Facilitating Anti-Cancer Combinatorial Drug Discovery by Targeting Epistatic Disease Genes


Abstract
1. Introduction
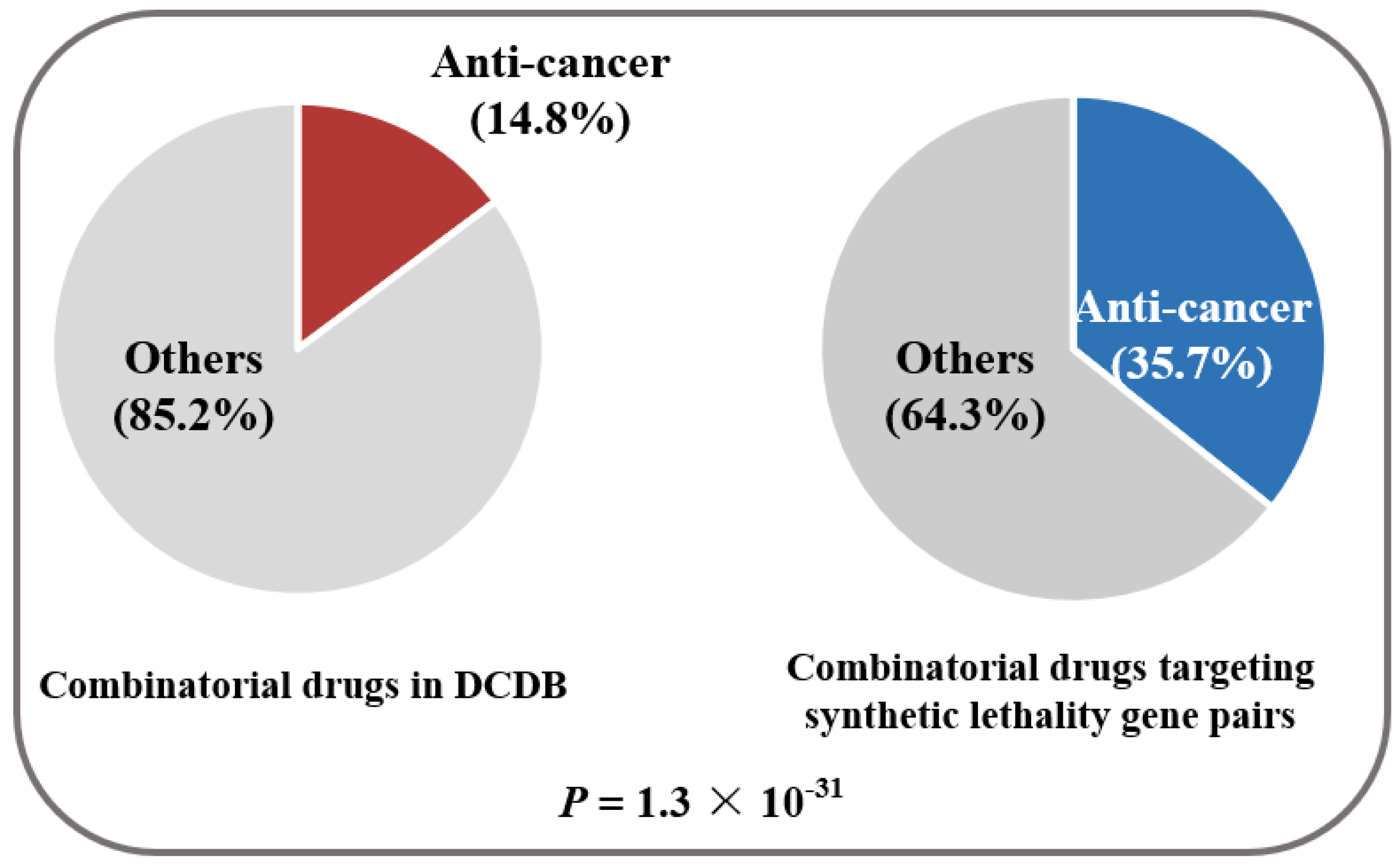
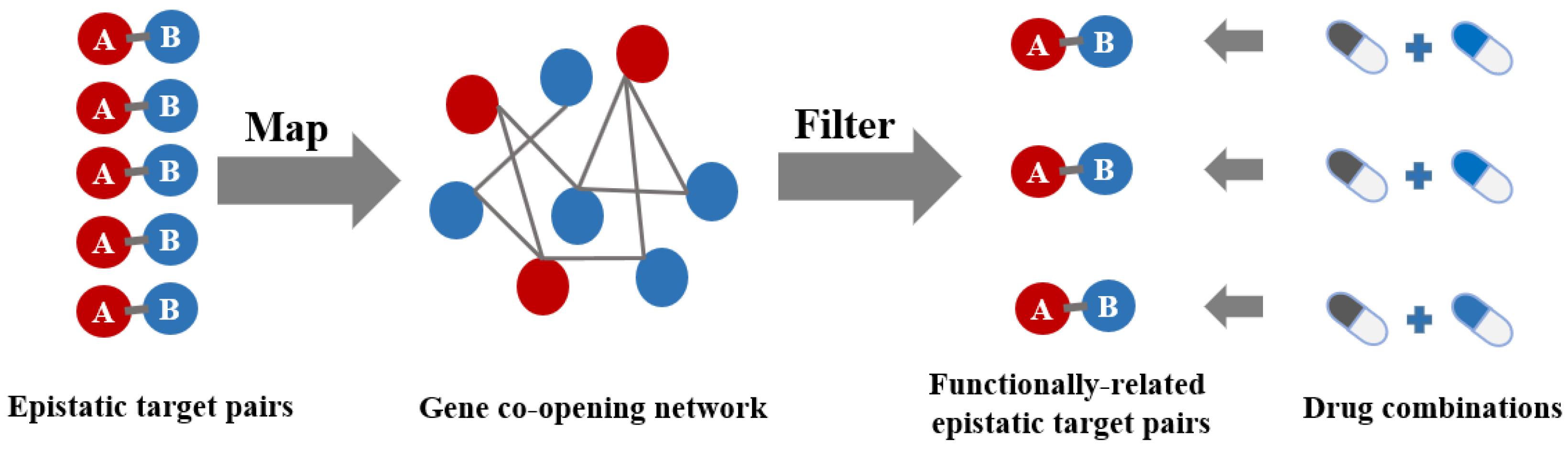
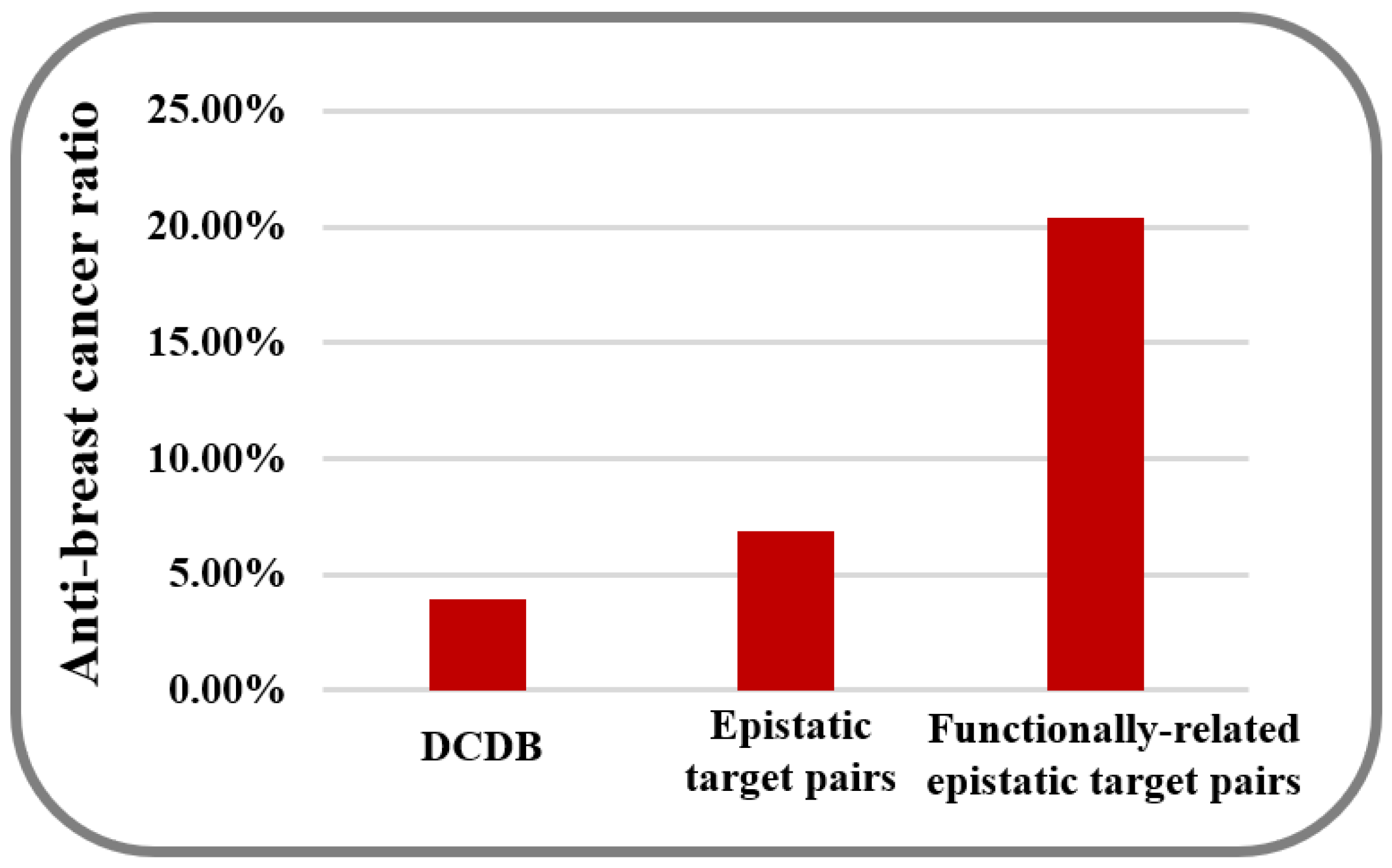
2. Results and Discussion
3. Materials and Methods
3.1. Breast Cancer-Related GWAS Dataset
3.2. Breast Cancer Genes
3.3. Information on Drugs
3.4. Information on Combinatorial Drugs
3.5. Gene Co-Opening Network
3.6. Genetic Epistasis Detection in GWAS
3.7. Permutation Test
3.8. Cytotoxicity Assays
3.8.1. Cell culture and Reagents
3.8.2. Cytotoxicity Assays
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, D.; Liu, X.; Yang, Y.; Yang, H.; Lu, P. Systematic synergy modeling: Understanding drug synergy from a systems biology perspective. BMC Syst. Biol. 2015, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Perez, J.; Kurzrock, R. Combining targeted therapies: Practical issues to consider at the bench and bedside. Oncologist 2010, 15, 37–50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frangione, M.L.; Lockhart, J.H.; Morton, D.T.; Pava, L.M.; Blanck, G. Anticipating designer drug-resistant cancer cells. Drug Discov. Today 2015, 20, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, Q.; Yu, G.; Gai, W.; Li, Y.; Chen, X. Dcdb 2.0: A major update of the drug combination database. Database (Oxford) 2014, 2014, bau124. [Google Scholar] [CrossRef] [PubMed]
- Feala, J.D.; Cortes, J.; Duxbury, P.M.; Piermarocchi, C.; McCulloch, A.D.; Paternostro, G. Systems approaches and algorithms for discovery of combinatorial therapies. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.; Yang, J.; Karan, C.; Menden, M.P.; Costello, J.C.; Tang, H.; Xiao, G.; Li, Y.; Allen, J.; Zhong, R.; et al. A community computational challenge to predict the activity of pairs of compounds. Nat. Biotechnol. 2014, 32, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, R.R.; Dubé, M.P.; Rouleau, G.A.; Orr, A.C.; Samuels, M.E. Human monogenic disorders-A source of novel drug targets. Nat. Rev. Genet. 2006, 7, 249. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Fu, L.Y.; Zhang, H.Y. Can medical genetics and evolutionary biology inspire drug target identification? Trends Mol. Med. 2012, 18, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Sanseau, P.; Agarwal, P.; Barnes, M.R.; Pastinen, T.; Richards, J.B.; Cardon, L.R.; Mooser, V. Use of genome-wide association studies for drug repositioning. Nat. Biotechnol. 2012, 30, 317. [Google Scholar] [CrossRef] [PubMed]
- Plenge, R.M.; Scolnick, E.M.; Altshuler, D. Validating therapeutic targets through human genetics. Nat Rev. Drug Discov. 2013, 12, 581. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Xiong, L.; Chen, J.; Zhang, H.Y. Genetics-directed drug discovery for combating mycobacterium tuberculosis infection. J. Biomol. Struct. Dyn. 2017, 35, 616. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.G.; Marcotte, E.M. A probabilistic view of gene function. Nat. Genet. 2004, 36, 559. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Wang, Z.Y.; Chu, X.Y.; Zhang, H.Y. Evolutionary and genetic features of drug targets. Med. Res. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Jeng, E.E.; Hess, G.T.; Morgens, D.W.; Li, A.; Bassik, M.C. Synergistic drug combinations for cancer identified in a crispr screen for pairwise genetic interactions. Nat. Biotechnol. 2017, 35, 463. [Google Scholar] [CrossRef] [PubMed]
- Jing, G.; Hui, L.; Jie, Z. Synlethdb: Synthetic lethality database toward discovery of selective and sensitive anticancer drug targets. Nucleic Acids Res. 2016, 44, D1011–D1017. [Google Scholar]
- Wagner, A.H.; Coffman, A.C.; Ainscough, B.J.; Spies, N.C.; Skidmore, Z.L.; Campbell, K.M.; Krysiak, K.; Pan, D.; McMichael, J.F.; Eldred, J.M.; et al. Dgidb 2.0: Mining clinically relevant drug-gene interactions. Nucleic Acids Res. 2016, 44, D1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. Drugbank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Qin, C.; Li, Y.H.; Tao, L.; Zhou, J.; Yu, C.Y.; Xu, F.; Chen, Z.; Zhu, F.; Chen, Y.Z. Therapeutic target database update 2016: Enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res. 2016, 44, D1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Kraft, P.; Jacobs, K.B.; Cox, D.G.; Yeager, M.; Hankinson, S.E.; Wacholder, S.; Wang, Z.; Welch, R.; Hutchinson, A.; et al. A genome-wide association study identifies alleles in fgfr2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007, 39, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Bush, W.S.; Dudek, S.M.; Ritchie, M.D. Parallel multifactor dimensionality reduction: A tool for the large-scale analysis of gene-gene interactions. Bioinformatics 2006, 22, 2173. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.S.; Yang, C.; Wan, X.; Yu, W. Gboost: A gpu-based tool for detecting gene-gene interactions in genome-wide case control studies. Bioinformatics 2011, 27, 1309–1310. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation plink: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- SchüPbach, T.; Xenarios, I.; Bergmann, S.; Kapur, K. Fastepistasis: A high performance computing solution for quantitative trait epistasis. Bioinformatics 2010, 26, 1468–1469. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Yang, C.; Yang, Q.; Xue, H.; Tang, N.L.S.; Yu, W. Predictive rule inference for epistatic interaction detection in genome-wide association studies. Bioinformatics 2010, 26, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Kelly, R.; Romdhane, R. Antepiseeker: Detecting epistatic interactions for case-control studies using a two-stage ant colony optimization algorithm. BMC Res Notes 2010, 3, 117. [Google Scholar]
- Wright, M.N.; Ziegler, A. Ranger: A fast implementation of random forests for high dimensional data in c++ and r. J. Stat. Softw. 2017, 077, 1–7. [Google Scholar] [CrossRef]
- Zhang, Y. A novel bayesian graphical model for genome-wide multi-snp association mapping. Genet. Epidemiol. 2012, 36, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.G.; Barnes, K.C.; Bright, T.J.; Wang, S.A. The genetic association database. Nat. Genet. 2004, 36, 431. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online mendelian inheritance in man (omim), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, D514–D517. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. Clinvar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–985. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Liu, Z.; Wang, P.; Wong, M.P.; Nelson, M.R.; Kocher, J.P.; Yeager, M.; Sham, P.C.; Chanock, S.J.; Xia, Z.; et al. Gwasdb v2: An update database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 2016, 44, D869–876. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The nhgri gwas catalog, a curated resource of snp-trait associations. Nucleic Acids Res. 2014, 42, D1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.F.; Chen, C.Y.; Su, S.C.; Li, C.Y.; Wu, K.J.; Wang, H.W.; Cheng, W.C. Driverdbv2: A database for human cancer driver gene research. Nucleic Acids Res. 2016, 44, D975–979. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. Cosmic: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, X.; Thijssen, B.; Das, J.; Lipkin, S.M.; Yu, H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat. Biotechnol. 2012, 30, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Aronson, A.R. Effective mapping of biomedical text to the umls metathesaurus: The metamap program. Proc. AMIA Symp. 2001, 2001, 17. [Google Scholar]
- Liu, C.C.; Tseng, Y.T.; Li, W.; Wu, C.Y.; Mayzus, I.; Rzhetsky, A.; Sun, F.; Waterman, M.; Chen, J.J.W.; Chaudhary, P.M. Diseaseconnect: A comprehensive web server for mechanism-based disease-disease connections. Nucleic Acids Res. 2014, 42, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zarin, D.A.; Tse, T.; Williams, R.J.; Califf, R.M.; Ide, N.C. The clinicaltrials.Gov results database--update and key issues. N. Engl. J. Med. 2011, 364, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S. Synergistic Inhibition of Breast Cell Proliferation by Several Drugs and the Underlying Mechanisms. Master’s Thesis, Shandong University of Technology, Zibo, China, 4 April 2017. [Google Scholar]
- Wu, Y.S.; Quan, Y.; Zhang, D.X.; Liu, D.W.; Zhang, X.Z. Synergistic inhibition of breast cancer cell growth by an epigenome-targeting drug and a tyrosine kinase inhibitor. Biol. Pharm. Bull. 2017, 40, 1747–1753. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, D.; Wu, B.; Quan, Y.; Liu, D.; Li, Y.; Zhang, X. Synergistic activity of an antimetabolite drug and tyrosine kinase inhibitors against breast cancer cells. Chem. Pharm. Bull. (Tokyo) 2017, 65, 768. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, W.; Wang, M.; Sun, J.; Wang, Y.; Jiang, R. Gene co-opening network deciphers gene functional relationships. Mol. Biosyst. 2017, 13, 2428–2439. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The nih roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Belmont, J.W.; Hardenbol, P.; Willis, T.D.; Yu, F.; Yang, H.; Ch’Ang, L.Y.; Huang, W.; Liu, B.; Shen, Y. The international hapmap project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.H.; Hemani, G.; Haley, C.S. Detecting epistasis in human complex traits. Nat. Rev. Genet. 2014, 15, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.B.; Ehrenreich, I.M. Higher-order genetic interactions and their contribution to complex traits. Trends Genet. 2015, 31, 34. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F. Epistasis and quantitative traits: Using model organisms to study gene-gene interactions. Nat. Rev. Genet. 2014, 15, 22–33. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the combinatorial drugs in Table 4 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | Model | Version | Cost (Days) 9 | SNP Pairs | Gene Pairs |
|---|---|---|---|---|---|
| GBOOST 1 | Regression | - | <1 | 670,084 | 143,008 |
| PLINK 2 | Regression | 1.9 | <1 | 427,444 | 14,850 |
| FastEpistasis 3 | Regression | 2.05 | <1 | 498,482 | 48,189 |
| pMDR 4 | Data Mining | 3.0.2 | <1 | 500 | 0 |
| AntEpiSeeker 5 | Data Mining | 1 | >30 | 0 | 0 |
| SNPRuler 6 | Machine learning | - | ~21 | 2 | 0 |
| Ranger 7 | Machine learning | 0.5.0 | ~2 | 0 | 0 |
| BEAM3 8 | Beyesian | 1 | ~9 | 0 | 0 |
| Software | Clinically Active Ratio 1/P 2,3 | Approval Ratio 1/P 2,3 |
|---|---|---|
| GBOOST | 76/985 (7.72%)/P < 1 × 10−4 | 21/985 (2.13%)/P < 1 × 10−4 |
| PLINK | 20/181 (11.05%)/P < 1 ×1 0−4 | 5/181 (2.76%)/P < 1 × 10−4 |
| FastEpistasis | 26/364 (7.14%)/P < 1 × 10−4 | 7/364 (1.92%)/P < 1 × 10−4 |
| Software | Clinically Anti-Breast Cancer Ratio 1 | Background Ratio 1 | p2 |
|---|---|---|---|
| GBOOST | 41/617 (6.6%) | 53/1363 (3.9%) | 1.2 × 10−6 |
| PLINK | 31/270 (11.5%) | 53/1363 (3.9%) | 8.0 × 10−11 |
| FastEpistasis | 36/355 (10.1%) | 53/1363 (3.9%) | 2.5 × 10−10 |
| Combinatorial Drugs | Combination Index 1 | Software |
|---|---|---|
| Dasatinib + Vorinostat | 0.439 | BOOST/FastEpistasis |
| Gefitinib + Vorinostat | 0.502 | BOOST/FastEpistasis |
| Cladribine + Dasatinib | 0.539 | BOOST/FastEpistasis |
| Dasatinib + Gefitinib | 0.628 | BOOST/PLINK |
| Cladribine + Gefitinib | 0.723 | BOOST |
| Gefitinib + Sorafenib | 1.288 | BOOST/PLINK |
| Cladribine + Sorafenib | >1 | BOOST/FastEpistasis |
| Everolimus + Sorafenib | >1 | BOOST/PLINK |
| Sorafenib + Vorinostat | >1 | BOOST/PLINK/FastEpistasis |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quan, Y.; Liu, M.-Y.; Liu, Y.-M.; Zhu, L.-D.; Wu, Y.-S.; Luo, Z.-H.; Zhang, X.-Z.; Xu, S.-Z.; Yang, Q.-Y.; Zhang, H.-Y. Facilitating Anti-Cancer Combinatorial Drug Discovery by Targeting Epistatic Disease Genes. Molecules 2018, 23, 736. https://doi.org/10.3390/molecules23040736
Quan Y, Liu M-Y, Liu Y-M, Zhu L-D, Wu Y-S, Luo Z-H, Zhang X-Z, Xu S-Z, Yang Q-Y, Zhang H-Y. Facilitating Anti-Cancer Combinatorial Drug Discovery by Targeting Epistatic Disease Genes. Molecules. 2018; 23(4):736. https://doi.org/10.3390/molecules23040736
Chicago/Turabian StyleQuan, Yuan, Meng-Yuan Liu, Ye-Mao Liu, Li-Da Zhu, Yu-Shan Wu, Zhi-Hui Luo, Xiu-Zhen Zhang, Shi-Zhong Xu, Qing-Yong Yang, and Hong-Yu Zhang. 2018. "Facilitating Anti-Cancer Combinatorial Drug Discovery by Targeting Epistatic Disease Genes" Molecules 23, no. 4: 736. https://doi.org/10.3390/molecules23040736
APA StyleQuan, Y., Liu, M.-Y., Liu, Y.-M., Zhu, L.-D., Wu, Y.-S., Luo, Z.-H., Zhang, X.-Z., Xu, S.-Z., Yang, Q.-Y., & Zhang, H.-Y. (2018). Facilitating Anti-Cancer Combinatorial Drug Discovery by Targeting Epistatic Disease Genes. Molecules, 23(4), 736. https://doi.org/10.3390/molecules23040736