A Comparative Study of Human Saposins



Abstract
1. Introduction
2. Results
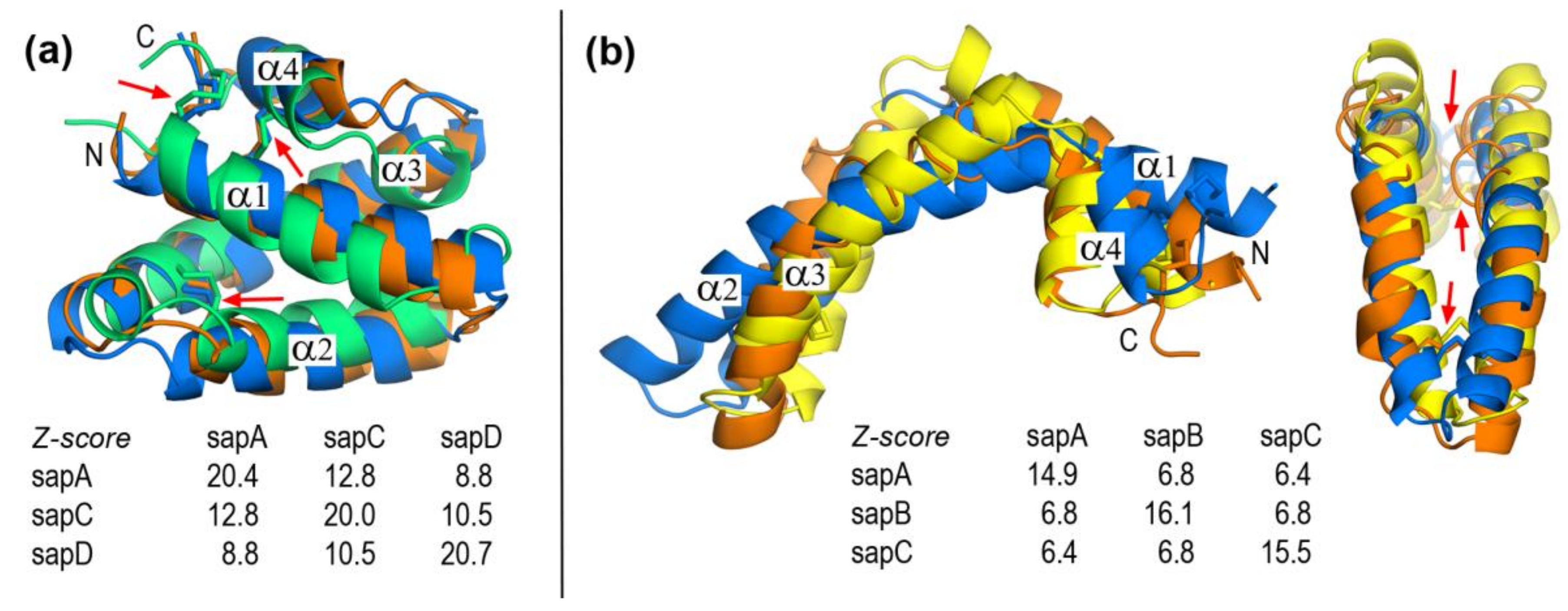
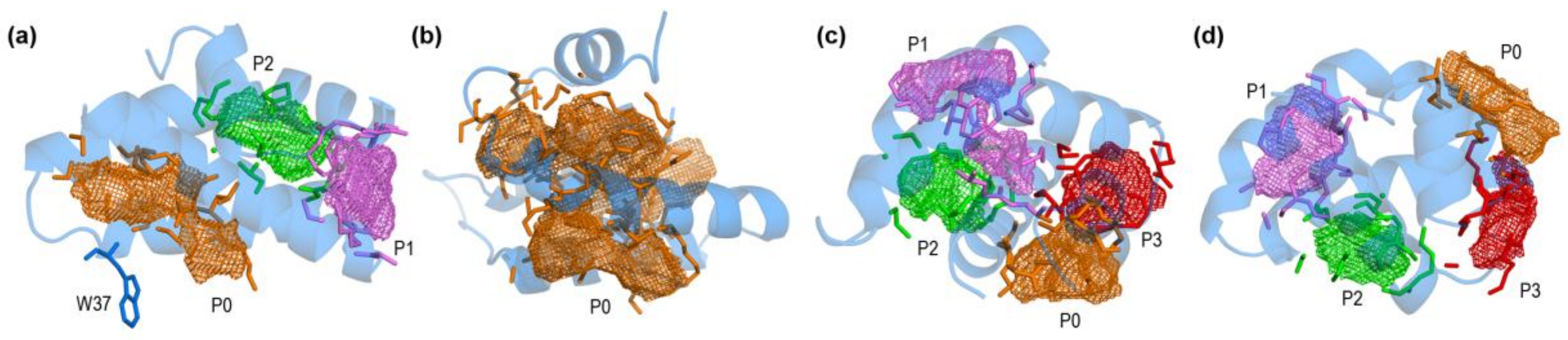
2.1. Closed and Open Forms of Human Saposins: Structural and Surface Features
2.2. Closed and Open Forms of Human Saposins: Electrostatic Features
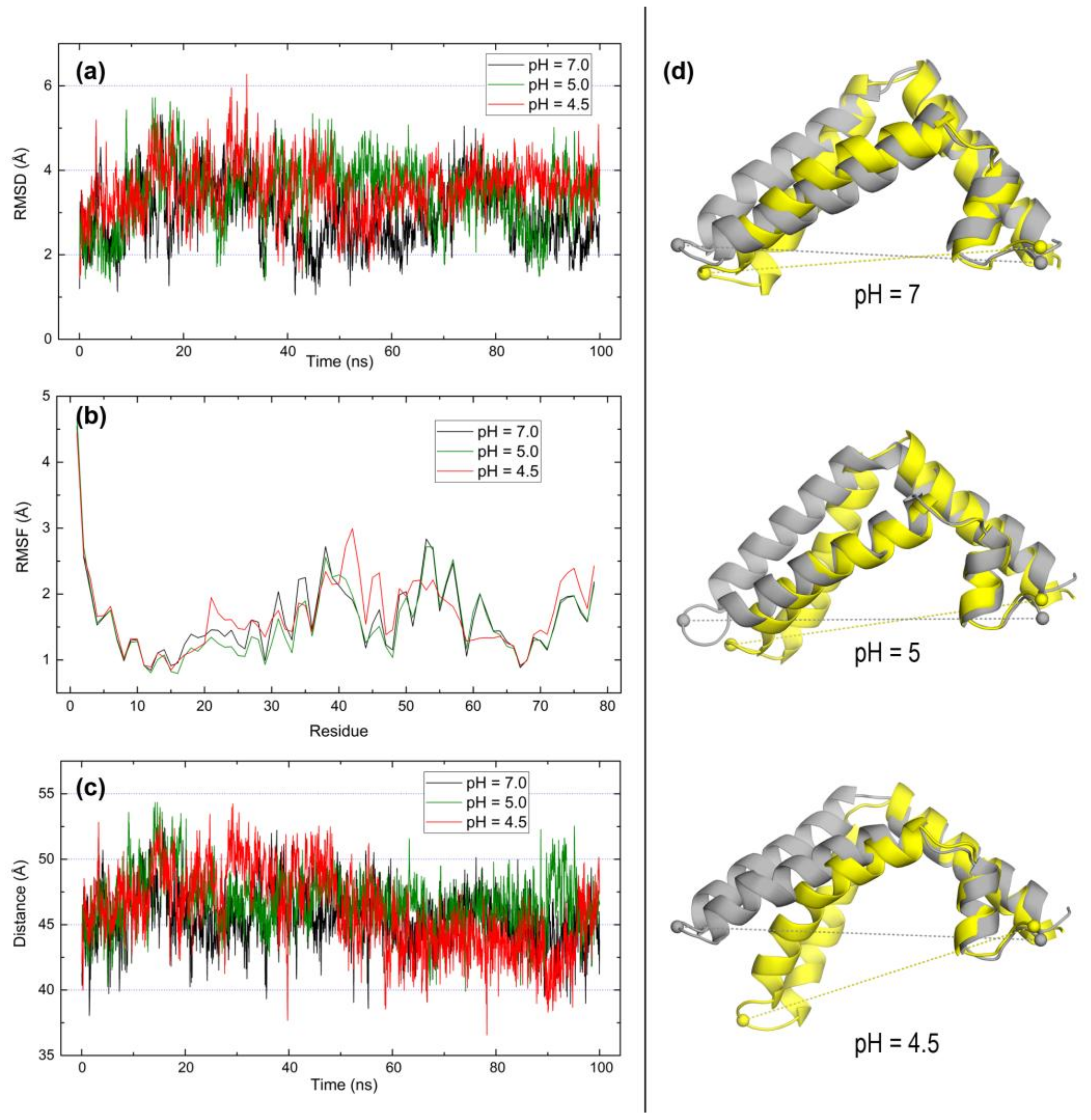
2.3. Open Forms of Human Saposins: Dynamic Features
3. Discussion
4. Materials and Methods
4.1. Structural Analyses
4.2. Structural Homology Modeling
4.3. Calculations of pKa and Protonation States
4.4. Poisson-Boltzmann Electrostatic Potentials
4.5. Molecular Dynamics Calculations
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barral, D.C.; Brenner, M.B. CD1 antigen presentacion: How it works. Nat. Rev. Immunol. 2007, 7, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.R.; Garg, S.; Brenner, M.B. Antigen presentation by CD1 lipids, T cells, and NKT cells in microbial immunity. Adv. Immunol. 2009, 102, 1–94. [Google Scholar] [PubMed]
- Van Rhijn, I.; Godfrey, D.I.; Rossjohn, J.; Moody, D.B. Lipid and small-molecule display by CD1 and MR1. Nat. Rev. Immunol. 2015, 15, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Cresswell, P. Saposins facilitate CD1-restricted presentation of an exogenous lipid antigen to T cells. Nat. Immunol. 2004, 5, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Cantu, C.; Sagiv, Y.; Schrantz, N.; Kulkarni, A.B.; Qi, X.; Mahuran, D.J.; Morales, C.R.; Grabowski, G.A.; Benlagha, K.; et al. Editing of CD1d-bound lipid antigens by endosomal lipid transfer proteins. Science 2004, 303, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Sandhoff, K. Principles of lysosomal membrane digestion: Stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 2005, 21, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.C.; Giddens, M.M.; Coleman, B.M.; Hall, R.A. The protective role of prosaposin and its receptors in the nervous system. Brain Res. 2014, 1585, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Leon, L.; Tatituri, R.V.V.; Grenha, R.; Sun, Y.; Barral, D.C.; Minnaard, A.J.; Bhowruth, V.; Veerapen, N.; Besra, G.S.; Kasmar, A.; et al. Saposins utilize two strategies for lipid transfer and CD1 antigen presentation. Proc. Natl. Acad. Sci. USA 2012, 109, 4357–4364. [Google Scholar] [CrossRef] [PubMed]
- Alattia, J.R.; Shaw, J.E.; Yip, C.M.; Privé, G.G. Direct visualization of saposin remodeling of lipid bilayers. J. Mol. Biol. 2006, 362, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H.; Sandhoff, K. Sphingolipids and lysosomal pathologies. Biochim. Biophys. Acta 2014, 1841, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Popovic, K.; Holyoake, J.; Pomès, R.; Privé, G.G. Structure of saposin a lipoprotein discs. Proc. Natl. Acad. Sci. USA 2012, 109, 2908–2912. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Qi, X.; Tsang, P.; Kang, S.J.; Ilarionov, P.A.; Besra, G.S.; Gumperz, J.; Cresswell, P. Saposin B is the dominant saposin that facilitates lipid binding to human CD1d molecules. Proc. Natl. Acad. Sci. USA 2007, 104, 5551–5556. [Google Scholar] [CrossRef] [PubMed]
- Ahn, V.E.; Faull, K.F.; Whitelegge, J.P.; Fluharty, A.L.; Privé, G.G. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. USA 2003, 100, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Alattia, J.R.; Shaw, J.E.; Yip, C.M.; Privé, G.G. Molecular imaging of membrane interfaces reveals mode of β-glucosidase activation by saposin C. Proc. Natl. Acad. Sci. USA 2007, 104, 17394–17399. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.; Schultz-Heienbrok, R.; Behlke, J.; Remmel, N.; Allings, C.; Sandhoff, K.; Saenger, W.; Maier, T. Crystal structures of human saposins C and D: Implications for lipid recognition and membrane interactions. Structure 2008, 16, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Atrian, S.; López-Viñas, E.; Gómez-Puertas, P.; Chabás, A.; Vilageliu, L.; Grinberg, D. An evolutionary and structure-based docking model for glucocerebrosidase-saposin C and glucocerebrosidase-substrate interactions: Relevance for Gaucher disease. Proteins 2008, 80, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Popovic, K.; Privé, G.G. Structures of the human ceramide activator protein saposin D. Acta Crystallogr. D 2008, 64, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Winau, F.; Schwierzeck, V.; Hurwitz, R.; Remmel, N.; Sieling, P.A.; Modlin, R.L.; Porcelli, S.A.; Brinkmann, V.; Sugita, M.; Sandhoff, K.; et al. Saposin C is required for lipid presentation by human CD1b. Nat. Immunol. 2004, 5, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Vartabedian, V.F.; Savage, P.B.; Teyton, L. The processing and presentation of lipids and glycolipids to the immune system. Immunol. Rev. 2016, 272, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Malinina, L.; Patel, D.J.; Brown, R.E. How α-helical motifs form functionally diverse lipid-binding compatments. Annu. Rev. Biochem. 2017, 86, 609–636. [Google Scholar] [CrossRef] [PubMed]
- José-Estanyol, M.; Gomis-Rüth, F.X.; Puidomènech, P. The eight-cysteine motif, a versatile structure in plant proteins. Plant Physiol. Biochem. 2004, 42, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, G.; Sánchez-Monge, R.; Barber, D.; Díaz-Perales, A. Plant non-specific lipid transfer proteins: An interface between plant defence and human allergy. Biochim. Biophys. Acta 2007, 1771, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.B.; Cheung, R.C.F.; Wong, J.H.; Ye, X. Lipid transfer proteins. Biopolymers 2012, 98, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Tordesillas, L.; Cubells-Baeza, N.; Gómez-Casado, C.; Berin, C.; Esteban, V.; Barcik, W.; O’Mahony, L.; Ramirez, C.; Pacios, L.F.; Garrido-Arandia, M.; et al. Mechanisms underlying induction of allergic sensitization by Pru p 3. Clin. Exp. Allergy 2017, 47, 1398–1408. [Google Scholar] [CrossRef] [PubMed]
- Ahn, V.E.; Leyko, P.; Alattia, J.R.; Chen, L.; Privé, G.G. Crystal structures of saposins A and C. Protein Sci. 2006, 15, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.H.; Read, R.J.; Deane, J.E. Structure of human saposin A at lysosomal pH. Acta Crystallogr. F 2015, 71, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Huta, B.P.; Mehlenbacher, M.R.; Nie, Y.; Lai, X.; Zubieta, C.; Bou-Abdallah, F.; Doyle, R.P. The lysosomal protein saposin B binds chloroquine. Chem. Med. Chem. 2016, 11, 277–282. [Google Scholar] [CrossRef] [PubMed]
- De Alba, E.; Weiler, S.; Tjandra, N. Solution structure of human saposin C: PH-dependent interaction with phospholipid vesicles. Biochemistry 2003, 42, 14729–14740. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.A.; De Alba, E.; Tjandra, N. Solution structure of human saposin C in a detergent environment. J. Mol. Biol. 2005, 346, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Laakso, L.M. Dali server update. Nucl. Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. Dali: A network tool for structure comparison. Trends Biochem. Sci. 1995, 20, 478–480. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucl. Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M. Analyzing the topology of active sites: On the predictions of pockets and sub-pockets. J. Chem. Inf. Model. 2010, 50, 2042–2052. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, T.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Johanssom, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Grabowski, G.A. Differential membrane interactions of saposins A and C: Implications for the functional specificity. J. Biol. Chem. 2001, 276, 27010–27017. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. Propka 3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Godzik, A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 2003, 19 (Suppl. 2), ii246–ii255. [Google Scholar] [CrossRef] [PubMed]
- Shyndyalov, I.N.; Bourne, P.E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998, 11, 739–747. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-Align: A protein structure alignment algorithm based on the TM-score. Nucl. Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.M.; Ciaffoni, F.; Tatti, M.; Salvioli, R.; Barca, A.; Tognozzi, D.; Scerch, C. pH-dependent conformational properties of saposins and their interactions with phospholipid membranes. J. Biol. Chem. 1995, 27, 30576–30580. [Google Scholar] [CrossRef]
- John, M.; Wendeler, M.; Heller, M.; Sandhoff, K.; Kessler, H. Characterization of human saposins by NMR spectroscopy. Biochemistry 2006, 45, 5206–5216. [Google Scholar] [CrossRef] [PubMed]
- Ciaffoni, F.; Tatti, M.; Boe, A.; Salvioli, R.; Fluharty, A.; Sonnino, S.; Vaccaro, A.M. Saposin B binds and transfers phospholipids. J. Lipid Res. 2006, 47, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Henseler, M.; Klein, C.; Suzuki, K.; Harzer, K.; Sandhoff, K. Sphingolipid activator protein (sap-D) stimulates the lysosomal degradation of ceramide in vivo. Biochem. Biophys. Res. Commun. 1994, 200, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Linke, T.; Wilkening, G.; Sadeghlar, F.; Mozcall, H.; Bernardo, K.; Schuchman, E.; Sandhoff, K. Interfacial regulation of acid ceramidase activity. Stimulation of ceramide degradation by lysosomal lipids and sphingolipid activator proteins. J. Biol. Chem. 2001, 276, 5760–5768. [Google Scholar] [CrossRef] [PubMed]
- Ciaffoni, F.; Salvioli, R.; Tatti, M.; Arancia, G.; Crateri, P.; Vaccaro, A.M. Saposin D solubilizes anionic phospholipid-containing membranes. J. Biol. Chem. 2001, 276, 31583–31589. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, version 2.0; Schrödinger, LLC: New York, NY, USA, 2017.
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL Wokspace: A web-based environment for protein structure modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucl. Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N. PDB2PQR: An automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatic calculations. Nucl. Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Czodroowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucl. Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [PubMed]
- McKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Applications to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2017, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi1 and chi2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Glykos, N.M. Carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Koukos, P.I.; Glykos, N.M. Grcarma: A fully automated task-oriented interface for the analysis of molecular dynamics trajectories. J. Comput. Chem. 2013, 34, 2310–2312. [Google Scholar] [CrossRef] [PubMed]
- Prlic, A.; Bliven, S.; Rose, P.W.; Bluhm, W.F.; Bizon, C.; Godzik, A.; Bourne, P.E. Pre-calculated protein structure alignments at the RCSB PDB website. Bioinformatics 2010, 26, 2983–2985. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | PDB Id | Title | Struct. | Conf. 1 | State | HET 2 | Occ 3 | Year | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Saposin A | 2DOB | Human SapA | Xray | Closed | Monom | Ca2+ | No | 2006 | [25] |
| 4UEX | Human SapA at lysosomal pH | Xray | Closed | Monom | - | Yes | 2015 | [26] | |
| 4DDJ | SapA in complex with LDAO | Xray | Open | Dimer | LDAO | No | 2012 | [11] | |
| Saposin B | 1N69 | Human SapB with PEH | Xray | Open | Dimer | PEH | No | 2003 | [13] |
| 4V2O | Human SapB in complex with CLQ | Xray | Open | Dimer | CLQ | Yes | 2016 | [27] | |
| Saposin C | 1M12 | Human SapC | NMR | Closed | Dimer | - | No | 2003 | [28] |
| 1SN6 | Human SapC in SDS micelles | NMR | Open | Monom | - | No | 2005 | [29] | |
| 2GTG | Human SapC | Xray | Closed | Monom | - | No | 2006 | [25] | |
| 2QYP | Orthorhombic crystal of SapC dimer | Xray | Open | Dimer | - | No | 2008 | [15] | |
| 2Z9A | Human SapC dimer | Xray | Open | Dimer | - | No | 2008 | [15] | |
| Saposin D | 2RB3 | Human SapD | Xray | Closed | Dimer | - | Yes | 2008 | [15] |
| 2R0R | K9E variant of human SapD | Xray | Closed | Dimer | SO42− | Yes | 2008 | [15] | |
| 2R1Q | Iodinated SapD in space group C2221 | Xray | Closed | Dimer | IYR | No | 2008 | [15] | |
| 3BQP | Orthorhombic human SapD | Xray | Closed | Monom | Mg2+ | Yes | 2008 | [17] | |
| 3BQQ | Triclinic human SapD | Xray | Closed | Monom | - | Yes | 2008 | [17] |
| Type | SapA | SapB | SapC | SapD |
|---|---|---|---|---|
| SapA | 100.0 | 23.08 | 39.74 | 36.71 |
| SapB | 23.08 | 100.0 | 16.00 | 22.37 |
| SapC | 39.74 | 16.00 | 100.0 | 34.18 |
| SapD | 36.71 | 22.37 | 34.18 | 100.0 |
| Type | PDB Id | Conf.2 | State | Number of | qT 3 | qT 1 at | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E | D | R | K | H | pH = 7.0 | pH = 5.0 | pH = 4.5 | |||||
| SapA | 2DOB | Closed | Monom | 8 | 7 | 1 | 6 | 0 | −8 | −8 | −8 | −3 |
| 4UEX | Closed | Monom | 8 | 7 | 1 | 6 | 0 | −8 | −8 | −8 | −2 | |
| 4DDJ | Open | Dimer1 4 | 8 | 7 | 1 | 6 | 0 | −8 | −8 | −6 | −1 | |
| SapB | 1N69 | Open | Dimer2 4 | 5 | 6 | 2 | 3 | 2 | −6 | −9 (−6, −5) | −6 (−4, −4) | +1 (−1, 0) |
| 4V2O | Open | Dimer2 | 5 | 6 | 2 | 3 | 2 | −6 | −10 (−6, −4) | −8 (−4, −4) | −2 (−1, −1) | |
| Model | Closed | Monom | 5 | 6 | 2 | 3 | 2 | −6 | −6 | −4 | −1 | |
| SapC | 1M12 | Closed | Monom | 11 | 5 | 1 | 7 | 1 | −8 | −8 | −4 | +2 |
| 1SN6 | Open | Monom | 11 | 5 | 1 | 7 | 1 | −8 | −8 | −6 | +3 | |
| 2GTG | Closed | Monom | 11 | 5 | 0 | 7 | 1 | −9 | −9 | −8 | −1 | |
| 2QYP | Open | Dimer2 | 11 | 5 | 0 | 7 | 1 | −9 | −17 (−9, −8) | −6 (−6, −6) | +5 (+4, +2) | |
| 2Z9A | Open | Dimer2 | 11 | 5 | 0 | 7 | 1 | −9 | −18 (−9, −9) | −8 (−7, −6) | +1 (0, 0) | |
| SapD | 2RB3 | Closed | Dimer2 | 8 | 4 | 1 | 7 | 0 | −4 | −8 (−4, −4) | −6 (−3, −3) | 0 (0, −2) |
| 2R0R 5 | Closed | Dimer2 | 9 | 4 | 1 | 6 | 0 | −6 | −12 (−6, −6) | −8 (−5, −5) | −2 (−2, −2) | |
| 2R1Q | Closed | Dimer2 | 8 | 4 | 1 | 7 | 0 | −4 | −8 (−4, −4) | −2 (−3, −3) | +3 (0, 0) | |
| 3BQP | Closed | Monom | 8 | 5 | 1 | 7 | 0 | −5 | −5 | −4 | 0 | |
| 3BQQ | Closed | Monom | 8 | 5 | 1 | 7 | 0 | −5 | −5 | −3 | −1 | |
| Model | Open | Monom | 8 | 4 | 1 | 7 | 0 | −4 | −4 | −2 | +3 | |
| pH | FATCAT Rigid | CE | TM-Align | |||
|---|---|---|---|---|---|---|
| p-Value | RMSD1 | Z-Score | RMSD 1 | TM-Score | RMSD 1 | |
| Saposin A | ||||||
| 7 | 4.02 × 10−5 | 6.48 | 4.25 | 5.75 | 0.407 | 3.41 |
| 5 | 2.08 × 10−9 | 3.71 | 4.74 | 3.99 | 0.579 | 3.26 |
| 4.5 | 1.01 × 10−8 | 4.00 | 4.58 | 4.22 | 0.563 | 2.62 |
| Saposin B | ||||||
| 7 | 6.93 × 10−12 | 2.89 | 5.04 | 2.79 | 0.683 | 2.77 |
| 5 | 5.34 × 10−11 | 3.13 | 4.89 | 3.01 | 0.643 | 2.82 |
| 4.5 | 2.38 × 10−9 | 3.77 | 4.58 | 3.51 | 0.544 | 3.08 |
| Saposin C | ||||||
| 7 | 3.22 × 10−5 | 5.87 | 3.89 | 2.81 | 0.463 | 3.39 |
| 5 | 1.40 × 10−8 | 3.09 | 4.89 | 2.66 | 0.686 | 2.83 |
| 4.5 | 2.44 × 10−8 | 3.21 | 4.89 | 3.44 | 0.582 | 3.29 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido-Arandia, M.; Cuevas-Zuviría, B.; Díaz-Perales, A.; Pacios, L.F. A Comparative Study of Human Saposins. Molecules 2018, 23, 422. https://doi.org/10.3390/molecules23020422
Garrido-Arandia M, Cuevas-Zuviría B, Díaz-Perales A, Pacios LF. A Comparative Study of Human Saposins. Molecules. 2018; 23(2):422. https://doi.org/10.3390/molecules23020422
Chicago/Turabian StyleGarrido-Arandia, María, Bruno Cuevas-Zuviría, Araceli Díaz-Perales, and Luis F. Pacios. 2018. "A Comparative Study of Human Saposins" Molecules 23, no. 2: 422. https://doi.org/10.3390/molecules23020422
APA StyleGarrido-Arandia, M., Cuevas-Zuviría, B., Díaz-Perales, A., & Pacios, L. F. (2018). A Comparative Study of Human Saposins. Molecules, 23(2), 422. https://doi.org/10.3390/molecules23020422