
VpStyA1/VpStyA2B of Variovorax paradoxus EPS: An Aryl Alkyl Sulfoxidase Rather than a Styrene Epoxidizing Monooxygenase

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
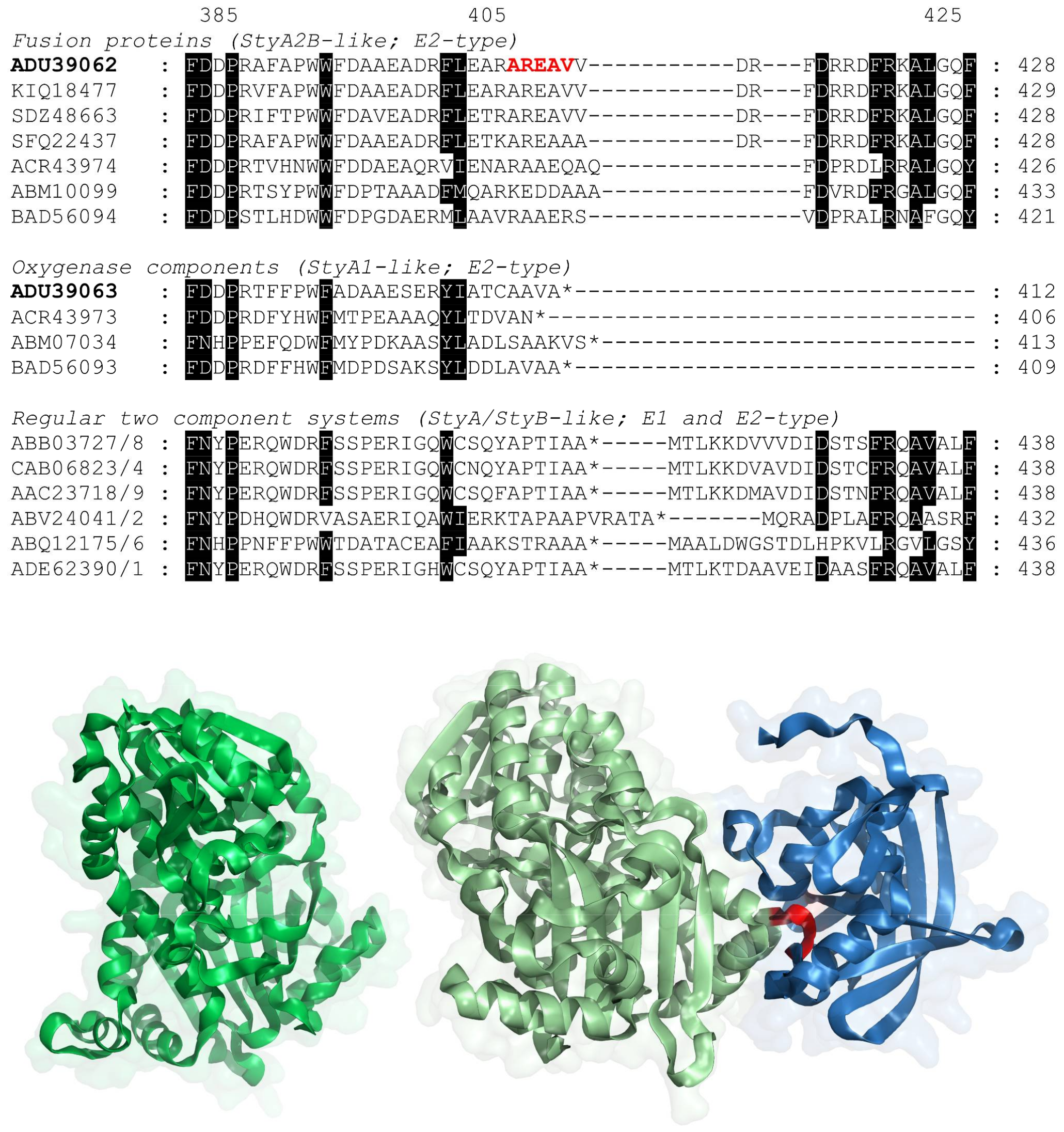
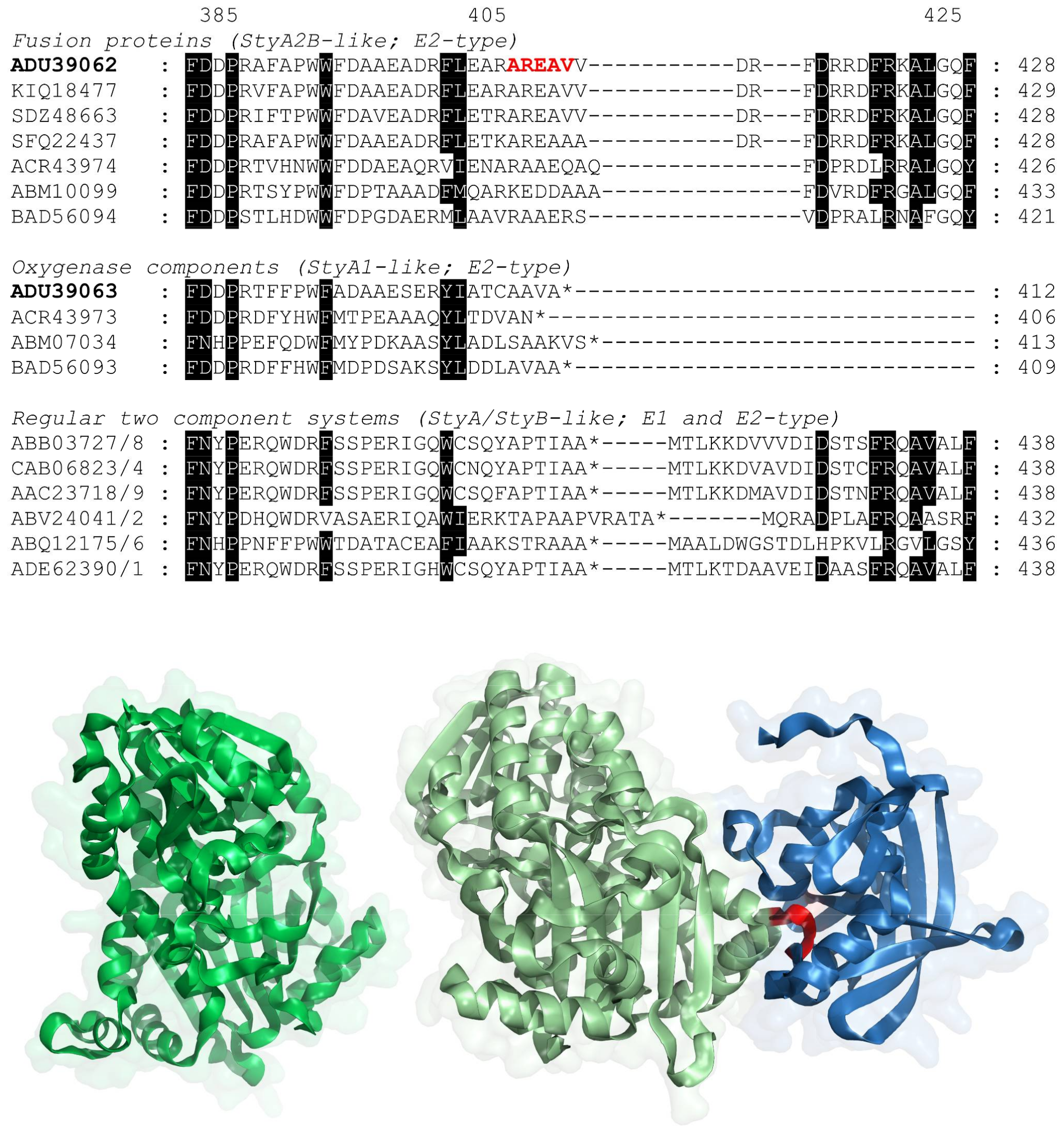
2.1. Evolution of VpStyA1/VpStyA2B from Strain EPS
2.2. Molecular Genetic Work and Enzyme Production
2.3. Reductase Activity of VpStyA2B and VpStyA2B-Mutants
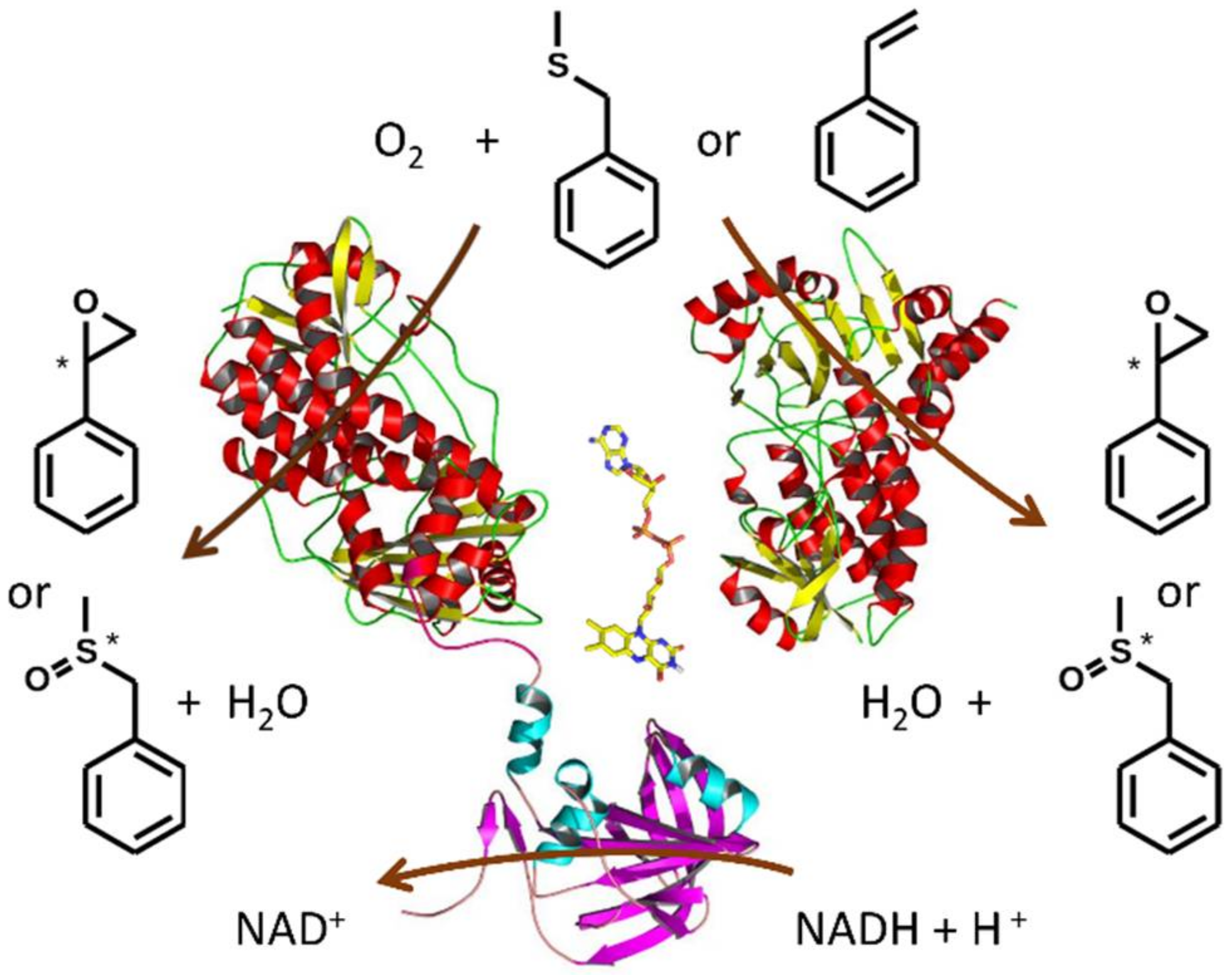
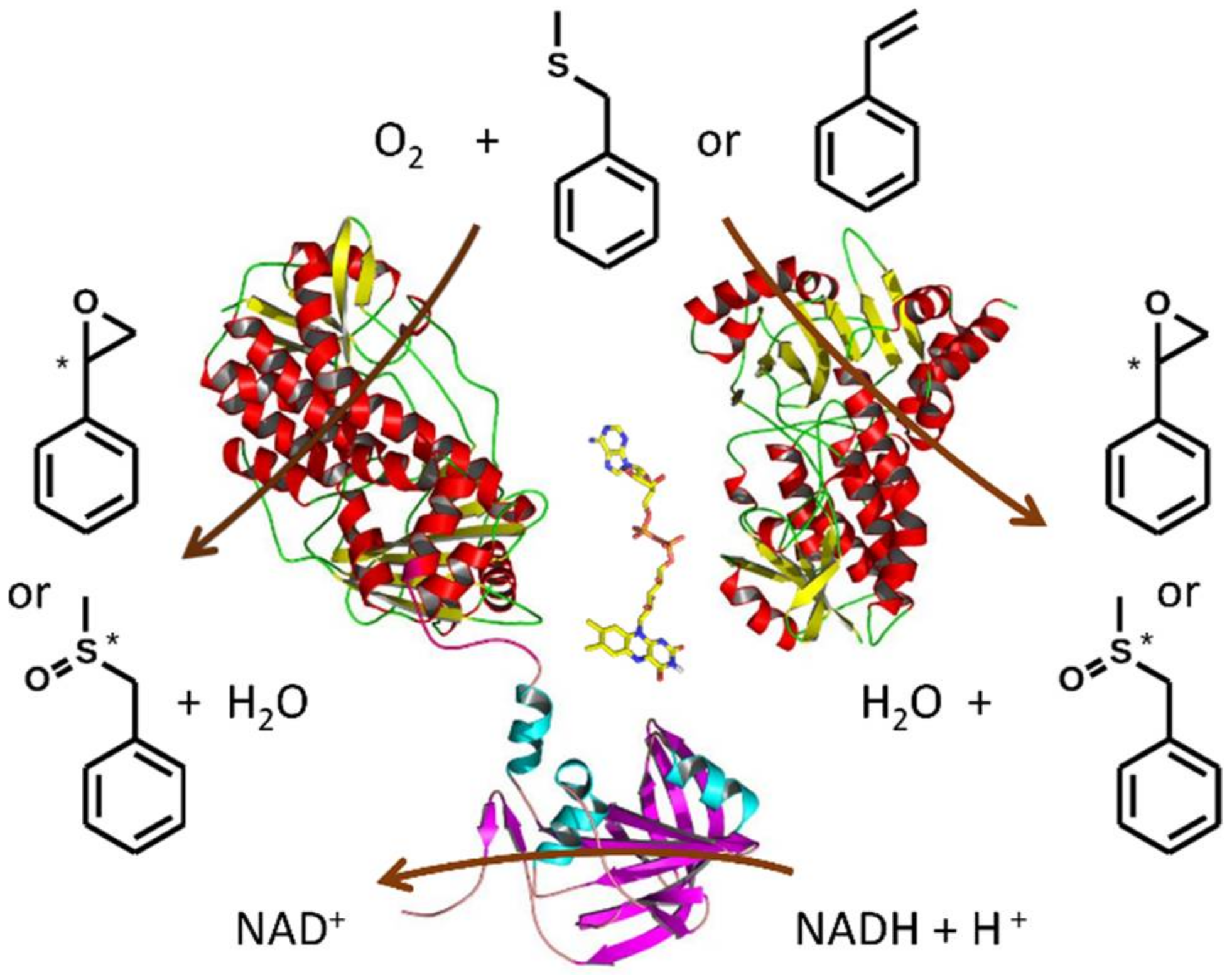
2.4. Monooxygenase Activity of VpStyA1, VpStyA2B and VpStyA2B-Mutants
2.5. Biotransformation of Sulfides
3. Materials and Methods
3.1. Synthesis of Sulfoxides
3.2. Nucleotides, Sequence Analysis and Molecular Modelling
3.3. Bacterial Strains and Cultivation
3.4. Molecular Genetic Work
3.5. Protein Purification and Quantification
3.6. Enzyme Assays and Product Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huijbers, M.M.E.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Montersino, S.; Tischler, D.; Gassner, G.T.; van Berkel, W.J.H. Catalytic and structural features of flavoprotein hydroxylases and epoxidases. Adv. Synth. Catal. 2011, 353, 2301–2319. [Google Scholar] [CrossRef]
- Van Berkel, W.J.H.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 124, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Kaschabek, S.R. Microbial Styrene Degradation: From Basics to Biotechnology. In Microbial Degradation of Xenobiotics; Singh, S.N., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 67–99. ISBN 978-3-642-23789-8. [Google Scholar]
- Tischler, D.; Eulberg, D.; Lakner, S.; Kaschabek, S.R.; van Berkel, W.J.H.; Schlömann, M. Identification of a novel self-sufficient styrene monooxygenase from Rhodococcus opacus 1CP. J. Bacteriol. 2009, 191, 4996–5009. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Kermer, R.; Gröning, J.A.D.; Kaschabek, S.R.; van Berkel, W.J.H.; Schlömann, M. StyA1 and StyA2B from Rhodococcus opacus 1CP: A multifunctional styrene monooxygenase system. J. Bacteriol. 2010, 192, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Toda, H.; Imae, R.; Komio, T.; Itoh, N. Expression and characterization of styrene monooxygenases of Rhodococcus sp. ST-5 and ST-10 for synthesizing enantiopure (S)-epoxides. Appl. Microbiol. Biotechnol. 2012, 96, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, F.; Lin, P.-C.; Witholt, B.; Schmid, A. Stereospecific biocatalytic epoxidation: The first example of direct regeneration of a FAD-dependent monooxygenase for catalysis. J. Am. Chem. Soc. 2003, 125, 8209–8217. [Google Scholar] [CrossRef] [PubMed]
- Riedel, A.; Heine, T.; Westphal, A.H.; Conrad, C.; Rathsack, P.; van Berkel, W.J.H.; Tischler, D. Catalytic and hydrodynamic properties of styrene monooxygenases from Rhodococcus opacus 1CP are modulated by cofactor binding. AMB Express 2015, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Van Hellemond, E.W.; Janssen, D.B.; Fraaije, M.W. Discovery of a novel styrene monooxygenase originating from the metagenome. Appl. Environ. Microbiol. 2007, 73, 5832–5839. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D. Pathways for the degradation of styrene. In Microbial Styrene Degradation; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1, pp. 7–22. ISBN 978-3-319-24862-2. [Google Scholar]
- Heine, T.; Zimmerling, J.; Ballmann, A.; Kleeberg, S.B.; Rückert, C.; Busche, T.; Winkler, A.; Kalinowski, J.; Poetsch, A.; Scholtissek, A.; et al. On the enigma of glutathione dependent styrene degradation in Gordonia rubripertincta CWB2. Appl. Environ. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.-H.; Chen, H.-P.; Shu, H.-Y.; Lee, S.-W. Detoxification of indole by an indole-induced flavoprotein oxygenase from Acinetobacter baumannii. PLoS ONE 2015, 10, e0138798. [Google Scholar] [CrossRef] [PubMed]
- Sadauskas, M.; Vaitekūnas, J.; Gasparavičiūtė, R.; Meškys, R. Indole biodegradation in Acinetobacter sp. strain O153: Genetic and biochemical characterization. Appl. Environ. Microbiol. 2017, 83, e01453-17. [Google Scholar] [CrossRef] [PubMed]
- Kantz, A.; Chin, F.; Nallamothu, N.; Nguyen, T.; Gassner, G.T. Mechanism of flavin transfer and oxygen activation by the two-component flavoenzyme styrene monooxygenase. Arch. Biochem. Biophys. 2005, 442, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Kantz, A.; Gassner, G.T. Nature of the reaction intermediates in the flavin adenine dinucleotide-dependent epoxidation mechanism of styrene monooxygenase. Biochemistry 2011, 50, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.; Kantz, A.; Gassner, G.T.; Sazinsky, M.H. Structure and mechanism of styrene monooxygenase reductase: New insight into the FAD-transfer reaction. Biochemistry 2013, 52, 6063–6075. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Schlömann, M.; van Berkel, W.J.H.; Gassner, G.T. FAD C(4a)-hydroxide stabilized in a naturally fused styrene monooxygenase. FEBS Lett. 2013, 587, 3848–3852. [Google Scholar] [CrossRef] [PubMed]
- Ukaegbu, U.E.; Kantz, A.; Beaton, M.; Gassner, G.T.; Rosenzweig, A.C. Structure and ligand binding properties of the epoxidase component of styrene monooxygenase. Biochemistry 2010, 49, 1678–1688. [Google Scholar] [CrossRef] [PubMed]
- Heine, T.; Tucker, K.; Okonkwo, N.; Assefa, B.; Conrad, C.; Scholtissek, A.; Schlömann, M.; Gassner, G.T.; Tischler, D. Engineering styrene monooxygenase for biocatalysis: Reductase-epoxidase fusion proteins. Appl. Biochem. Biotechnol. 2017, 181, 1590–1610. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Gröning, J.A.D.; Kaschabek, S.R.; Schlömann, M. One-component styrene monooxygenases: An evolutionary view on a rare class of flavoproteins. Appl. Biochem. Biotechnol. 2012, 167, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Tischler, D.; Riedel, A.; Schwabe, R.; Siegel, L.; Friebel, K.; Heine, T.; Gröning, J.A.D.; Kaschabek, S.R.; Schlömann, M. Evolution der Styrol-Monooxygenase StyA1/StyA2B aus Variovorax paradoxus EPS und seine biotechnologische Anwendung. Chem. Ing. Tech. 2014, 86, 1401–1426. [Google Scholar] [CrossRef]
- Gröning, J.A.D.; Kaschabek, S.R.; Schlömann, M.; Tischler, D. A mechanistic study on SMOB-ADP1: An NADH: flavin oxidoreductase of the two-component styrene monooxygenase of Acinetobacter baylyi ADP1. Arch. Microbiol. 2014, 196, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Heine, T.; Scholtissek, A.; Westphal, A.H.; van Berkel, W.J.H.; Tischler, D. N-terminus determines activity and specificity of styrene monooxygenase reductases. BBA Proteins Proteom. 2017, 1865, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug. Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the structural biology of protein-protein interactions. Protein Sci. 2013, 22, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Amet, N.; Wang, W.; Shen, W.C. Human growth hormone-transferrin fusion protein for oral delivery in hypophysectomized rats. J. Control. Release 2010, 141, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Bae, J.W.; Han, J.H.; Lee, E.Y.; Lee, S.-G.; Park, S. Characterization of styrene catabolic genes of Pseudomonas putida SN1 and construction of a recombinant Escherichia coli containing styrene monooxygenase gene for the production of (S)-styrene oxide. J. Microbiol. Biotechnol. 2006, 16, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Corrado, M.L.; Knaus, T.; Mutti, F. Chimeric styrene monooxygenase with increased efficiency in asymmetric biocatalytic epoxidation. ChemBioChem 2018. [Google Scholar] [CrossRef] [PubMed]
- Otto, K.; Hofstetter, K.; Röthlisberger, M.; Witholt, B.; Schmid, A. Biochemical characterization of StyAB from Pseudomonas sp. strain VLB120 as a two-component flavin-diffusible monooxygenase. J. Bacteriol. 2004, 186, 5292–5302. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Numata, T.; Oae, S. Mild and facile preparation of sulfoxides from sulfides using titanium (III) chloride/hydrogen peroxide. Synthesis 1981, 1981, 204–206. [Google Scholar] [CrossRef]
- Anderson, J.L.; Ding, J.; McCulla, R.D.; Jenks, W.S.; Armstrong, D.W. Separation of racemic sulfoxides and sulfinate esters on four derivatized cyclodextrin chiral stationary phases using capillary gas chromatography. J. Chrom. A 2002, 946, 197–208. [Google Scholar] [CrossRef]
- Van den Heuvel, R.H.H.; Westphal, A.H.; Heck, A.J.R.; Walsh, M.A.; Rovida, S.; van Berkel, W.J.H.; Mattevi, A. Structural studies on flavin reductase PheA2 reveal binding of NAD in an unusual folded conformation and support novel mechanism of action. J. Biol. Chem. 2004, 279, 12860–12867. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 3, ISBN 978-0879695767. [Google Scholar]
- Miller, J.H. Experiments in Molecular Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1972; pp. 431–433. ISBN 978-0879691066. [Google Scholar]
- Puigbo, P.; Guzman, E.; Romeu, A.; Garcia-Vallve, S. OPTIMIZER: A web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007, 35, W126–W131. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Schlömann, M.; Tischler, D. Biochemical characterization of an azoreductase from Rhodococcus opacus 1CP possessing methyl red degradation ability. J. Mol. Catal. B Enzym. 2016, 130, 9–17. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds or plasmids are available from the authors. |

{kind=link}
{kind=link}
| Donor 1/Acceptor (µM) 2 | Km (μM) | Vmax (U mg−1) | kcat (s−1) | kcat Km−1 (s−1 μM−1) |
|---|---|---|---|---|
| NADH (7.9–164)/FAD (70) | 24.0 ± 4.0 | 16.2 ± 0.8 | 17.9 ± 0.9 | 0.72 |
| NADH (164)/FAD (6.3–78.8) | 33.6 ± 4.0 | 20.2 ± 1.0 | 22.3 ± 1.1 | 0.64 |
| NADH (164)/FMN (4.1–90.2) | 45.9 ± 6.8 | 26.0 ± 1.8 | 28.7 ± 1.9 | 0.57 |
| NADH (164 µM)/Riboflavin (6.3–88.2) | 37.7 ± 7.2 | 31.3 ± 2.7 | 34.6 ± 2.9 | 0.88 |
| VpStyA2B-Variant | Donor NADH 1 | Acceptor FAD 1 | ||||
|---|---|---|---|---|---|---|
| Km (μM) | Vmax (U mg−1) | kcat Km−1 (s−1 μM−1) | Km (μM) | Vmax (U mg−1) | kcat Km−1 (s−1 μM−1) | |
| wildtype VpStyA2B | 24.0 | 16.2 | 0.72 | 33.6 | 20.2 | 0.64 |
| 408-TIVVV | 37.7 | 3.3 | 0.09 | 5.1 | 3.1 | 0.65 |
| 408-AAAAA | 28.1 | 8.9 | 0.34 | 1.8 | 8.8 | 5.21 |
| 408-HHHHH | 44.2 | 2.5 | 0.06 | 14.5 | 3.1 | 0.23 |
| 408-WYHHH | 20.1 | 7 | 0.37 | 3.2 | 6.9 | 2.30 |
| 408-WYHHHHH | 40.6 | 13.8 | 0.36 | 7.4 | 13.2 | 1.90 |
| 408-GQWCSQY | 47.6 | 3.8 | 0.09 | 14.5 | 3.7 | 0.27 |
| Substrate | Styrene | Indole | ||
|---|---|---|---|---|
| SMO 1 | Vmax (mU mg−1) | ETY 2 (%) | Vmax (mU mg−1) + Extra RoStyBart 3 | Plate Screening 4 |
| wildtype VpStyA1 | n.a. | n.a. | 460 | +++ |
| wildtype VpStyA2B | 159 | 1 | 140 | ++ |
| 408-TIVVV | 135 | 4.4 | 327 | + |
| 408-AAAAA | 260 | 3 | 260 | +++ |
| 408-HHHHH | <1 | <1 | 4 | + |
| 408-WYHHH | 1.7 | <1 | 7.3 | + |
| 408-WYHHHHH | <1 | <1 | 1.6 | + |
| 408-GQWCSQY | <1 | <1 | 3.1 | + |
| Substrate | Product | VpStyA1 Conversion (%) | ee (%) | VpStyA2B Conversion (%) | ee (%) |
|---|---|---|---|---|---|
| PMS | 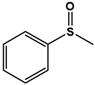 | 43.6 ± 1.9 | 98 (S) | 6.3 ± 0.3 | 64 (S) |
| 4F-PMS | 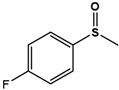 | 59.8 ± 0.4 | 99 (S) | 4.3 ± 0.7 | 84 (S) |
| 4Cl-PMS | 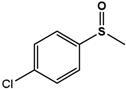 | 38.5 ± 1.7 | 99 (S) | 4.4 ± 0.4 | 96 (S) |
| 4Br-PMS | 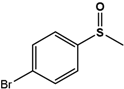 | 32.4 ± 4.7 | 99 (S) | 6 ± 0.7 | 96 (S) |
| BMS | 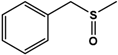 | 95 ± 0.5 | 97 (S) | 13 ± 0.9 | n.d. (S) |
| Styrene | 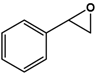 | 9.9 ± 0.4 | 98.2 (S) | 0.5 ± 0.1 | 45 (S) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tischler, D.; Schwabe, R.; Siegel, L.; Joffroy, K.; Kaschabek, S.R.; Scholtissek, A.; Heine, T. VpStyA1/VpStyA2B of Variovorax paradoxus EPS: An Aryl Alkyl Sulfoxidase Rather than a Styrene Epoxidizing Monooxygenase. Molecules 2018, 23, 809. https://doi.org/10.3390/molecules23040809
Tischler D, Schwabe R, Siegel L, Joffroy K, Kaschabek SR, Scholtissek A, Heine T. VpStyA1/VpStyA2B of Variovorax paradoxus EPS: An Aryl Alkyl Sulfoxidase Rather than a Styrene Epoxidizing Monooxygenase. Molecules. 2018; 23(4):809. https://doi.org/10.3390/molecules23040809
Chicago/Turabian StyleTischler, Dirk, Ringo Schwabe, Lucas Siegel, Kristin Joffroy, Stefan R. Kaschabek, Anika Scholtissek, and Thomas Heine. 2018. "VpStyA1/VpStyA2B of Variovorax paradoxus EPS: An Aryl Alkyl Sulfoxidase Rather than a Styrene Epoxidizing Monooxygenase" Molecules 23, no. 4: 809. https://doi.org/10.3390/molecules23040809
APA StyleTischler, D., Schwabe, R., Siegel, L., Joffroy, K., Kaschabek, S. R., Scholtissek, A., & Heine, T. (2018). VpStyA1/VpStyA2B of Variovorax paradoxus EPS: An Aryl Alkyl Sulfoxidase Rather than a Styrene Epoxidizing Monooxygenase. Molecules, 23(4), 809. https://doi.org/10.3390/molecules23040809