3.2. General Procedure A: Steglich Esterification of Carboxylic Acids with N-Hydroxyphthalimide 1, 26–34
A 100 mL Schlenk flask equipped with a stir bar and a septum was flushed with argon and charged with dry THF (30 mL). The carboxylic acid (1.00 equiv.), N,N’-dicyclohexylcarbodiimide (1.20 equiv.), 4-dimethylaminopyridine (0.10 equiv.) were added. After stirring for one minute, N-hydroxyphthalimide (1.10 equiv.) was added and the reaction mixture was stirred for 24 h at room temperature. The precipitating dicyclohexyl urea was filtered off and the solution was concentrated by evaporation of the solvent. Flash column chromatography afforded the desired product.
N-(Cyclohexylcarbonyloxy)phthalimide (1). This compound was prepared according to General Procedure A using cyclohexanecarboxylic acid (1.00 g, 7.80 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 5/1) afforded the title compound (2.12 g, 7.74 mmol, 99%) as a colorless solid. Rf = 0.38 (SiO2, cHex/EtOAc 5/1). IR (ATR): (cm−1) 2935, 2858, 1786, 1742, 1372, 1186, 1142, 1001, 697. 1H-NMR, COSY (400 MHz, CDCl3): δ/ppm 7.89–7.86 (m, 2H, H-3,6), 7.79–7.77 (m, 2H, H-4,5), 2.73 (tt, J = 11.0, 3.7 Hz, 1H, H-1′), 2.12–2.07 (m, 2H, H’), 1.85–1.80 (m, 2H, H’), 1.69–1.63 (m, 4H, H-2′,6′), 1.42–1.31 (m, 2H, H4′). 13C, HSQC, HMBC (100.6 MHz, CDCl3): δ/ppm 171.9 (CO), 163.2 (2C, CO), 134.8 (2C, C-4,5), 129.1 (2C, Cq), 124.0 (2C, C-3,6), 40.6 (CH), 28.9 (CH2), 25.6 (CH2), 25.2 (CH2). Mp: 69.1–70.3 °C. FD-MS: m/z 273.0 (100%, [M]+). EA: anal. C 65.84, H 5.61, N 5.26%, calcd for C15H15NO4, C 65.92, H 5.53, N 5.13%.
N-(Hexanoyloxy)phthalimide (26). This compound was prepared according to General Procedure A using hexanoic acid (1.00 g, 8.60 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 5/1) afforded the title compound (1.89 g, 7.23 mmol, 84%) as a colorless solid. Rf = 0.32 (SiO2, cHex/EtOAc 10/1). IR (ATR): [cm−1] 2957, 1816, 1788, 1740, 1467, 1186, 1065, 878, 670. 1H-NMR, COSY (400 MHz, CDCl3): δ/ppm 7.90–7.86 (m, 2H, H-3,6), 7.81–7.76 (m, 2H, H-4,5), 2.68 (t, J = 7.4 Hz, 2H, H-2′), 1.82 (q, J = 7.6 Hz, 2H, H-3′), 1.48–1.40 (m, 4H, H-4′,5′), 0.95 (t, J = 7.4 Hz, 3H, H-6′). 13C, HSQC, HMBC (100.6 MHz, CDCl3): δ/ppm 169.8 (CO), 162.1 (2C, CO), 134.8 (2C, C-4,5), 129.1 (2C, Cq), 139.9 (2C, C-3,6), 31.0 (C-2′,4′), 24.4 (C-3′), 22.2 (C-5′), 13.8 (C-6′). Mp: 41.2–42.9 °C. FD-MS: m/z 261.0 (100%, [M]+). EA: anal. C 64.46, H 5.57, N 5.51%, calcd for C14H15NO4, C 64.36, H 5.79, N 5.36%.
N-(Pivaloyloxy)phthalimide (
27). This compound was prepared according to General Procedure A using pivalic acid (1.00 g, 9.79 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 5/1) afforded the title compound (2.40 g, 9.70 mmol, 99%) as a colorless solid.
Rf = 0.61 (SiO
2,
cHex/EtOAc 3/1). IR (ATR):
[cm
−1] 2936, 1784, 1743, 1486, 1369, 1186, 1062, 1023, 878,697.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 7.86–7.84 (m, 2H, H-3,6), 7.77–7.74 (m, 2H, H-4,5), 1.41 (s, 9H, C(CH
3)
3).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 174.5(CO), 162.2 (2C, CO), 134.8 (2C, C-4,5), 129.1 (2C, C
q), 123.9 (2C, C-2,6), 38.5 (C
q), 27.1 (CH
3). Mp: 76.5–79.3 °C. FD-MS:
m/
z 247.4 (100%, [M]
+). The spectral data are consistent with those reported in the literature [
32].
N-(N-tert-Butoxycarbonyl-l-valinoyloxy)phthalimide (28). This compound was prepared according to General Procedure A using N-(tert-butoxycarbonyl)-l-valine (1.50 g, 6.90 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 9/1) afforded the title compound (1.71 g, 4.72 mmol, 68%) as a colorless solid. Rf = 0.17 (SiO2, cHex/EtOAc 9/1). IR (ATR): [cm−1] 2973, 1788, 1742, 1708, 1366, 1184, 1136, 1069, 876, 696. 1H-NMR, COSY (400 MHz, CDCl3): δ/ppm 7.89 (dd, J = 5.5, 3.1 Hz, 2H, H-3,6), 7.80 (dd, J = 5.5, 3.1 Hz, 2H, H-4,5), 5.04 (d, J = 9.0 Hz, 1H, NH), 4.67 (dd, J = 9.0, 4.8 Hz, 1H, C*H), 2.28–2.34 (m, 1H, CH(CH3)2), 1.47 (s, 9H, (CH3)3), 1.14 (s, 3H, CH3), 1.10 (s, 3H, CH3). 13C, HSQC, HMBC (100.6 MHz, CDCl3): δ/ppm 169.9 (CO), 161.7 (2C, CO), 155.3 (CO), 134.8 (2C, C-4,5), 129.0 (2C, Cq), 124.0 (2C, C-3,6), 80.6 (Cq(CH3), 57.0 (C*H), 31.8 (CH(CH3)2), 28.2 (3C, Cq(CH3)3), 18.7 (CH3), 17.3 (CH3). Mp: 79.6–80.9 °C. ESI-MS (pos): m/z 363.1 (100%, [M + H]+). ESI-HRMS (pos): m/z calculated for [C18H14N2O6]+ m/z 363.1551, found 363.1567.
N-(Isobutyryloxy)phthalimide (
29). This compound was prepared according to General Procedure A using isobutyric acid (1.00 g, 11.4 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 5/1) afforded the title compound (2.48 g, 10.6 mmol, 93%) as a colorless solid.
Rf = 0.52 (SiO
2,
cHex/EtOAc 2/1). IR (ATR):
[cm
−1] 2879, 1845, 1811, 1739, 1467, 1359, 1048, 967, 877, 739.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 7.89–7.86 (m, 2H, H-3,6), 7.79–7.76 (m, 2H, H-4,5), 2.95 (sept,
J = 7.0 Hz, 1H, C
H), 1.38 (s, 3H, C
H3). 1.36 (s, 3H, C
H3).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 173.2 (CO), 162.2 (2C, CO), 134.8 (2C, C-4,5), 129.1 (2C, C
q), 124.0 (2C, C-3,6), 31.9 (CH), 19.0 (2C, CH
3). Mp: 66.4–67.7 °C. FD-MS:
m/
z 233.4 (100%, [M]
+). The spectral data are consistent with those reported in the literature [
33].
N-(2-Methylpentanoyloxy)phthalimide (30). This compound was prepared according to General Procedure A using 2-methylpentanoic acid (1.00 g, 8.60 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 5/1) afforded the title compound (1.48 g, 5.36 mmol, 62%) as a colorless solid. Rf = 0.57 (SiO2, cHex/EtOAc 3/1). IR (ATR): [cm−1] 2978, 2875, 1844, 1783, 1481, 1062, 1023, 878, 805, 696. Mp: 29.8–31.2 °C. 1H-NMR, COSY (300 MHz, CDCl3): δ/ppm 7.87–7.84 (m, 2H, H-3,6), 7.78–7.75 (m, 2H, H-4,5), 2.87–2.82 (m, 1H, H-2′), 1.84–1.80 (m, 1H, H-3a’), 1.64–1.56 (m, 1H, H-3b’), 1.54–1.44 (m, 2H, H-4′), 1.33 (d, J = 7.0 Hz, 3H, CHCH3), 0.96 (t, J = 7.1 Hz, 3H, H-5′). 13C, HSQC, HMBC (75.4 MHz, CDCl3): δ/ppm 172.9 (CO), 162.1 (2C, CO), 134.8 (2C, C-4,5), 129.1 (2C, Cq), 124.0 (2C, C-3,6), 37.0 (C-2′), 35.9 (C-3′), 20.2 (C-4′), 17.0 (CHCH3), 14.0 (C-5′). FD-MS: m/z 261.3 (100%, [M]+). EA: anal. C 64.12, H 5.78, N 5.66%, calcd for C14H15NO4, C 64.36, H 5.79, N 5.36%.
N-(2-Ethylbutyryloxy)phthalimide (31). This compound was prepared according to General Procedure A using 2-ethylbutyric acid (1.00 g, 8.60 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 5/1) afforded the title compound (1.48 g, 5.36 mmol, 62%) as a colorless oil. Rf = 0.58 (SiO2, cHex/EtOAc 3/1). IR (ATR): [cm−1] 2968, 2879, 1811, 1742, 1466, 1369, 1135, 967, 677, 696. 1H-NMR, COSY (300 MHz, CDCl3): δ/ppm 7.86–7.83 (m, 2H, H-3,6), 7.77–7.74 (m, 2H, H-4,5), 2.58 (tt, J = 8.6, 5.5 Hz, 1H, H-3′), 1.78–1.71 (m, 4H, H-2′,4′), 1.05 (t, J = 7.5 Hz, 6H, H-1′,5′). 13C, HSQC, HMBC (75.4 MHz, CDCl3): δ/ppm 172.3 (CO), 162.1 (2C, CO), 134.8 (2C, C-4,5), 129.0 (2C, Cq), 123.9 (2C, C-3,6), 46.5 (C-3′), 25.3 (C-2′,4′), 11.6 (C-1′,5′). FD-MS: m/z 261.4 (100%, [M]+). EA: anal. C 63.98, H 5.93, N 5.35%, calcd for C14H15NO4, C 64.36, H 5.79, N 5.36%.
N-(Pent-4-enyl-2-carbonyloxy)phthalimide (32). This compound was prepared according to General Procedure A using 2-methyl-4-pentenoic acid (1.00 g, 8.76 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 9/1) afforded the title compound (2.16 g, 8.34 mmol, 95%) as a colorless oil. Rf = 0.37 (SiO2, cHex/EtOAc 8/1). IR (ATR): [cm−1] 2938, 1785, 1739, 1459, 1325, 1135, 1081, 967, 924, 696. 1H-NMR, COSY (300 MHz, CDCl3): δ/ppm 7.87–7.82 (m, 2H, H-3,6), 7.79–7.74 (m, 2H, H-4,5), 5.90–5.80 (m, 1H, H-4′), 5.20–5.11 (m, 2H, H-5′), 2.91 (sept, J = 6.9 Hz, 1H, H-2′), 2.57 (app dtt, J = 14.5, 6.9, 1.2 Hz, 1H, H-3a’), 2.37 (m, 1H, H-3b’), 1.34 (d, J = 6.9 Hz, 3H, H-5′). 13C, HSQC, HMBC (75.4 MHz, CDCl3): δ/ppm 172.1 (CO), 162.0 (2C, CO), 134.8 (2C, C-4,5), 134.0 (C-4′), 129.0 (2C, Cq), 124.0 (2C, C-3,6), 118.3 (C-5′), 37.5 (C-2′), 36.9 (C-3′), 16.4 (C-1′). ESI-MS (pos): m/z 260.1 (100%, [M + H]+), 282.1 (54%, [M + Na]+). ESI-HRMS (pos): m/z calculated for [C14H14NO4]+ m/z 260.0917, found 260.0917.
N-(Benzoyloxy)phthalimide (
33). This compound was prepared according to General Procedure A using benzoic acid (0.50 g, 4.09 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 3/1) afforded the title compound (1.01 g, 3.80 mmol, 93%) as a colorless solid.
Rf = 0.29 (SiO
2,
cHex/EtOAc 3/1). IR (ATR):
[cm
−1] 1771, 1741, 1600, 1453, 1372, 1236, 1186, 1003, 877, 659.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 8.21–8.18 (m, 2H, H-2′,6′), 7.94–7.92 (m, 2H, H-3,6), 7.83–7.80 (m, 2H, H-4,5), 7.70 (pseudo tt,
J = 7.7, 1.2 Hz, 1H, H-4′), 7.57–7.51 (m, 2H, H-3′,5′).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 162.9 (CO), 162.2 (2C, CO), 135.0 (C-4′), 134.9 (2C, C-4,5), 130.8 (2C, C-2′,6′), 129.2 (2C, C
q), 129.0 (2C, C-3′,5′), 125.4 (C
q), 124.2 (2C, C-3,6). Mp: 174.3–176.9 °C. FD-MS:
m/
z 267.0 (100%, [M]
+). The spectral data are consistent with those reported in the literature [
34].
N-(2-Phenylacetyloxy)phthalimide (
34). This compound was prepared according to General Procedure A using phenylacetic acid (0.25 g, 1.84 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 3/1) afforded the title compound (408 mg, 1.45 mmol, 79%) as a colorless solid.
Rf = 0.36 (SiO
2,
cHex/EtOAc 3/1). IR (ATR):
[cm
−1] 1815, 1788, 1742, 1498, 1359, 1133, 1065, 878, 728, 519.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 7.89–7.84 (m, 2H, H-3,6), 7.80–7.76 (m, 2H, H-4,5), 7.40–7.38 (m, 3H, H-2′,4′,6′), 7.36–7.31 (m, 2H, H-3′,5′), 4.00 (s, 2H, CH
2).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 167.9 (CO), 161.9 (2C, CO), 134.9 (2C, C-4,5), 131.6 (C
q), 129.4 (2C, C-2′,6′), 129.0 (4C, C
q, C-3′,5′), 127.9 (C-4′), 124.1 (2C, C-3,6), 37.8 (CH
2). Mp: 174.8–178.0 °C. FD-MS:
m/
z 281.4 (100%, [M]
+). The spectral data are consistent with those reported in the literature [
35].
3.3. General Procedure B: Coupling of N-Hydroxyphthalimide Esters with Heterocycles 3, 5–18
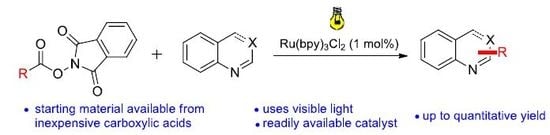
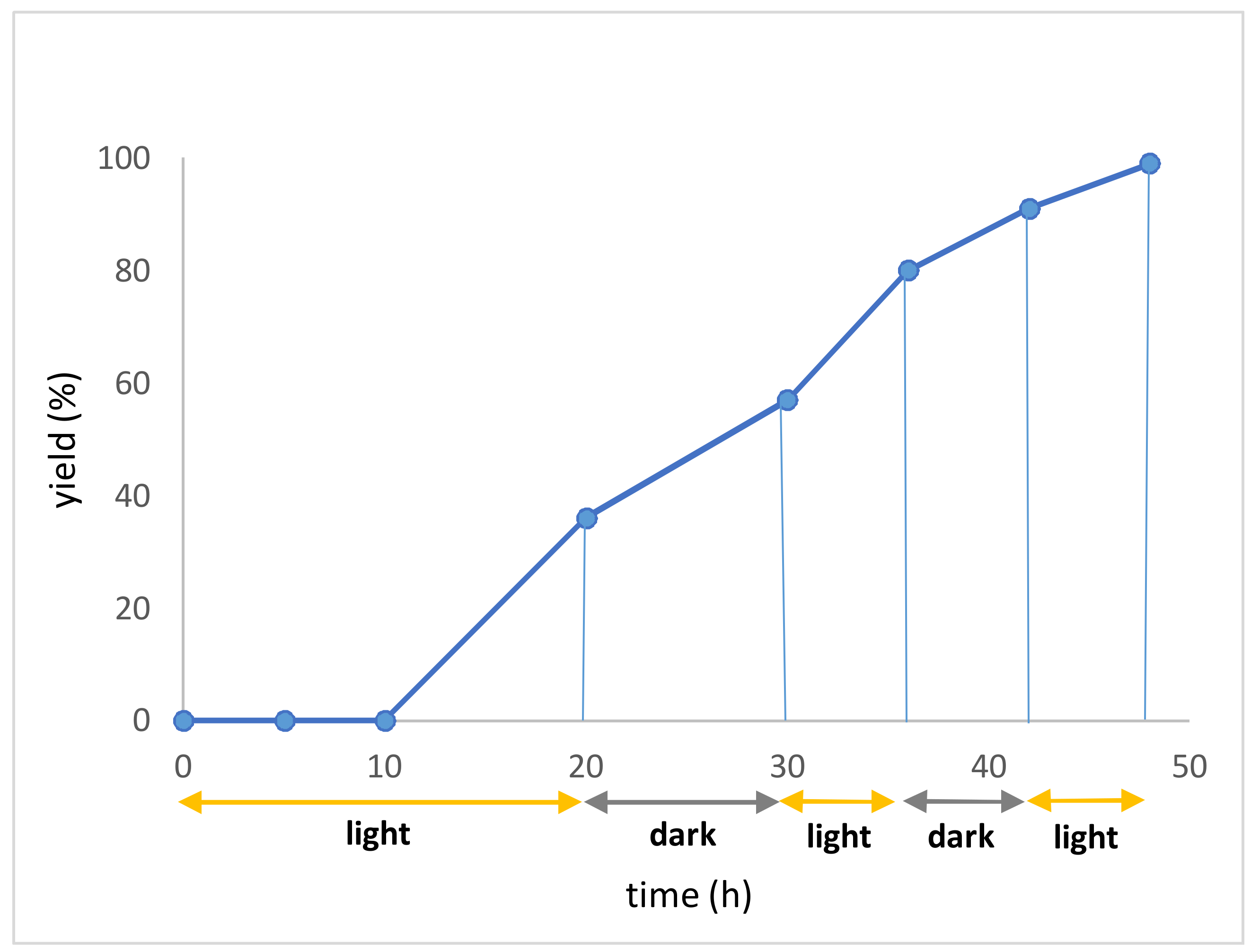
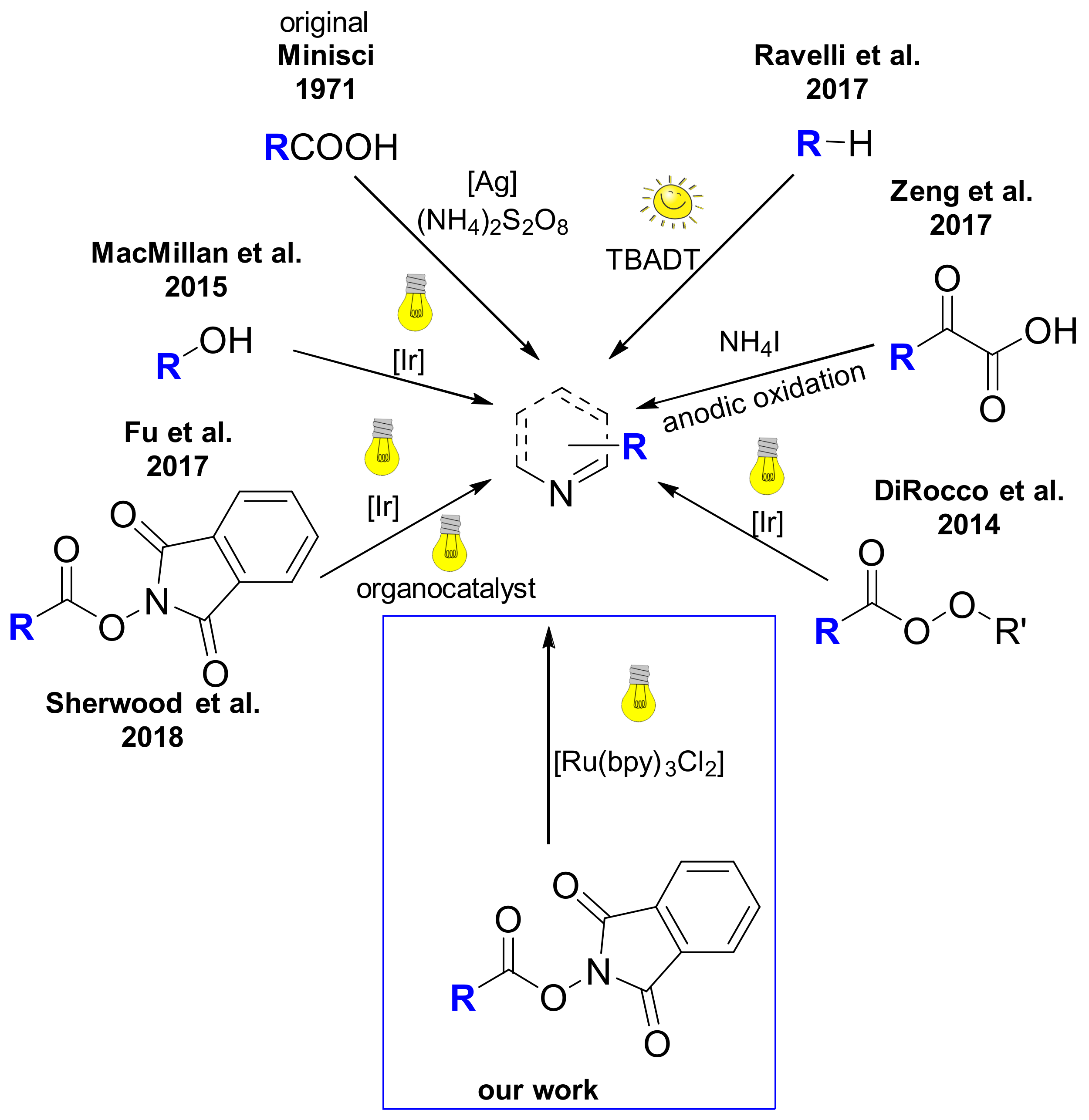
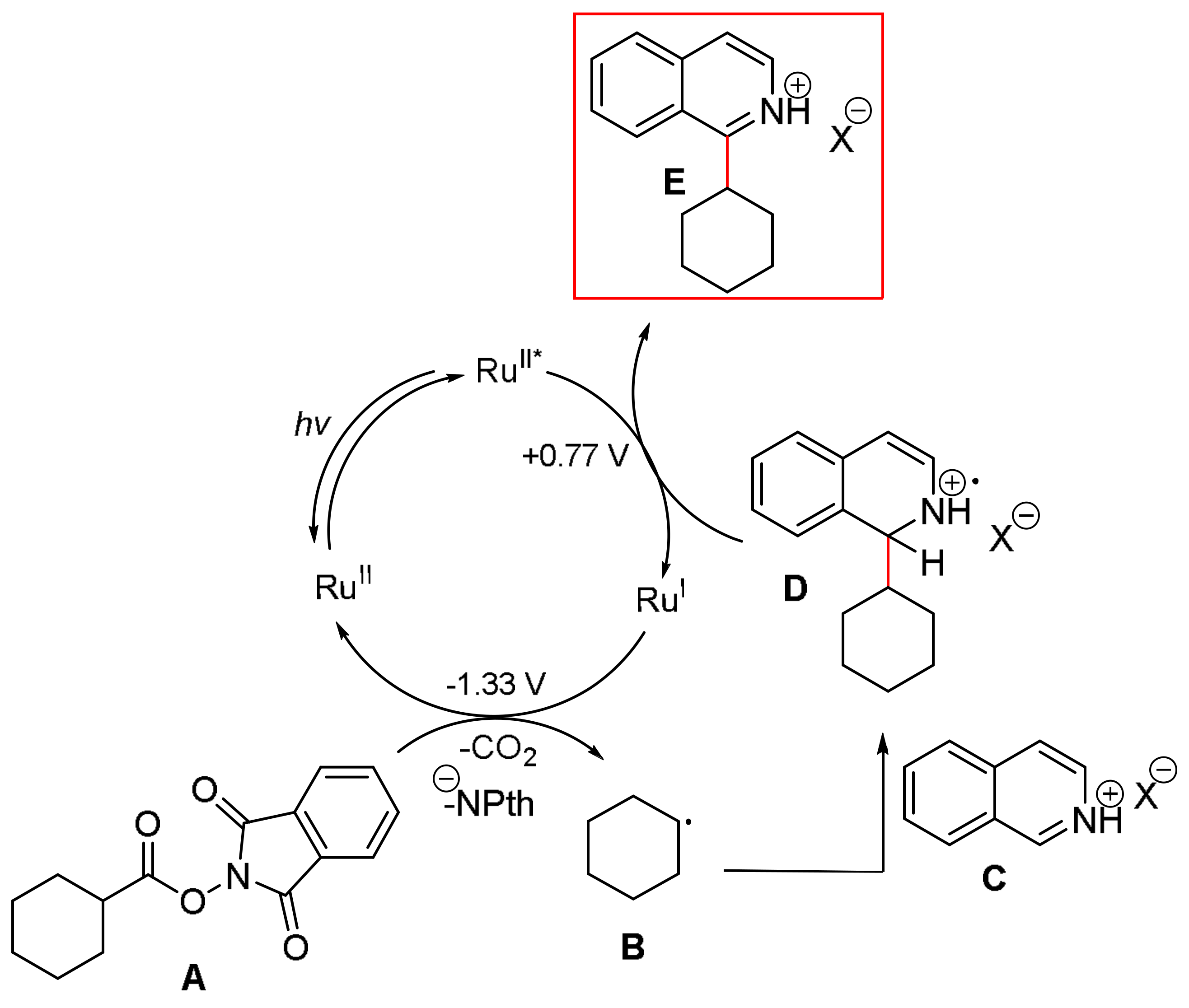
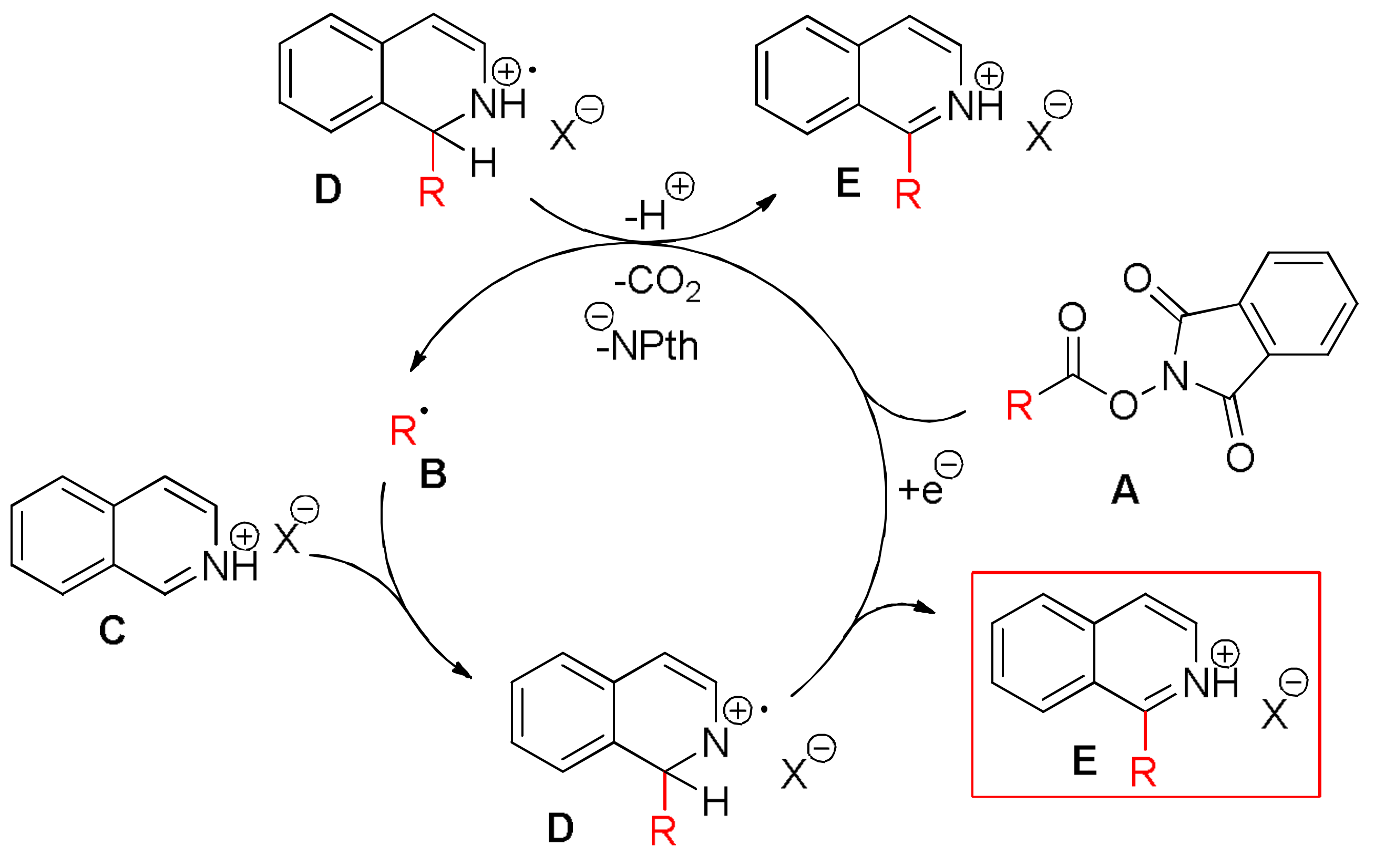
A 10 mL vial equipped with a stirrer bar and a septum was flushed with argon and charged with dry N,N’-dimethylformamid. The solvent was previously degassed by ultrasonication (see Gerneral Information). The N-hydroxyphthalimide ester (0.30 mmol, 1.50 equiv.), the heterocyclic compound (0.20 mmol, 1 equiv.), p-toluenesulfonic acid monohydrate (0.30 mmol, 1.50 equiv.) and Ru(bpy)3Cl2 (2 μmol, 1 mol %) were added, followed by argon sparging for 1 min. The mixture was irradiated with a blue LED module from a distance of 5 cm for 48 h. A saturated aqueous NaHCO3 solution (30 mL) was added, followed by extraction with dichloromethane (3 × 15 mL). The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. Flash column chromatography afforded the desired product.
1-(Cyclohexyl)isoquinoline (
3). This compound was prepared according to General Procedure B using
N-(cyclohexycarbonyloxy)phthalimide (82.00 mg, 0.30 mmol) and Isoquinoline (25.8 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 14/1) afforded the title compound (42.30 mg, 0.2 mmol, quant.) as a yellow oil.
Rf = 0.32 (SiO
2,
cHex/EtOAc 14/1). IR (ATR):
[cm
−1] 2926, 2852, 1562, 1449, 1391, 1335, 993, 822, 744, 675.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 8.48 (d,
J = 5.6 Hz, 1H, H-3), 8.22 (d,
J = 8.1 Hz, 1H, H-8), 7.84–7.81 (m, 1H, H-5), 7.64 (mc, 1H, H-6), 7.58 (ddd,
J = 8.1, 6.8, 1.3 Hz, 1H, H-7), 7.48 (d,
J = 5.6, 0.8 Hz, 1H, H-4), 3.56 (tt,
J = 11.6, 3.3 Hz, 1H, H-1′), 2.00–1.93 (m, 4H, H’), 1.88–1.80 (m, 4H, H‘), 1.58–1.33 (m, 2H, H-4′).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 165.7 (C
q-1), 141.9 (C-3), 136.4 (C
q-4a), 129.5 (C-6), 127.5 (C-5), 126.3 (2C, C-7,C
q-8a), 124.7 (C-8), 118.9 (C-4), 41.5 (C-1′), 32.6 (2C, C-3′,5′), 26.9 (2C, C-2′,6′), 26.3 (C-4′). ESI-MS (pos):
m/
z 212.1 (100%, [M + H]
+). The spectral data are consistent with those reported in the literature [
36].
1-(Propan-2-yl)isoquinoline (
5). This compound was prepared according to General Procedure B using
N-(isobutyryloxy)phthalimide (70.00 mg, 0.30 mmol) and isoquinoline (25.8 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 10/1) afforded the title compound (9.50 mg, 5.55 × 10
−5 mol, 28%) as a colorless oil.
Rf = 0.44 (SiO
2,
cHex/EtOAc 8/1). IR (ATR):
[cm
−1] 3631, 1719, 1655, 1586, 1543, 1008, 903,869, 822, 727.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 8.49 (d,
J = 5.7 Hz, 1H, H-3), 8.23 (app dq,
J = 8.3, 0.8 Hz, 1H, H-8), 7.83–7.80 (m, 1H, H-5), 7.62 (m, 2H, H-6,7), 7.49 (dd,
J = 5.7, 0.8 Hz, 1H, H-4), 3.96 (sept,
J = 6.9 Hz, 1H, H-2′), 1.46 (s, 3H, CH
3), 1.44 (s, 3H, CH
3).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 166.5 (C
q-1), 142.0 (C-3), 136.5 (C
q-4a), 129.7 (C-6), 127.7 (C-5), 127.0 (C-7), 126.4 (C
q-8a), 124.9 (C-8), 119.1 (C-4), 31.1 (C-2′), 22.4 (2C, CH
3). ESI-MS (pos):
m/
z 172.1 (100%, [M + H]
+). The spectral data are consistent with those reported in the literature [
37].
1-(Pentan-3-yl)isoquinoline (
6). This compound was prepared according to General Procedure B using
N-(2-ethylbutyryloxy)phthalimide (52.3 mg, 0.30 mmol) and isoquinoline (25.8 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 8/1) afforded the title compound (22.3 mg, 1.12 × 10
−4 mol, 56%) as a colorless oil.
Rf = 0.47 (SiO
2,
cHex/EtOAc 8/1). IR (ATR):
[cm
−1] 3050, 2960, 2929, 2872, 1585, 1458, 1313, 821, 745, 682.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 8.53 (d,
J = 5.7 Hz, 1H, H-3), 8.25 (ddd,
J = 8.4, 1.4, 0.7 Hz, 1H, H-8), 7.83–7.80 (m, 1H, H-5), 7.62 (mc, 2H, H-6,7), 7.48 (dd,
J = 5.7, 0.9 Hz, 1H, H-4), 3.54 (tt,
J = 8.3, 5.5 Hz, 1H, H-3′), 1.98–1.80 (m, 4H, H-2′,4′), 0.79 (t,
J = 7.4 Hz, 6H, H-1′,5′).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 165.2 (C
q-1), 142.2 (C-3), 136.4 (C
q-4a), 129.6 (C-6), 128.3 (C
q-8a), 127.6 (C-5), 126.9 (C-7), 125.1 (C-8), 118.9 (C-4), 44.9 (C-3′), 28.3 (2C, C-2′,4′), 12.5 (C-1′,5′). ESI-MS (pos):
m/
z 200.1 (100%, [M + H]
+). The spectral data are consistent with those reported in the literature [
38].
1-(Pentan-2-yl)isoquinoline (
7). This compound was prepared according to General Procedure B using
N-(2-methylpentanoyloxy)phthalimide (78.40 mg, 0.30 mmol) and isoquinoline (25.8 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 8/1) afforded the title compound (33.1 mg, 1.66 × 10
−4 mol, 83%) as a colorless oil.
Rf = 0.37 (SiO
2,
cHex/EtOAc 8/1). IR (ATR):
[cm
−1] 3050, 2957, 2929, 2870, 1585, 1456, 1309, 821, 745, 683.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 8.50 (d,
J = 5.7 Hz, 1H, H-3), 8.23 (app dq,
J = 8.0, 0.8 Hz, 1H, H-8), 7.83–7.80 (m, 1H, H-5), 7.62 (mc, 2H, H-6,7), 7.48 (dd,
J = 5.7, 0.8 Hz, 1H, H-4), 3.82 (sept,
J = 6.9 Hz, 1H, H-2′), 2.01–1.94 (m, 1H, H-3′), 1.75–1.67 (m, 1H, H-3′), 1.41 (d,
J = 6.9 Hz, 3H, H-1′), 1.36–1.25 (m, 2H, H-4′), 0.90 (t,
J = 7.3 Hz, 3H, H-5′).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 166.2 (C
q-1), 142.1 (C-3), 136.5 (C
q-4a), 129.7 (C-6), 127.7 (C-5), 127.0 (C-7), 126.9 (C
q-8a), 124.9 (C-8), 118.9 (C-4), 39.1 (C-3′), 35.0 (C-2′), 21.1 (C-4′), 20.7 (C-1′), 14.4 (C-5′). ESI-MS (pos):
m/
z 200.1 (100%, [M + H]
+). The spectral data are consistent with those reported in the literature [
37].
1-(Pent-4-en-2-yl)isoquinoline (8). This compound was prepared according to General Procedure B using N-(pent-4-enyl-2-carbonyloxy)phthalimide (77.7 mg, 0.30 mmol) and isoquinoline (25.8 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 8/1) afforded the title compound (33.1 mg, 1.68 × 10−4 mol, 84%) as a yellow oil. Rf = 0.47 (SiO2, cHex/EtOAc 8/1). IR (ATR): [cm−1] 3052, 2969, 2928, 1639, 1622, 1374, 994, 913, 822, 798. 1H-NMR, COSY (300 MHz, CDCl3): δ/ppm 8.50 (d, J = 5.8 Hz, 1H, H-3), 8.23–8.20 (m, 1H, H-8), 7.83–7.80 (m, 1H, H-5), 7.62 (mc, 2H, H-6,7), 7.49 (dd, J = 5.8, 0.9 Hz, 1H, H-4), 5.83 (ddd, J = 17.1, 10.2, 7.7 Hz, 1H, H-4′), 5.08–5.02 (m, 2H, H-5′), 3.87 (sept, J = 6.8 Hz, 1H, H-2′), 2.79–2.74 (m, 1H, H-3′), 2.50 (mc, 1H, H-3′), 1.43 (d, J = 6.8 Hz, 1H, H-1′). 13C, HSQC, HMBC (75.4 MHz, CDCl3): δ/ppm 165.2 (Cq-1), 142.0 (C-3), 137.4 (C-4′), 136.5 (Cq-4a), 129.7 (C-6), 127.7 (C-5), 127.0 (C-7), 126.7 (Cq-8a), 124.8 (C-8), 119.1 (C-4), 116.6 (C-5′), 40.9 (C-3′), 36.2 (C-2′), 20.3 (C-1′). ESI-MS (pos): m/z 198.1 (100%, [M + H]+). ESI-HRMS (pos): m/z calculated for [C14H15N]+ m/z 198.1277, found 198.1278.
1-(Pentan-1-yl)isoquinoline (
9). This compound was prepared according to General Procedure B using
N-(hexanoyloxy)phthalimide (78.4 mg, 0.30 mmol) and isoquinoline (25.8 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 15/1) afforded the title compound (6.6 mg, 3.31 × 10
−5 mol, 17%) as a yellow oil.
Rf = 0.35 (SiO
2,
cHex/EtOAc 5/1). IR (ATR):
[cm
−1] 2926, 2852, 1562, 1449, 1391, 1335, 993, 822, 744, 675.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 8.43 (d,
J = 5.8 Hz, 1H, H-3), 8.16 (d,
J = 8.5, 1.1 Hz, 1H, H-8), 7.83–7.80 (m, 1H, H-5), 7.63 (mc, 2H, H-6,7), 7.50 (dd,
J = 5.8, 0.9 Hz, 1H, H-4), 3.32–3.26 (m, 2H, H-1′), 1.92–1.84 (m, 2H, H-2′), 1.50–1.40 (m, 2H, H-3‘,4′), 0.92 (t,
J = 7.1 Hz, 3H, H-5′).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 162.2 (C
q-1), 142.2 (C-3), 136.4 (C
q-4a), 129.9 (C-6), 127.5 (C-5), 127.1 (C-7), 125.5 (C-8), 124.1 (C
q-8a), 119.3 (C-4), 35.7 (C-1′), 32.3 (C-3′), 29.7 (C-2′), 22.8 (C-4′), 14.2 (C-5′). ESI-MS (pos):
m/
z 200.1 (100%, [M + H]
+). The spectral data are consistent with those reported in the literature [
37].
tert-Butyl-[1-(isoquinolin-2-yl)-2-methylpropyl]carbamate (11). This compound was prepared according to General Procedure B using N-(N-tert-butoxycarbonyl-l-valinyloxy)phthalimide (108.7 mg, 0.30 mmol) and isoquinoline (25.8 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 8/1) afforded the title compound (41.9 mg, 1.39 × 10−4 mol, 70%) as a colorless solid. Rf = 0.31 (SiO2, cHex/EtOAc 8/1). Mp: 108.3–109.9 °C. IR (ATR): [cm−1] 3631, 2969, 2931, 1718, 1508, 1459, 1170, 903, 727, 649. 1H-NMR, COSY (300 MHz, CDCl3): δ/ppm 8.46 (d, J = 5.7 Hz, 1H, H-3), 8.24 (d, J = 8.3 Hz, 1H, H-8), 7.84–7.81 (m, 1H, H-5), 7.65 (mc, 2H, H-6,7), 7.55 (dd, J = 5.7, 0.9 Hz, 1H, H-4), 6.07 (d, J = 9.1 Hz, 1H, NH), 5.54 (dd, J = 9.1, 5.7 Hz, 1H, H-2′), 2.25–2.16 (m, 1H, H-3′), 1.44 (s, 9H, C(CH3)3), 0.96 (d, J = 6.8 Hz, 1H, CH3), 0.87 (d, J = 6.8 Hz, 1H, CH3). 13C, HSQC, HMBC (75.4 MHz, CDCl3): δ/ppm 160.4 (Cq-1), 156.2 (CO), 141.4 (C-3), 136.4 (Cq-4a), 130.1 (C-6), 127.5 (2C, C-5,7), 126.2 (Cq-8a), 125.0 (C-8), 120.1 (C-4), 79.2 (Cq(CH3)3), 55.6 (C-2′), 34.9 (CH(CH3)2), 28.6 (3C, (CH3)3), 20.5 (CH3), 17.6 (CH3) . ESI-MS (pos): m/z 301.2 (100%, [M + H]+). ESI-HRMS (pos): m/z calculated for [C18H24N2O2]+ m/z 301.1911, found 301.1909.
2-Cyclohexyl-1,3-benzothiazole (
14). This compound was prepared according to General Procedure B using
N-(cyclohexycarbonyloxy)phthalimide (82.0 mg, 0.30 mmol) and 1,3-benzothiazole (27.0 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 8/1) afforded the title compound (37.8 mg, 1.74 × 10
−4 mol, 87%) as a colorless oil.
Rf = 0.48 (SiO
2,
cHex/EtOAc 8/1). IR (ATR):
[cm
−1] 2926, 2851, 1514, 1448, 1437, 1244, 1014, 757, 728, 668.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 7.97 (dt,
J = 8.2, 1.0 Hz, 1H, H-4), 7.86–7.83 (m, 1H, H-7), 7.44 (ddd,
J = 8.3, 7.2, 1.3 Hz, 1H, H-5), 7.33 (ddd,
J = 8.3, 7.2, 1.3 Hz, 1H, H-6), 3.10 (tt,
J = 11.6, 3.6 Hz, 1H, H-1′), 2.23–2.18 (m, 2H, H-2′), 1.88–1.85 (m, 2H, H-3′), 1.76–1.67 (m, 3H, H-4′,6′), 1.52–1.38 (m, 3H, H-4′,5′).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 177.7 (C
q-2), 153.2 (C
q-3a), 134.2 (C
q-7a), 125.9 (C-5), 124.6 (C-6), 122.7 (C-4), 121.7 (C-7), 43.6 (C-1′), 33.6 (2C, C-2′,6′), 26.2 (2C, C-3′,5′), 25.7 (C-4′). ESI-MS (pos):
m/
z 218.0 (100%, [M + H]
+). The spectral data are consistent with those reported in the literature [
39].
2-(Pentan-2-yl)-1,3-benzothiazole (
15). This compound was prepared according to General Procedure B using
N-(2-methylbutyryloxy)phthalimide (78.4 mg, 0.30 mmol) and 1,3-benzothiazole (27.0 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 40/1) afforded the title compound (29.8 mg, 1.45 × 10
−4 mol, 73%) as a colorless oil.
Rf = 0.39 (SiO
2,
cHex/EtOAc 30/1). IR (ATR):
[cm
−1] 2933, 1786, 1741, 1516, 1466, 1263, 1157, 1069, 1026, 877.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 7.98 (ddd,
J = 8.1, 1.3, 0.6 Hz, 1H, H-4), 7.85 (ddd,
J = 7.9, 1.3, 0.6 Hz, 1H, H-7), 7.44 (ddd,
J = 8.1, 7.3, 1.3 Hz, 1H, H-5), 7.33 (ddd,
J = 7.9, 7.3, 1.3 Hz, 1H, H-6), 3.30 (sept,
J = 7.0 Hz, 1H, H-1′), 1.91–1.81 (m, 1H, H-3′), 1.77–1.69 (m, 1H, H-3′), 1.45 (d,
J = 6.9 Hz, 3H, H-1′), 1.39–1.34 (m, 2H, H-4′), 0.93 (t,
J = 7.3 Hz, 3H, H-5′).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 178.3 (C
q-2), 153.2 (C
q-3a), 134.2 (C
q-7a), 125.9 (C-5), 124.7 (C-6), 122.7 (C-4), 121.7 (C-7), 40.0 (C-3′), 39.4 (C-2′), 21.3 (C-1′), 20.7 (C-4′), 14.1 (C-5′). ESI-MS (pos):
m/
z 206.0 (100%, [M + H]
+). The spectral data are consistent with those reported in the literature [
40].
2-(Pent-4-en-2-yl)-1,3-benzothiazole (16). This compound was prepared according to General Procedure B using N-(pent-4-enyl-2-carbonyloxy)phthalimide (77.7 mg, 0.30 mmol) and 1,3-benzothiazole (27.0 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 40/1) afforded the title compound (35.4 mg, 1.74 × 10−4 mol, 87%) as a colorless oil. Rf = 0.52 (SiO2, cHex/EtOAc 10/1). IR (ATR): [cm−1] 3073, 2972, 2928, 2870, 1517, 1455, 1437, 916, 758, 729. 1H-NMR, COSY (300 MHz, CDCl3): δ/ppm 7.99 (ddd, J = 8.1, 1.2, 0.6 Hz, 1H, H-4), 7.85 (ddd, J = 7.9, 1.3, 0.7 Hz, 1H, H-7), 7.45 (ddd, J = 8.1, 7.2, 1.3 Hz, 1H, H-5), 7.34 (ddd, J = 7.9, 7.2, 1.3 Hz, 1H, H-6), 5.81 (dddd, J = 16.9, 10.1, 7.4, 6.6 Hz, 1H, H-4′), 5.12–5.07 (m, 2H, H-5′), 3.38 (sept, J = 7.0 Hz, 1H, H-2′), 2.69 (app dtt, J = 14.6, 6.6, 1.3 Hz, 1H, H-3′), 2.48 (app dtt, J = 14.6, 6.6, 1.3 Hz, 1H, H-3′), 1.47 (d, J = 6.9 Hz, 3H, H-1′). 13C, HSQC, HMBC (75.4 MHz, CDCl3): δ/ppm 177.1 (Cq-2), 153.2 (Cq-3a), 135.5 (C-4′), 134.8 (Cq-7a), 126.0 (C-5), 124.8 (C-6), 122.8 (C-4), 121.7 (C-7), 117.5 (C-5′), 41.7 (C-3′), 39.3 (C-2′), 20.6 (C-1′). ESI-MS (pos): m/z 204.0 (100%, [M + H]+). ESI-HRMS (pos): m/z calculated for [C12H13NS]+ m/z 204.0841, found 204.0839.
2-Cyclohexyl-pyrazine (
17). This compound was prepared according to General Procedure B using
N-(cyclohexycarbonyloxy)phthalimide (82.0 mg, 0.30 mmol) and pyrazine (16.2 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO
2,
cHex/EtOAc 5/1) afforded the title compound (13.2 mg, 8.14 × 10
−5 mol, 41%) as a colorless oil.
Rf = 0.30 (SiO
2,
cHex/EtOAc 5/1). IR (ATR):
[cm
−1] 3052, 2926, 2852, 1736, 1468, 1449, 1184, 1060, 1016, 839.
1H-NMR, COSY (300 MHz, CDCl
3): δ/ppm 8.48–8.39 (m, 2H, H-5,6), 7.88–7.77 (m, 1H, H-3), 2.74 (tt,
J = 12.1, 3.4 Hz, 1H, H-1′), 1.97–1.86 (m, 4H, H-2′,6′), 1.59–1.31 (m, 6H, H-3′,4′,5′).
13C, HSQC, HMBC (75.4 MHz, CDCl
3): δ/ppm 161.7 (C
q-1), 143.9 (CH), 143.6 (CH), 134.3 (C-3), 44.1 (C-1′), 32.5 (C-2′,6′), 26.4 (C-3′,5′), 25.9 (C-4′). ESI-MS (pos):
m/
z 163.0 (100%, [M + H]
+). The spectral data are consistent with those reported in the literature [
37].
tert-Butyl-[2-methyl-1-(pyrazin-2-yl)propyl]carbamate (18). This compound was prepared according to General Procedure B using N-(N-tert-butoxycarbonyl-l-valinyloxy)phthalimide (108.7 mg, 0.30 mmol) and pyrazine (16.2 mg, 0.20 mmol). Purifications of the crude product by flash column chromatography (SiO2, cHex/EtOAc 5/1) afforded the title compound (20.3 mg, 8.1 × 10−5 mol, 40%) as a colorless oil. Rf = 0.14 (SiO2, cHex/EtOAc 3/1). IR (ATR): [cm−1] 3057, 2968, 2931, 1702, 1522, 1275, 1171, 1017, 744, 667, 548. 1H-NMR, COSY (300 MHz, CDCl3): δ/ppm 8.52–8.51 (m, 2H, H-5,6), 8.47 (d, J = 2.4 Hz, 1H, H-3), 5.50 (d, J = 8.9 Hz, 1H, NH), 4.64 (t, J = 7.8 Hz, 1H, H-1′), 2.11 (sept, J = 6.8 Hz, 1H, H-2′), 1.43 (s, 9H, (CH3)3), 0.94 (d, J = 6.8 Hz, 1H, CH3), 0.87 (d, J = 6.8 Hz, 1H, CH3). 13C, HSQC, HMBC (75.4 MHz, CDCl3): δ/ppm 156.0 (CO), 155.7 (Cq-2), 144.1 (2C, C-5,6), 143.4 (C-3), 79.7 (Cq(CH3)3), 58.5 (C-1′), 33.9 (C-2′), 28.5 (3C, (CH3)3), 19.3 (CH3), 18.3 (CH3). ESI-MS (pos): m/z 252.1 (100%, [M + H]+), 274.1 (17%, [M + Na]+). ESI-HRMS (pos): m/z calculated for [C13H21N3O2]+ m/z 252.1703, found 252.1707.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


