A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Novel smHDAC8 Inhibitor(s) Identified from the Virtual Screening
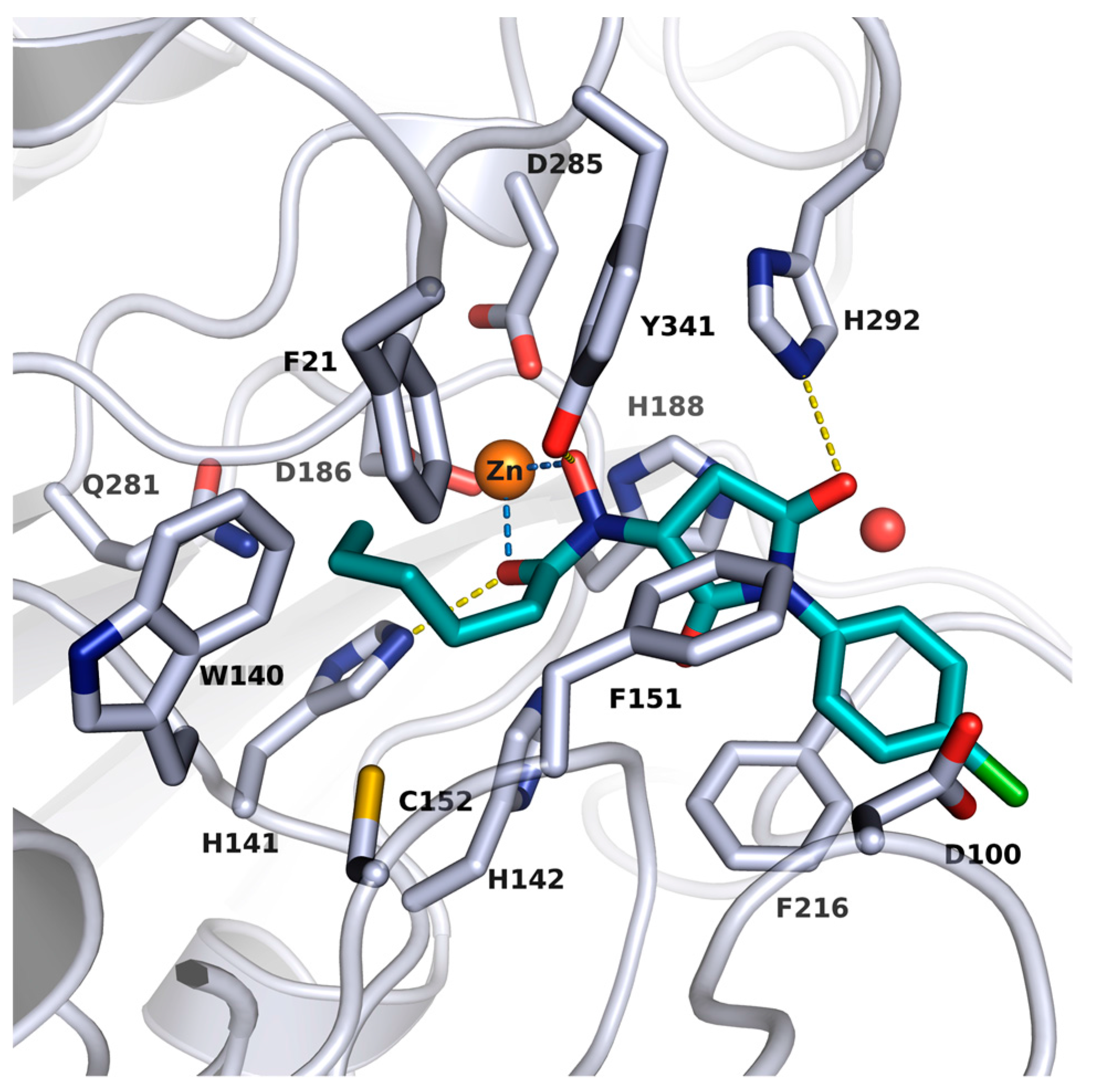
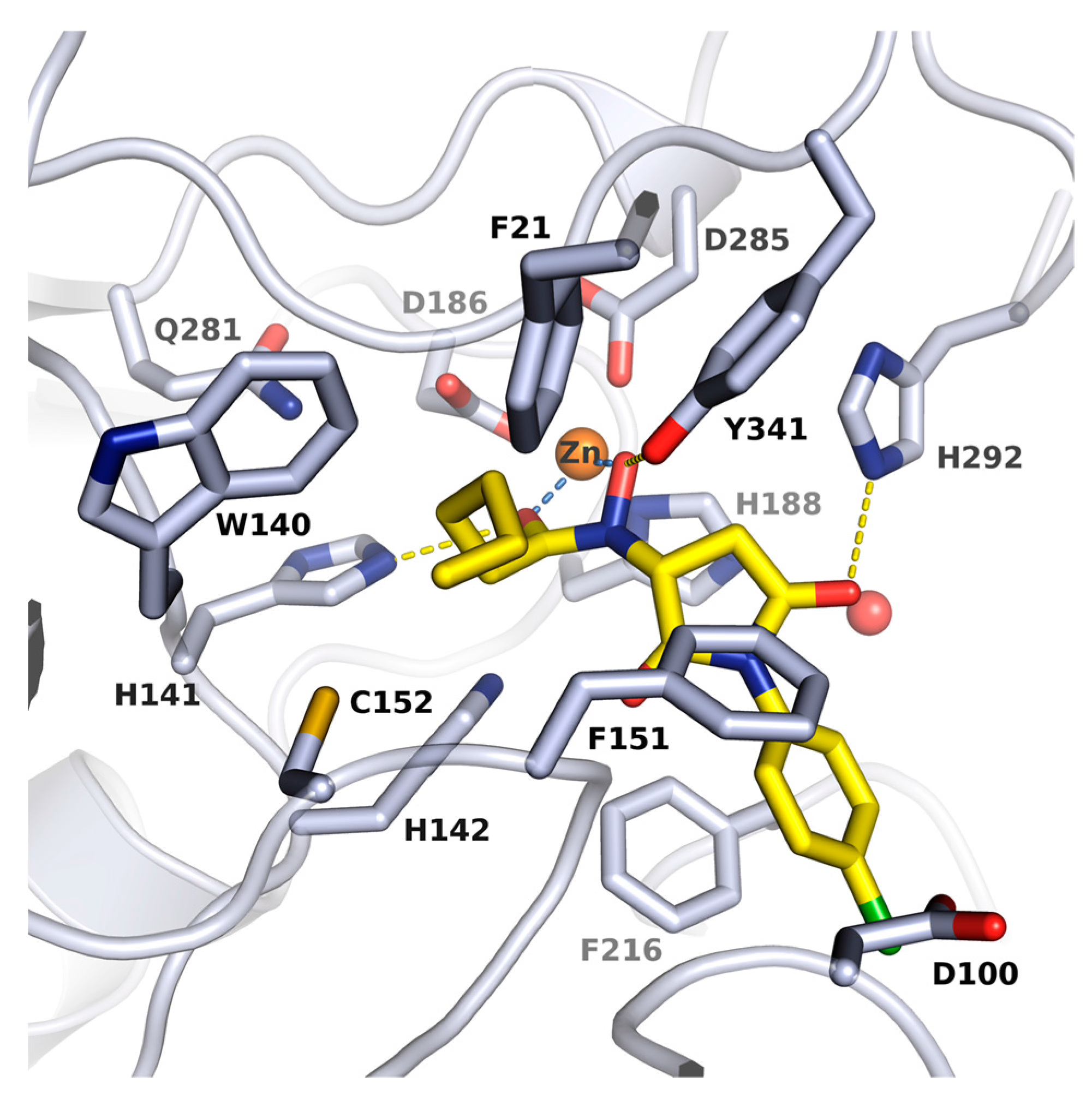
2.2. X-ray Structure of smHDAC8 in Complex with J1036
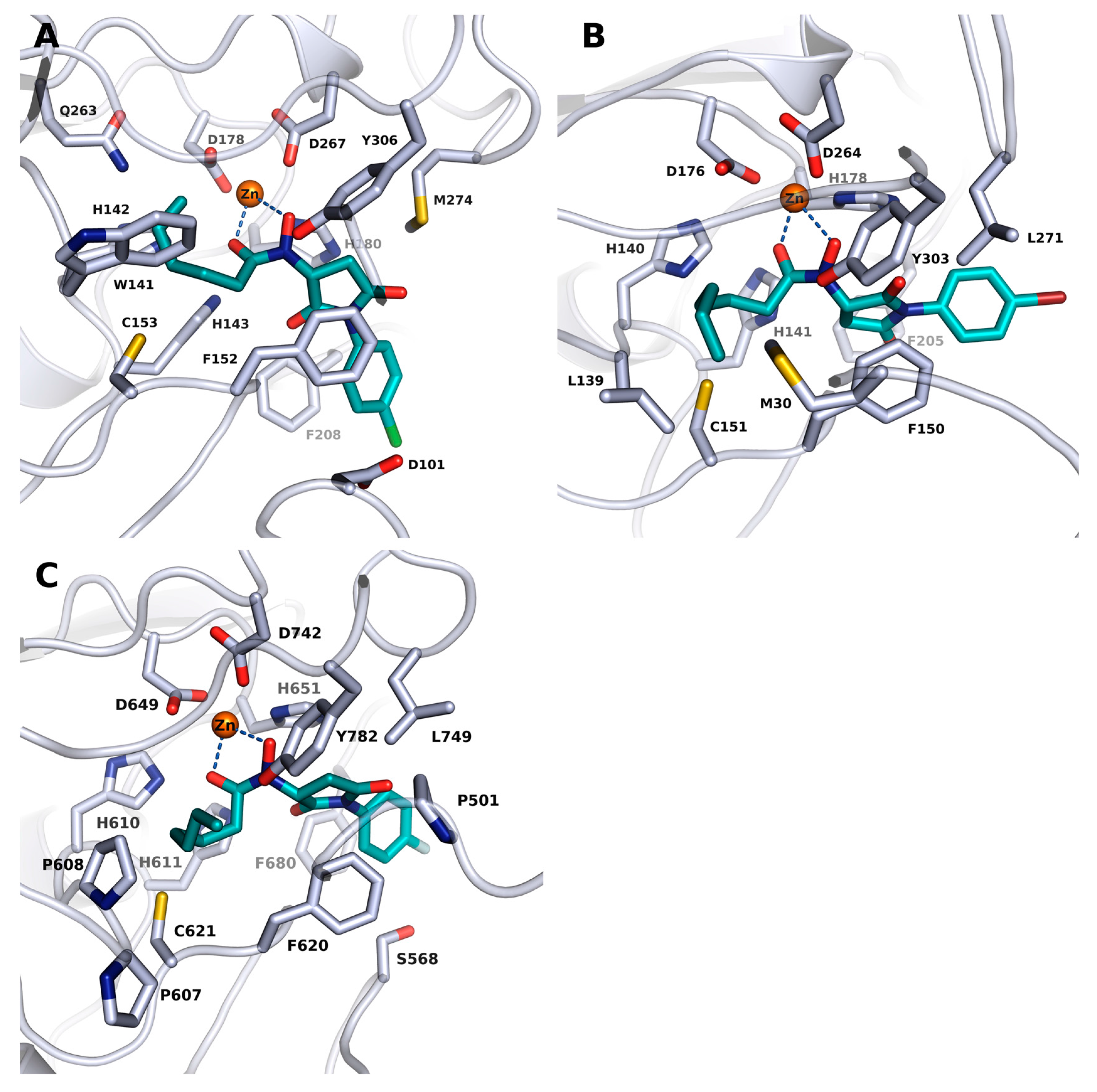
2.3. Docking into X-ray Structures of Human HDACs
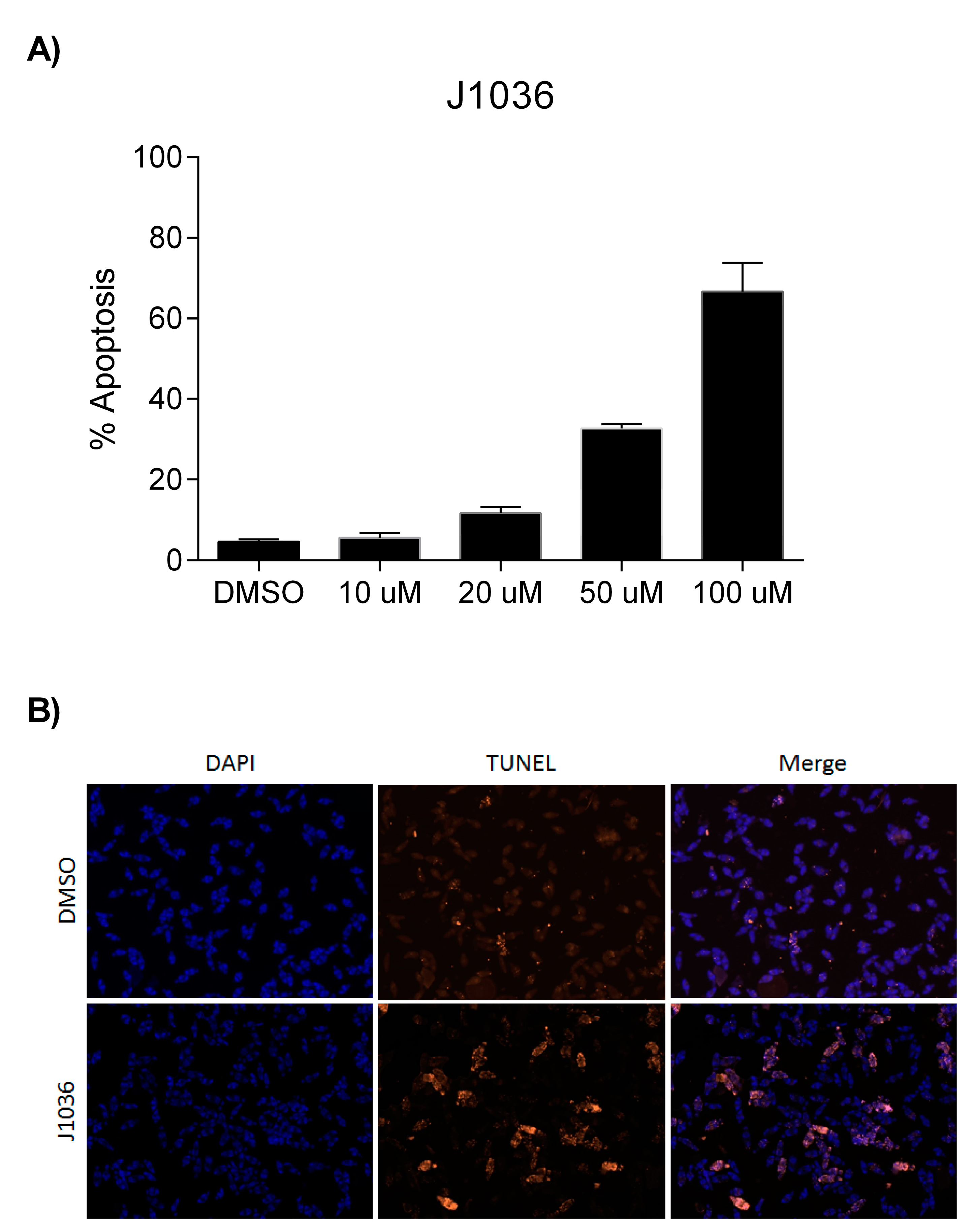
2.4. The n-Alkylhydroxamate Derivative J1036 Induces Apoptosis in S. mansoni Larvae
3. Materials and Methods
3.1. Computational Methods
3.1.1. Molecular Docking
Ligand Preparation
Protein Preparation
Virtual Screening
Docking to Human HDACs
3.2. Activity and Inhibition Assays
3.3. Phenotypic Screening
3.4. Crystallization and X-ray Structure Determination
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gray, D.J.; Ross, A.G.; Li, Y.S.; McManus, D.P. Diagnosis and management of schistosomiasis. BMJ 2011, 342, d2651. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J.; Kamath, A. Neglected tropical diseases in sub-saharan Africa: Review of their prevalence, distribution, and disease burden. PLoS Negl. Trop. Dis. 2009, 3, e412. [Google Scholar] [CrossRef] [PubMed]
- What Is Schistosomiasis? Available online: http://www.who.int/schistosomiasis/disease/en/ (accessed on 15 January 2018).
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Siqueira, L.D.P.; Fontes, D.A.F.; Aguilera, C.S.B.; Timoteo, T.R.R.; Angelos, M.A.; Silva, L.; de Melo, C.G.; Rolim, L.A.; da Silva, R.M.F.; Neto, P.J.R. Schistosomiasis: Drugs used and treatment strategies. Acta Trop. 2017, 176, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L.; Basso, A.; Guidi, A. Schistosomiasis control: Praziquantel forever? Mol. Biochem. Parasitol. 2014, 195, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.A.; Soliman, R.H. Schistosomiasis in Egypt: A never-ending story? Acta Trop. 2015, 148, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L. Praziquantel. Parasitol. Res. 2003, 90 (Suppl. 1), S3–S9. [Google Scholar] [PubMed]
- Doenhoff, M.J.; Cioli, D.; Utzinger, J. Praziquantel: Mechanisms of action, resistance and new derivatives for schistosomiasis. Curr. Opin. Infect. Dis. 2008, 21, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Doenhoff, M.J.; Pica-Mattoccia, L. Praziquantel for the treatment of schistosomiasis: Its use for control in areas with endemic disease and prospects for drug resistance. Expert Rev. Anti-Infect. Ther. 2006, 4, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Danso-Appiah, A.; De Vlas, S.J. Interpreting low praziquantel cure rates of Schistosoma mansoni infections in Senegal. Trends Parasitol. 2002, 18, 125–129. [Google Scholar] [CrossRef]
- Lawn, S.D.; Lucas, S.B.; Chiodini, P.L. Case report: Schistosoma mansoni infection: Failure of standard treatment with praziquantel in a returned traveller. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 100–101. [Google Scholar] [CrossRef]
- Melman, S.D.; Steinauer, M.L.; Cunningham, C.; Kubatko, L.S.; Mwangi, I.N.; Wynn, N.B.; Mutuku, M.W.; Karanja, D.M.; Colley, D.G.; Black, C.L.; et al. Reduced susceptibility to praziquantel among naturally occurring Kenyan isolates of Schistosoma mansoni. PLoS Negl. Trop. Dis. 2009, 3, e504. [Google Scholar] [CrossRef] [PubMed]
- Bonesso-Sabadini, P.I.; de Souza Dias, L.C. Altered response of strain of Schistosoma mansoni to oxamniquine and praziquantel. Mem. Inst. Oswaldo Cruz 2002, 97, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Couto, F.F.; Coelho, P.M.; Araujo, N.; Kusel, J.R.; Katz, N.; Jannotti-Passos, L.K.; Mattos, A.C. Schistosoma mansoni: A method for inducing resistance to praziquantel using infected Biomphalaria glabrata snails. Mem. Inst. Oswaldo Cruz 2011, 106, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Fallon, P.G.; Doenhoff, M.J. Drug-resistant schistosomiasis: Resistance to praziquantel and oxamniquine induced in Schistosoma mansoni in mice is drug specific. Am. J. Trop. Med. Hyg. 1994, 51, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.M.; Taha, S.A.; Farghaly, A.M.; el-Azony, A.S. Laboratory induced resistance to praziquantel in experimental schistosomiasis. J. Egypt. Soc. Parasitol. 1994, 24, 685–695. [Google Scholar] [PubMed]
- Hailu, G.S.; Robaa, D.; Forgione, M.; Sippl, W.; Rotili, D.; Mai, A. Lysine deacetylase inhibitors in parasites: Past, present, and future perspectives. J. Med. Chem. 2017, 60, 4780–4804. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, V.; Yang, C.; Huang, S.; Sullivan, W.J., Jr. Bromodomains in protozoan parasites: Evolution, function, and opportunities for drug development. Microbiol. Mol. Biol. Rev. 2017, 81. [Google Scholar] [CrossRef] [PubMed]
- Oger, F.; Dubois, F.; Caby, S.; Noel, C.; Cornette, J.; Bertin, B.; Capron, M.; Pierce, R.J. The class I histone deacetylases of the platyhelminth parasite Schistosoma mansoni. Biochem. Biophys. Res. Commun. 2008, 377, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Cosseau, C.; Grunau, C. Schistosoma mansoni: Developmental arrest of miracidia treated with histone deacetylase inhibitors. Exp. Parasitol. 2009, 121, 288–291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dubois, F.; Caby, S.; Oger, F.; Cosseau, C.; Capron, M.; Grunau, C.; Dissous, C.; Pierce, R.J. Histone deacetylase inhibitors induce apoptosis, histone hyperacetylation and up-regulation of gene transcription in Schistosoma mansoni. Mol. Biochem. Parasitol. 2009, 168, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Marek, M.; Kannan, S.; Hauser, A.T.; Moraes Mourao, M.; Caby, S.; Cura, V.; Stolfa, D.A.; Schmidtkunz, K.; Lancelot, J.; Andrade, L.; et al. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke Schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Maolanon, A.R.; Madsen, A.S.; Olsen, C.A. Innovative strategies for selective inhibition of histone deacetylases. Cell Chem. Biol. 2016, 23, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Keusch, J.J.; Wang, L.; Saito, M.; Hess, D.; Wang, X.; Melancon, B.J.; Helquist, P.; Gut, H.; Matthias, P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016, 12, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kozikowski, A.P. Why hydroxamates may not be the best histone deacetylase inhibitors—What some may have forgotten or would rather forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, L.; Dobler, M.R.; Radetich, B.; Zhu, Y.; Atadja, P.W.; Claiborne, T.; Grob, J.E.; McRiner, A.; Pancost, M.R.; Patnaik, A.; et al. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg. Med. Chem. 2011, 19, 4626–4634. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–46943. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Melesina, J.; Hauser, A.T.; Chakrabarti, A.; Heimburg, T.; Schmidtkunz, K.; Walter, A.; Marek, M.; Pierce, R.J.; Romier, C.; et al. Discovery of inhibitors of Schistosoma mansoni HDAC8 by combining homology modeling, virtual screening, and in vitro validation. J. Chem. Inf. Model. 2014, 54, 3005–3019. [Google Scholar] [CrossRef] [PubMed]
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F.; et al. Structure-based design and synthesis of novel inhibitors targeting HDAC8 from Schistosoma mansoni for the treatment of schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Nagano, Y.; Kouketsu, A.; Matsuura, A.; Maruyama, S.; Kurotaki, M.; Nakagawa, H.; Miyata, N. Novel inhibitors of human histone deacetylases: Design, synthesis, enzyme inhibition, and cancer cell growth inhibition of SAHA-based non-hydroxamates. J. Med. Chem. 2005, 48, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Kleinschek, A.; Meyners, C.; Digiorgio, E.; Brancolini, C.; Meyer-Almes, F.J. Potent and selective non-hydroxamate histone deacetylase 8 inhibitors. ChemMedChem 2016, 11, 2598–2606. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Miyata, N. Non-hydroxamate histone deacetylase inhibitors. Curr. Med. Chem. 2005, 12, 2867–2880. [Google Scholar] [CrossRef] [PubMed]
- Heltweg, B.; Dequiedt, F.; Marshall, B.L.; Brauch, C.; Yoshida, M.; Nishino, N.; Verdin, E.; Jung, M. Subtype selective substrates for histone deacetylases. J. Med. Chem. 2004, 47, 5235–5243. [Google Scholar] [CrossRef] [PubMed]
- Hildmann, C.; Wegener, D.; Riester, D.; Hempel, R.; Schober, A.; Merana, J.; Giurato, L.; Guccione, S.; Nielsen, T.K.; Ficner, R.; et al. Substrate and inhibitor specificity of class 1 and class 2 histone deacetylases. J. Biotechnol. 2006, 124, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017–2: LigPrep; Schrödinger, LLC: New York, NY, USA, 2017.
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-2: ConfGen; Schrödinger, LLC: New York, NY, USA, 2017.
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Christie, C.; Duarte, J.M.; Feng, Z.; Westbrook, J.; Young, J.; Zardecki, C. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018, 27, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), version 2016.08; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016.
- Schrödinger Release 2017-2: Schrödinger Suite 2017–2 Protein Preparation Wizard; Impact, version 7.5; Schrödinger, LLC: New York, NY, USA, 2017.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-2: Epik, version 4.0; Schrödinger, LLC: New York, NY, USA, 2017.
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-2: Glide; Schrödinger, LLC: New York, NY, USA, 2017.
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 201: Canvas; Schrödinger, LLC: New York, NY, USA, 2017.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Stolfa, D.A.; Marek, M.; Lancelot, J.; Hauser, A.T.; Walter, A.; Leproult, E.; Melesina, J.; Rumpf, T.; Wurtz, J.M.; Cavarelli, J.; et al. Molecular basis for the antiparasitic activity of a mercaptoacetamide derivative that inhibits histone deacetylase 8 (HDAC8) from the human pathogen Schistosoma mansoni. J. Mol. Biol. 2014, 426, 3442–3453. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D Biol. Crystallogr. 2010, 66 Pt 2, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd. | IBS Code | Structure | smHDAC8 IC50 (µM) | hsHDAC8 IC50 (µM) | hsHDAC1 IC50 (µM) | hsHDAC6 IC50 (µM) |
|---|---|---|---|---|---|---|
| J1036 | STOCK4S-53643 |  | 4.40 ± 0.17 | 0.49 ± 0.18 | 6.76 ± 0.97 | 5.02 ± 0.31 |
| J1057 | STOCK4S-48892 |  | 13.18 ± 1.85 | 2.62 ± 0.19 | 42.1 ± 2.20 | 6.20 ± 0.34 |
| J1058 | STOCK4S-78560 |  | 20.30 ± 2.78 | 3.99 ± 0.74 | 25.96 ± 2.40 | 6.20 ± 0.41 |
| J1060 | STOCK4S-27444 |  | 11.47 ± 0.91 | 1.80 ± 0.24 | 5.00 ± 0.42 | 0.86 ± 0.12 |
| J1061 | STOCK4S-02282 |  | 5.5 ± 0.7 | 7.69 ± 3.30 | 3.98 ± 0.45 | 2.65 ± 0.29 |
| J1063 | STOCK4S-11661 |  | 5.9 ± 1.6 | 7.72 ± 4.42 | 1.42 ± 0.13 | 0.77 ± 0.09 |
| J1064 | STOCK4S-11028 |  | 7.79 ± 0.28 | 2.08 ± 0.34 | 4.30 ± 0.46 | 0.60 ± 0.12 |
| J1065 | STOCK4S-31959 |  | 20.2 ± 2.7 | 3.96 ± 0.60 | 8.40 ± 0.28 | 1.57 ± 0.37 |
| J1066 | STOCK5S-25749 |  | 13% inhib. at 25 µM | n.d. | n.d. | n.d. |
| TH65 |  | 0.075 ± 0.025 | 0.026 ± 0.017 | 6.3 ± 2.1 | 0.390 ± 0.002 | |
| SAHA |  | 1.56 ± 0.20 | 0.40 ± 0.10 | 0.12 ± 0.01 | 0.104 ± 0.009 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simoben, C.V.; Robaa, D.; Chakrabarti, A.; Schmidtkunz, K.; Marek, M.; Lancelot, J.; Kannan, S.; Melesina, J.; Shaik, T.B.; Pierce, R.J.; et al. A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing. Molecules 2018, 23, 566. https://doi.org/10.3390/molecules23030566
Simoben CV, Robaa D, Chakrabarti A, Schmidtkunz K, Marek M, Lancelot J, Kannan S, Melesina J, Shaik TB, Pierce RJ, et al. A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing. Molecules. 2018; 23(3):566. https://doi.org/10.3390/molecules23030566
Chicago/Turabian StyleSimoben, Conrad V., Dina Robaa, Alokta Chakrabarti, Karin Schmidtkunz, Martin Marek, Julien Lancelot, Srinivasaraghavan Kannan, Jelena Melesina, Tajith B. Shaik, Raymond J. Pierce, and et al. 2018. "A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing" Molecules 23, no. 3: 566. https://doi.org/10.3390/molecules23030566
APA StyleSimoben, C. V., Robaa, D., Chakrabarti, A., Schmidtkunz, K., Marek, M., Lancelot, J., Kannan, S., Melesina, J., Shaik, T. B., Pierce, R. J., Romier, C., Jung, M., & Sippl, W. (2018). A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing. Molecules, 23(3), 566. https://doi.org/10.3390/molecules23030566