 0.6089 | 0.0146 | 0.9709 |
| Arg3484 | 11 | 64 | 0.3750 | 0.7826 | 0.7765 | 0.5171 | 0.0016 ** |
| Arg886 | 6 | 64 | 0.5156 | 0.4801 | 0.4763 | −0.0825 | 0.0093 ** |
| Arg3887 | 26 | 59 | 0.4464 | 0.9484 | 0.9399 | 0.5250 | 0.0007 ** |
| Arg20889 | 8 | 64 | 0.6094 | 0.6380 | 0.6331 | 0.0374 | 0.0004 ** |
| Arg1863 | 7 | 55 | 0.1455 | 0.4816 | 0.4772 | 0.6952 | 0.0001 ** |
| Arg384 | 16 | 62 | 0.7097 | 0.7578 | 0.7517 | 0.0559 | 0.0002 ** |
| Arg5649 | 11 | 64 | 0.4531 | 0.8108 | 0.8044 | 0.4367 | 0.0753 * |
| Argcon4899 | 20 | 59 | 0.5714 | 0.8465 | 0.8390 | 0.3189 | 0.0001 ** |
| Argcon14321 | 8 | 64 | 0.6094 | 0.7464 | 0.7406 | 0.1772 | 0.1467 |
| Argcon17958 | 17 | 64 | 0.7231 | 0.7659 | 0.7600 | 0.0486 | 0.0035 ** |
| Argcon8461 | 24 | 58 | 0.9074 | 0.9084 | 0.9000 | −0.0082 | 0.2887 |
| Argcon1615 | 14 | 57 | 0.5789 | 0.8871 | 0.8793 | 0.3416 | 0.0076 ** |
| Argcon8345 | 18 | 62 | 0.5484 | 0.8581 | 0.8512 | 0.3557 | 0.0012 ** |
| Argcon1452 | 16 | 38 | 0.5833 | 0.9221 | 0.9093 | 0.3585 | 0.0127 * |
| Argcon373 | 21 | 62 | 0.8548 | 0.9340 | 0.9265 | 0.0774 | 0.0016 ** |
| Argcon1132 | 11 | 62 | 0.6129 | 0.6808 | 0.6753 | 0.0924 | 0.0187 * |
| Argcon3813 | 8 | 63 | 0.3387 | 0.5507 | 0.5463 | 0.3800 | 0.9939 |
| Mean | 13.6 | 60.2 | 0.5535 | 0.7484 | 0.7419 | 0.2615 | |
| St, Dev | 0.1824 | 0.1517 | 0.15 | 0.0221 |
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variation |
|---|---|---|---|---|
| Among regions | 1 | 43.944 | 0.36434 | 2.17 |
| Among populations with regions | 4 | 48.2872 | 0.78612 | 2.24 |
| Within regions | 57 | 949.6497 | 15.47801 | 95.60 |
| Total | 62 | 997.937 | 16.26421 |
| Regions | East Norway | Sweden | West Norway |
|---|---|---|---|
| East Norway | 0.00000 | ||
| Sweden | 0.01577 | 0.00000 | |
| West Norway | 0.02522 | 0.01373 | 0.0000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elameen, A.; Eiken, H.G.; Fløystad, I.; Knudsen, G.; Hagen, S.B. Monitoring of the Apple Fruit Moth: Detection of Genetic Variation and Structure Applying a Novel Multiplex Set of 19 STR Markers. Molecules 2018, 23, 850. https://doi.org/10.3390/molecules23040850
Elameen A, Eiken HG, Fløystad I, Knudsen G, Hagen SB. Monitoring of the Apple Fruit Moth: Detection of Genetic Variation and Structure Applying a Novel Multiplex Set of 19 STR Markers. Molecules. 2018; 23(4):850. https://doi.org/10.3390/molecules23040850
Chicago/Turabian StyleElameen, Abdelhameed, Hans Geir Eiken, Ida Fløystad, Geir Knudsen, and Snorre B. Hagen. 2018. "Monitoring of the Apple Fruit Moth: Detection of Genetic Variation and Structure Applying a Novel Multiplex Set of 19 STR Markers" Molecules 23, no. 4: 850. https://doi.org/10.3390/molecules23040850
APA StyleElameen, A., Eiken, H. G., Fløystad, I., Knudsen, G., & Hagen, S. B. (2018). Monitoring of the Apple Fruit Moth: Detection of Genetic Variation and Structure Applying a Novel Multiplex Set of 19 STR Markers. Molecules, 23(4), 850. https://doi.org/10.3390/molecules23040850