
The Melatonin Signaling Pathway in a Long-Term Memory In Vitro Study



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
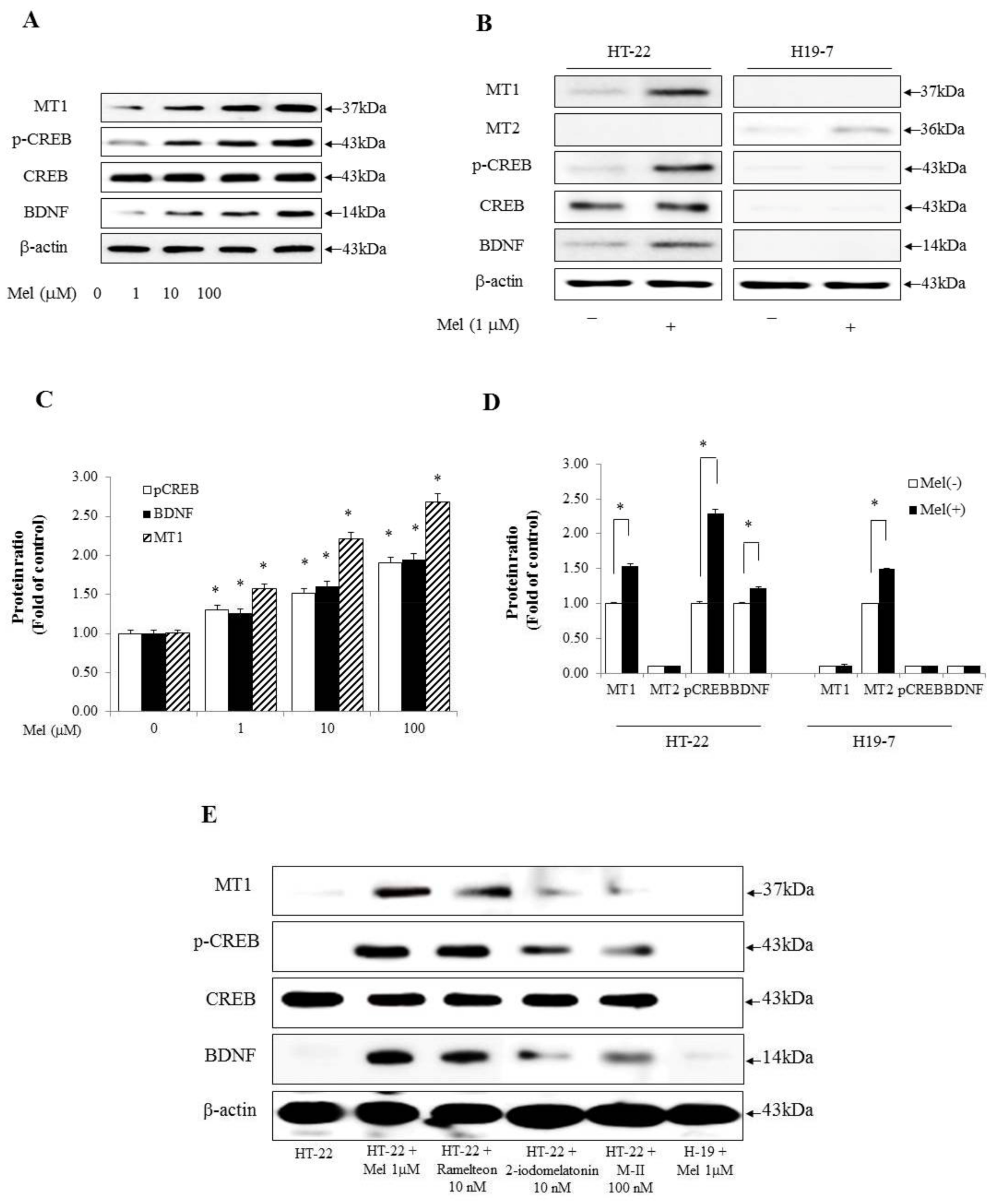
2.1. Melatonin Induces the p-CREB and BDNF Associated with Long-Term Memory Processing and Appears to Be Related to the Difference in Efficacy of Melatonin Receptor Agonists between HT-22 Cells and H19-7 Cells
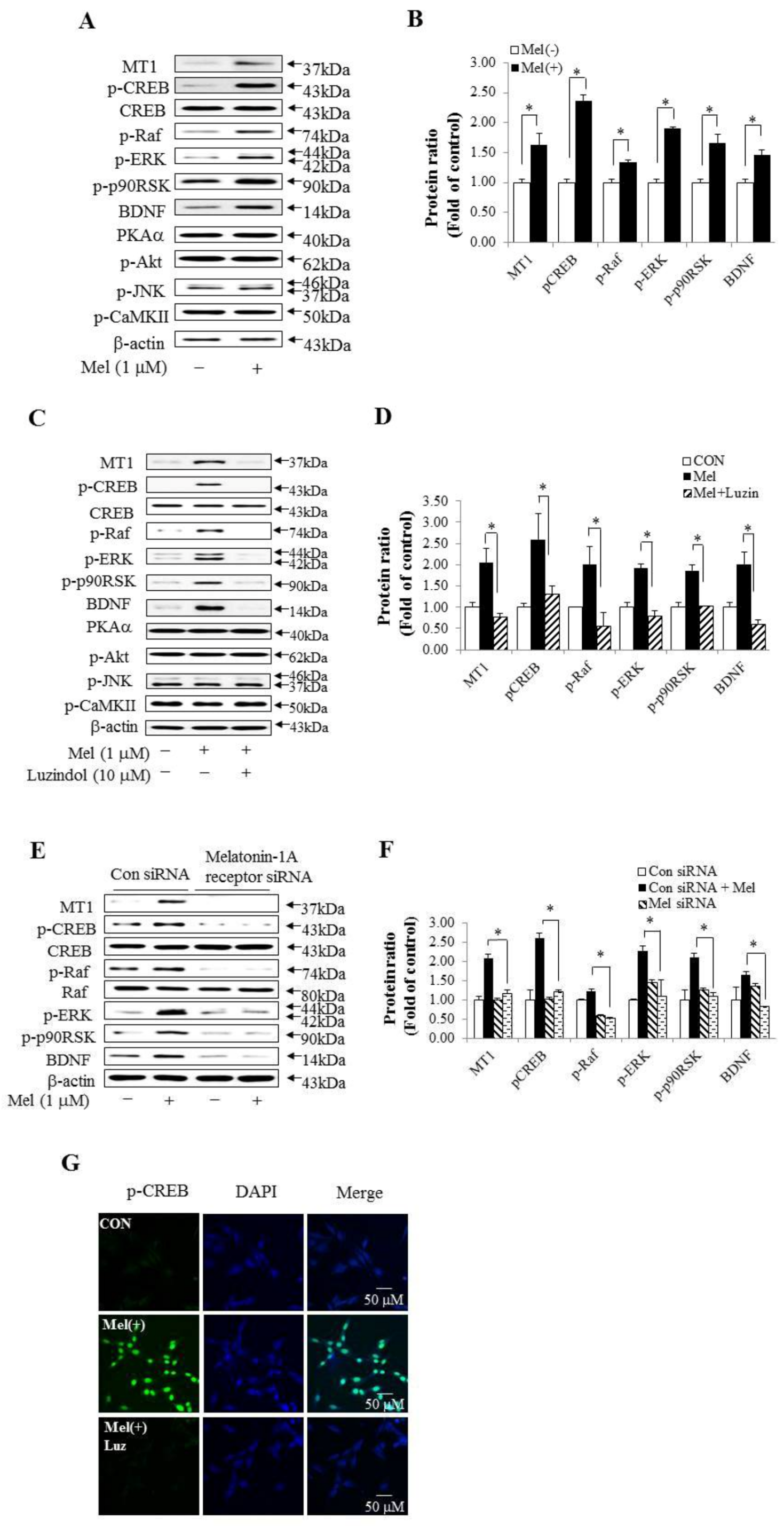
2.2. Melatonin May Be Related to the CREB Signaling Pathway in HT-22 Cells
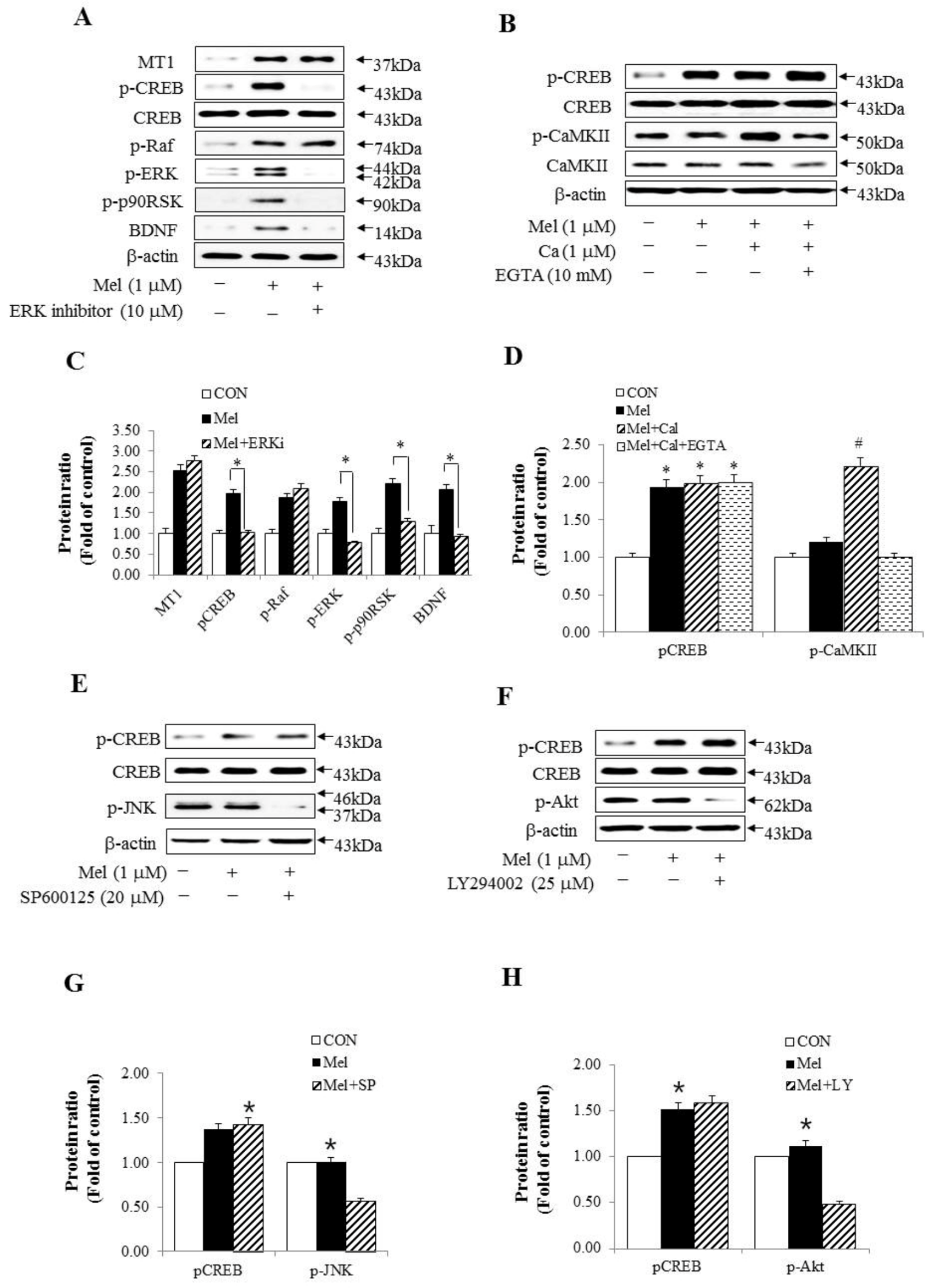
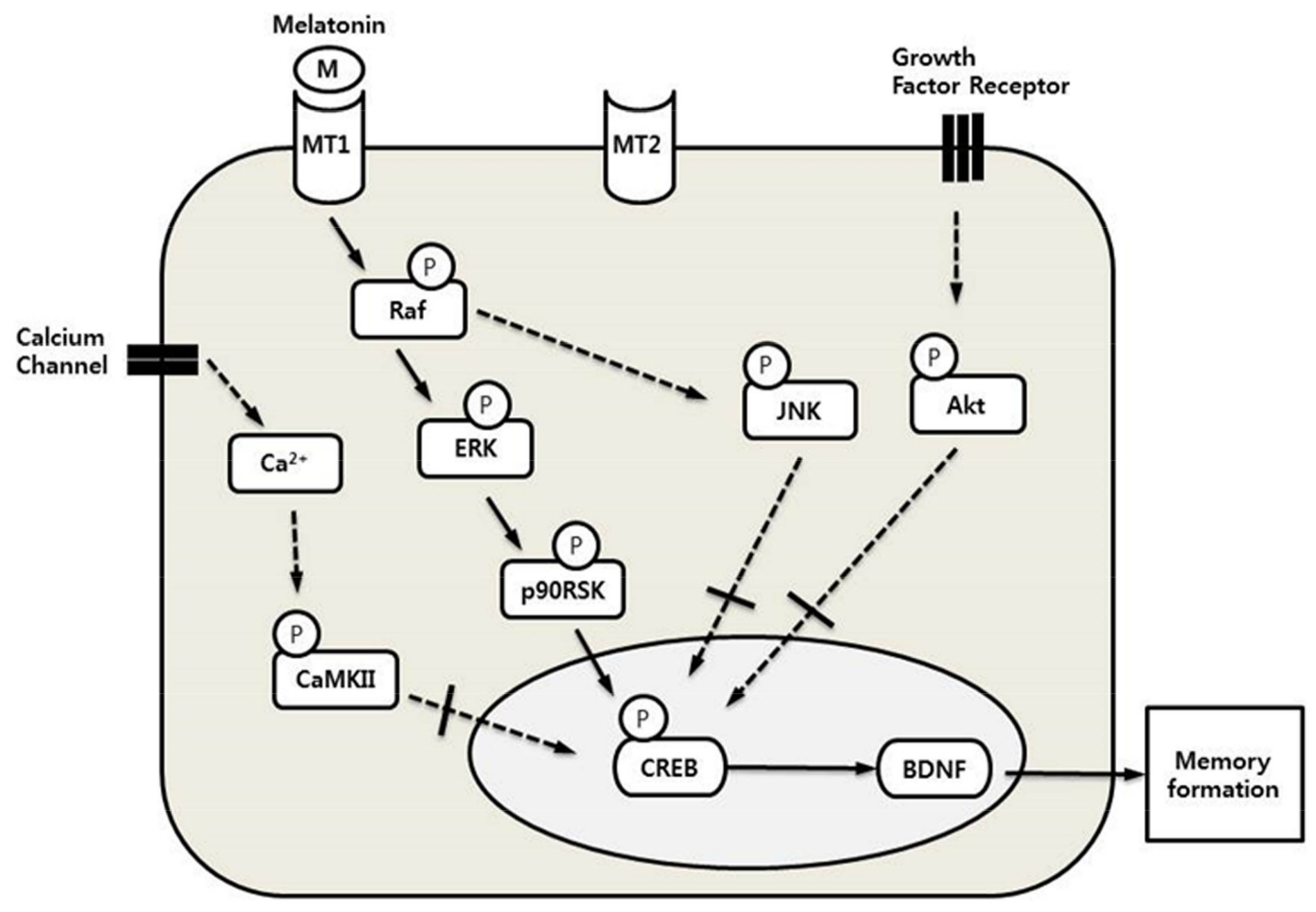
2.3. Other Signaling Pathways, Except for the Raf–ERK Pathway, Did Not Affect the Expression of pCREB through Melatonin
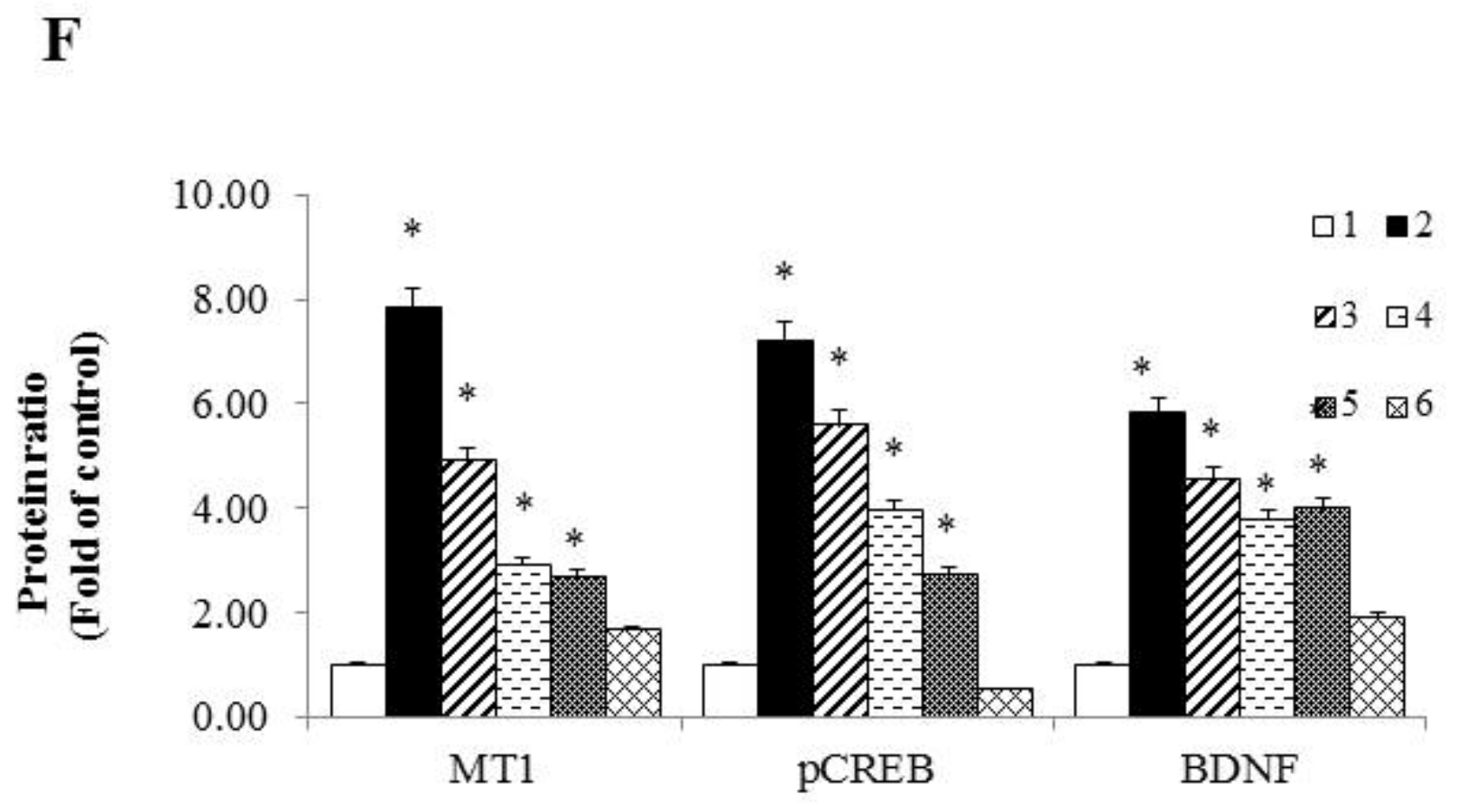
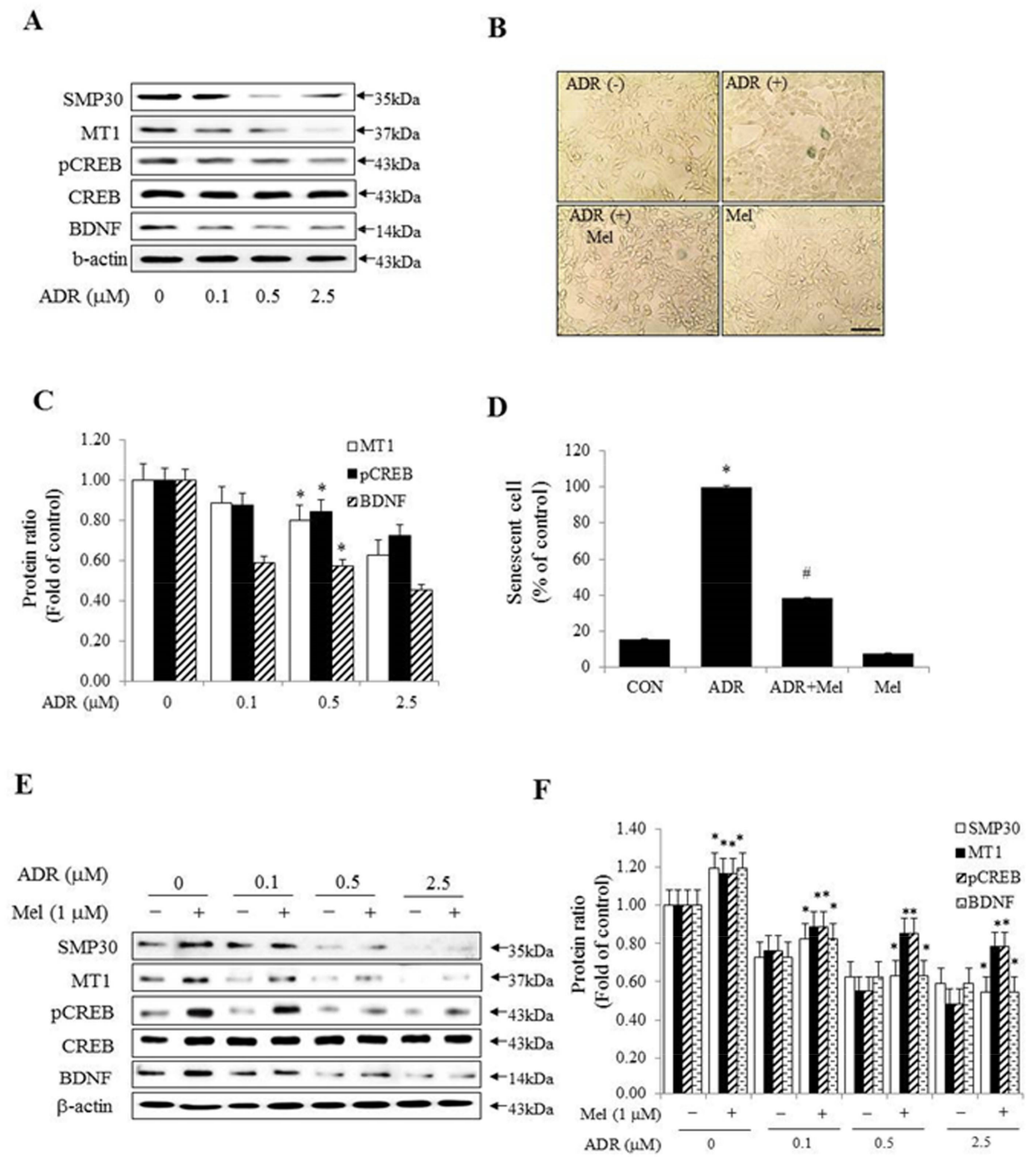
2.4. Melatonin Inhibited Senescence in ADR-Treated HT-22 Cells and Improved Memory Processing
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Senescence-Associated β-Galactosidase (SA-β-gal) Staining
4.4. Western Blot
4.5. Transfection of siRNA
4.6. Immunofluorescence
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hardeland, R. Melatonin and the theories of aging: A critical appraisal of melatonin’s role in antiaging mechanisms. J. Pineal Res. 2013, 55, 325–356. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.A. Decreased melatonin biosynthesis, calcium flux, pineal gland calcification and aging: A hypothetical framework. Gerontology 1993, 39, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. The pineal gland and melatonin in relation to aging: A summary of the theories and of the data. Exp. Gerontol. 1995, 30, 199–212. [Google Scholar] [CrossRef]
- O’Neal-Moffitt, G.; Pilli, J.; Kumar, S.S.; Olcese, J. Genetic deletion of MT(1)/MT(2) melatonin receptors enhances murine cognitive and motor performance. Neuroscience 2014, 277, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ni, C.; Li, Z.; Yang, N.; Zhou, Y.; Rong, X.; Qian, M.; Chui, D.; Guo, X. Prophylactic Melatonin Attenuates Isoflurane-induced Cognitive Impairment in Aged Rats through Hippocampal Melatonin Receptor 2—CREB Signalling. Basic Clin. Pharmacol. Toxicol. 2016, 120, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Bazwinsky-Wutschke, I.; Wolgast, S.; Muhlbauer, E.; Albrecht, E.; Peschke, E. Phosphorylation of cyclic AMP-response element-binding protein (CREB) is influenced by melatonin treatment in pancreatic rat insulinoma beta-cells (INS-1). J. Pineal Res. 2012, 53, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, B.; Angeloni, D.; Dominguez-Lopez, S.; Calderoni, S.; Mauro, A.; Fraschini, F.; Descarries, L.; Gobbi, G. Anatomical and cellular localization of melatonin MT1 and MT2 receptors in the adult rat brain. J. Pineal Res. 2015, 58, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Nonno, R.; Lucini, V.; Pannacci, M.; Mazzucchelli, C.; Angeloni, D.; Fraschini, F.; Stankov, B.M. Pharmacological characterization of the human melatonin Mel1a receptor following stable transfection into NIH3T3 cells. Br. J. Pharmacol. 1998, 124, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, C.; Pannacci, M.; Nonno, R.; Lucini, V.; Fraschini, F.; Stankov, B.M. The melatonin receptor in the human brain: Cloning experiments and distribution studies. Brain Res. Mol. Brain Res. 1996, 39, 117–126. [Google Scholar] [CrossRef]
- Wan, Q.; Man, H.Y.; Liu, F.; Braunton, J.; Niznik, H.B.; Pang, S.F.; Brown, G.M.; Wang, Y.T. Differential modulation of GABAA receptor function by Mel1a and Mel1b receptors. Nat. Neurosci. 1999, 2, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Musshoff, U.; Riewenherm, D.; Berger, E.; Fauteck, J.D.; Speckmann, E.J. Melatonin receptors in rat hippocampus: Molecular and functional investigations. Hippocampus 2002, 12, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Zhang, Y. Melatonin: A well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 2014, 57, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Yu, M.; Luo, Z.; Dai, M.; Han, J.; Xiu, R.; Yang, Z. Intracellular signaling pathways involved in cell growth inhibition of human umbilical vein endothelial cells by melatonin. J. Pineal Res. 2008, 44, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. The ageing pineal gland and its physiological consequences. Bioessays 1992, 14, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Pandi-Perumal, S.R.; Maestroni, G.J.; Esquifino, A.I.; Hardeland, R.; Cardinali, D.P. Role of melatonin in neurodegenerative diseases. Neurotox. Res. 2005, 7, 293–318. [Google Scholar] [CrossRef] [PubMed]
- Corrales, A.; Martinez, P.; Garcia, S.; Vidal, V.; Garcia, E.; Florez, J.; Sanchez-Barcelo, E.J.; Martinez-Cue, C.; Rueda, N. Long-term oral administration of melatonin improves spatial learning and memory and protects against cholinergic degeneration in middle-aged Ts65Dn mice, a model of Down syndrome. J. Pineal Res. 2013, 54, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Ma, C.; Qu, H.; Fan, W.; Pang, J.; He, H. Differential effects of melatonin on hippocampal neurodegeneration in different aged accelerated senescence prone mouse-8. Neuro Endocrinol. Lett. 2008, 29, 91–99. [Google Scholar] [PubMed]
- Flood, J.F.; Morley, J.E. Learning and memory in the SAMP8 mouse. Neurosci. Biobehav. Rev. 1998, 22, 1–20. [Google Scholar] [CrossRef]
- Yoo, D.Y.; Kim, W.; Lee, C.H.; Shin, B.N.; Nam, S.M.; Choi, J.H.; Won, M.H.; Yoon, Y.S.; Hwang, I.K. Melatonin improves D-galactose-induced aging effects on behavior, neurogenesis, and lipid peroxidation in the mouse dentate gyrus via increasing pCREB expression. J. Pineal Res. 2012, 52, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Bekinschtein, P.; Cammarota, M.; Igaz, L.M.; Bevilaqua, L.R.; Izquierdo, I.; Medina, J.H. Persistence of long-term memory storage requires a late protein synthesis- and BDNF- dependent phase in the hippocampus. Neuron 2007, 53, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J.; Kogan, J.H.; Frankland, P.W.; Kida, S. CREB and memory. Annu. Rev. Neurosci. 1998, 21, 127–248. [Google Scholar] [CrossRef] [PubMed]
- Bourtchuladze, R.; Frenguelli, B.; Blendy, J.; Cioffi, D.; Schutz, G.; Silva, A.J. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 1994, 79, 59–68. [Google Scholar] [CrossRef]
- Saura, C.A.; Valero, J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev. Neurosci. 2011, 22, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Delghandi, M.P.; Johannessen, M.; Moens, U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal. 2005, 17, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Wati, H.; Qiao, C.; Arita, J.; Kanba, S. Age-related disturbance of memory and CREB phosphorylation in CA1 area of hippocampus of rats. Brain Res. 2005, 1054, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Wu, M.; Citron, B.A.; Palazzo, R.E.; Festoff, B.W. Rapid tau aggregation and delayed hippocampal neuronal death induced by persistent thrombin signaling. J. Biol. Chem. 2003, 278, 37681–37689. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.S.; Li, B.; Lee, D.S.; Kim, K.H.; Lee, I.K.; Lee, K.R.; Kim, Y.C. Cytoprotective and anti-inflammatory effects of spinasterol via the induction of heme oxygenase-1 in murine hippocampal and microglial cell lines. Int. Immunopharmacol. 2010, 10, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Brossaud, J.; Roumes, H.; Moisan, M.P.; Pallet, V.; Redonnet, A.; Corcuff, J.B. Retinoids and glucocorticoids target common genes in hippocampal HT22 cells. J. Neurochem. 2013, 125, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, J.J.; Inda, C.; Senin, S.; Maccarrone, G.; Refojo, D.; Giacomini, D.; Turck, C.W.; Holsboer, F.; Arzt, E.; Silberstein, S. B-Raf and CRHR1 internalization mediate biphasic ERK1/2 activation by CRH in hippocampal HT22 Cells. Mol. Endocrinol. 2013, 27, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Morgan, P.J.; Barrett, P.; Howell, H.E.; Helliwell, R. Melatonin receptors: Localization, molecular pharmacology and physiological significance. Neurochem. Int. 1994, 24, 101–146. [Google Scholar] [CrossRef]
- Nonno, R.; Lucini, V.; Stankov, B.; Fraschini, F. 2-[125I]iodomelatonin binding sites in the bovine hippocampus are not sensitive to guanine nucleotides. Neurosci. Lett. 1995, 194, 113–116. [Google Scholar] [CrossRef]
- Nishiyama, K.; Hirai, K. In vitro comparison of duration of action of melatonin agonists on melatonin MT1 receptor: Possible link between duration of action and dissociation rate from receptor. Eur. J. Pharmacol. 2015, 757, 42–52. [Google Scholar] [CrossRef] [PubMed]
- McNulty, S.; Ross, A.W.; Barrett, P.; Hastings, M.H.; Morgan, P.J. Melatonin regulates the phosphorylation of CREB in ovine pars tuberalis. J. Neuroendocrinol. 1994, 6, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Richter, H.G.; Torres-Farfan, C.; Garcia-Sesnich, J.; Abarzua-Catalan, L.; Henriquez, M.G.; Alvarez-Felmer, M.; Gaete, F.; Rehren, G.E.; Seron-Ferre, M. Rhythmic expression of functional MT1 melatonin receptors in the rat adrenal gland. Endocrinology 2008, 149, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Yeung, T.L.; Yip, K.P.; Pradeep, S.; Balasubramanian, L.; Liu, J.; Wong, K.K.; Mangala, L.S.; Armaiz-Pena, G.N.; Lopez-Berestein, G.; et al. Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat. Commun. 2014, 5, 5092. [Google Scholar] [CrossRef] [PubMed]
- Gusek, W. Histology of the pineal gland in the elderly human. Aktuelle Gerontol. 1983, 13, 111–114. [Google Scholar] [PubMed]
- Schliebs, R.; Arendt, T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural Transm. 2006, 113, 1625–1644. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, A.; Mathias, R.S.; McCormick, J.A.; Dallman, M.F.; Pearce, D. Glucocorticoids prolong Ca(2+) transients in hippocampal-derived H19-7 neurons by repressing the plasma membrane Ca(2+)-ATPase-1. Mol. Endocrinol. 2002, 16, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, B.H.; Koshland, D.E., Jr. Induction and expression of long- and short-term neurosecretory potentiation in a neural cell line. Neuron 1990, 5, 875–880. [Google Scholar] [CrossRef]
- Dubocovich, M.L.; Rivera-Bermudez, M.A.; Gerdin, M.J.; Masana, M.I. Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front. Biosci. 2003, 8, d1093-108. [Google Scholar] [CrossRef] [PubMed]
- Pandi-Perumal, S.R.; Trakht, I.; Srinivasan, V.; Spence, D.W.; Maestroni, G.J.; Zisapel, N.; Cardinali, D.P. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Witt-Enderby, P.A.; Bennett, J.; Jarzynka, M.J.; Firestine, S.; Melan, M.A. Melatonin receptors and their regulation: Biochemical and structural mechanisms. Life Sci. 2003, 72, 2183–2198. [Google Scholar] [CrossRef]
- Drew, J.E.; Barrett, P.; Mercer, J.G.; Moar, K.M.; Canet, E.; Delagrange, P.; Morgan, P.J. Localization of the melatonin-related receptor in the rodent brain and peripheral tissues. J. Neuroendocrinol. 2001, 13, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Dubocovich, M.L.; Yun, K.; Al-Ghoul, W.M.; Benloucif, S.; Masana, M.I. Selective MT2 melatonin receptor antagonists block melatonin-mediated phase advances of circadian rhythms. FASEB J. 1998, 12, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.H.; Kandel, E.R.; Si, K. The persistence of long-term memory: A molecular approach to self-sustaining changes in learning-induced synaptic growth. Neuron 2004, 44, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.L.; Pittenger, C.; Morozov, A.; Martin, K.C.; Scanlin, H.; Drake, C.; Kandel, E.R. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron 2001, 32, 123–140. [Google Scholar] [CrossRef]
- Mizuno, M.; Yamada, K.; Maekawa, N.; Saito, K.; Seishima, M.; Nabeshima, T. CREB phosphorylation as a molecular marker of memory processing in the hippocampus for spatial learning. Behav. Brain Res. 2002, 133, 135–141. [Google Scholar] [CrossRef]
- Wong, S.T.; Athos, J.; Figueroa, X.A.; Pineda, V.V.; Schaefer, M.L.; Chavkin, C.C.; Muglia, L.J.; Storm, D.R. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 1999, 23, 787–798. [Google Scholar] [CrossRef]
- Orban, P.C.; Chapman, P.F.; Brambilla, R. Is the Ras-MAPK signalling pathway necessary for long-term memory formation? Trends Neurosci. 1999, 22, 38–44. [Google Scholar] [CrossRef]
- Chen, J.T.; Lu, D.H.; Chia, C.P.; Ruan, D.Y.; Sabapathy, K.; Xiao, Z.C. Impaired long-term potentiation in c-Jun N-terminal kinase 2-deficient mice. J. Neurochem. 2005, 93, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Horwood, J.M.; Dufour, F.; Laroche, S.; Davis, S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur. J. Neurosci. 2006, 23, 3375–3384. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Kandel, E.R. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn. Mem. 1994, 1, 74–82. [Google Scholar] [PubMed]
- Brindle, P.K.; Montminy, M.R. The CREB family of transcription activators. Curr. Opin. Genet. Dev. 1992, 2, 199–204. [Google Scholar] [CrossRef]
- Bito, H.; Deisseroth, K.; Tsien, R.W. CREB phosphorylation and dephosphorylation: A Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 1996, 87, 1203–1214. [Google Scholar] [CrossRef]
- Xie, H.; Rothstein, T.L. Protein kinase C mediates activation of nuclear cAMP response element-binding protein (CREB) in B lymphocytes stimulated through surface Ig. J. Immunol. 1995, 154, 1717–1723. [Google Scholar] [PubMed]
- Don, J.; Stelzer, G. The expanding family of CREB/CREM transcription factors that are involved with spermatogenesis. Mol. Cell. Endocrinol. 2002, 187, 115–124. [Google Scholar] [CrossRef]
- Du, K.; Montminy, M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998, 273, 32377–32379. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.J.; Manji, H.K.; Charney, D.S. Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology 2008, 33, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, C.; Duman, R.S. Stress, depression, and neuroplasticity: A convergence of mechanisms. Neuropsychopharmacology 2008, 33, 88–109. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.S.; Quamina, A. Extracellular receptor kinase and cAMP response element binding protein activation in the neonatal rat heart after perinatal cocaine exposure. Pediatr. Res. 2004, 56, 947–952. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yamamori, E.; Asai, M.; Yoshida, M.; Takano, K.; Itoi, K.; Oiso, Y.; Iwasaki, Y. Calcium/calmodulin kinase IV pathway is involved in the transcriptional regulation of the corticotropin-releasing hormone gene promoter in neuronal cells. J. Mol. Endocrinol. 2004, 33, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Thompson, M.A.; Greenberg, M.E. CREB: A Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 1991, 252, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Rawashdeh, O.; Maronde, E. The hormonal Zeitgeber melatonin: Role as a circadian modulator in memory processing. Front. Mol. Neurosci. 2012, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; von Gall, C.; Pieschl, R.L.; Gribkoff, V.K.; Stehle, J.H.; Reppert, S.M.; Weaver, D.R. Targeted disruption of the mouse Mel(1b) melatonin receptor. Mol. Cell. Biol. 2003, 23, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.S.; Rigon, A.P.; Leal, R.B.; Rossi, F.M. The activation of ERK1/2 and p38 mitogen-activated protein kinases is dynamically regulated in the developing rat visual system. Int. J. Dev. Neurosci. 2008, 26, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J., 3rd; Govindarajan, A.; Jung, H.Y.; Kang, H.; Tonegawa, S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 2004, 116, 467–479. [Google Scholar] [CrossRef]
- Soderling, T.R. Calcium/calmodulin-dependent protein kinase II: Role in learning and memory. Mol. Cell. Biochem. 1993, 127–128, 93–101. [Google Scholar] [CrossRef]
- Sun, C.Y.; Qi, S.S.; Lou, X.F.; Sun, S.H.; Wang, X.; Dai, K.Y.; Hu, S.W.; Liu, N.B. Changes of learning, memory and levels of CaMKII, CaM mRNA, CREB mRNA in the hippocampus of chronic multiple-stressed rats. Chin. Med. J. 2006, 119, 140–147. [Google Scholar] [PubMed]
- Maronde, E.; Schomerus, C.; Stehle, J.H.; Korf, H.W. Control of CREB phosphorylation and its role for induction of melatonin synthesis in rat pinealocytes. Biol. Cell. 1997, 89, 505–511. [Google Scholar] [CrossRef]
- Koch, M.; Mauhin, V.; Stehle, J.H.; Schomerus, C.; Korf, H.W. Dephosphorylation of pCREB by protein serine/threonine phosphatases is involved in inactivation of Aanat gene transcription in rat pineal gland. J. Neurochem. 2003, 85, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.N.; Liu, R.Y.; van Heerikhuize, J.; Hofman, M.A.; Swaab, D.F. Alterations in the circadian rhythm of salivary melatonin begin during middle-age. J. Pineal Res. 2003, 34, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Maestroni, G.J.; Cardinali, D.P.; Esquifino, A.I.; Perumal, S.R.; Miller, S.C. Melatonin, immune function and aging. Immun. Ageing 2005, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Richardson, B.A.; Johnson, L.Y.; Ferguson, B.N.; Dinh, D.T. Pineal melatonin rhythm: Reduction in aging Syrian hamsters. Science 1980, 210, 1372–1373. [Google Scholar] [CrossRef] [PubMed]
- Baydas, G.; Ozveren, F.; Akdemir, I.; Tuzcu, M.; Yasar, A. Learning and memory deficits in rats induced by chronic thinner exposure are reversed by melatonin. J. Pineal Res. 2005, 39, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Gorfine, T.; Zisapel, N. Melatonin and the human hippocampus, a time dependent interplay. J. Pineal Res. 2007, 43, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Kim, A.J.; Kim, H.J.; Jee, H.J.; Kim, M.; Yoo, Y.H.; Yun, J. Melatonin suppresses doxorubicin-induced premature senescence of A549 lung cancer cells by ameliorating mitochondrial dysfunction. J. Pineal Res. 2012, 53, 335–343. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sung, J.-Y.; Bae, J.-H.; Lee, J.-H.; Kim, Y.-N.; Kim, D.-K. The Melatonin Signaling Pathway in a Long-Term Memory In Vitro Study. Molecules 2018, 23, 737. https://doi.org/10.3390/molecules23040737
Sung J-Y, Bae J-H, Lee J-H, Kim Y-N, Kim D-K. The Melatonin Signaling Pathway in a Long-Term Memory In Vitro Study. Molecules. 2018; 23(4):737. https://doi.org/10.3390/molecules23040737
Chicago/Turabian StyleSung, Jin-Young, Ji-Hyun Bae, Jong-Ha Lee, Yoon-Nyun Kim, and Dae-Kwang Kim. 2018. "The Melatonin Signaling Pathway in a Long-Term Memory In Vitro Study" Molecules 23, no. 4: 737. https://doi.org/10.3390/molecules23040737
APA StyleSung, J.-Y., Bae, J.-H., Lee, J.-H., Kim, Y.-N., & Kim, D.-K. (2018). The Melatonin Signaling Pathway in a Long-Term Memory In Vitro Study. Molecules, 23(4), 737. https://doi.org/10.3390/molecules23040737