Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors

 and
and 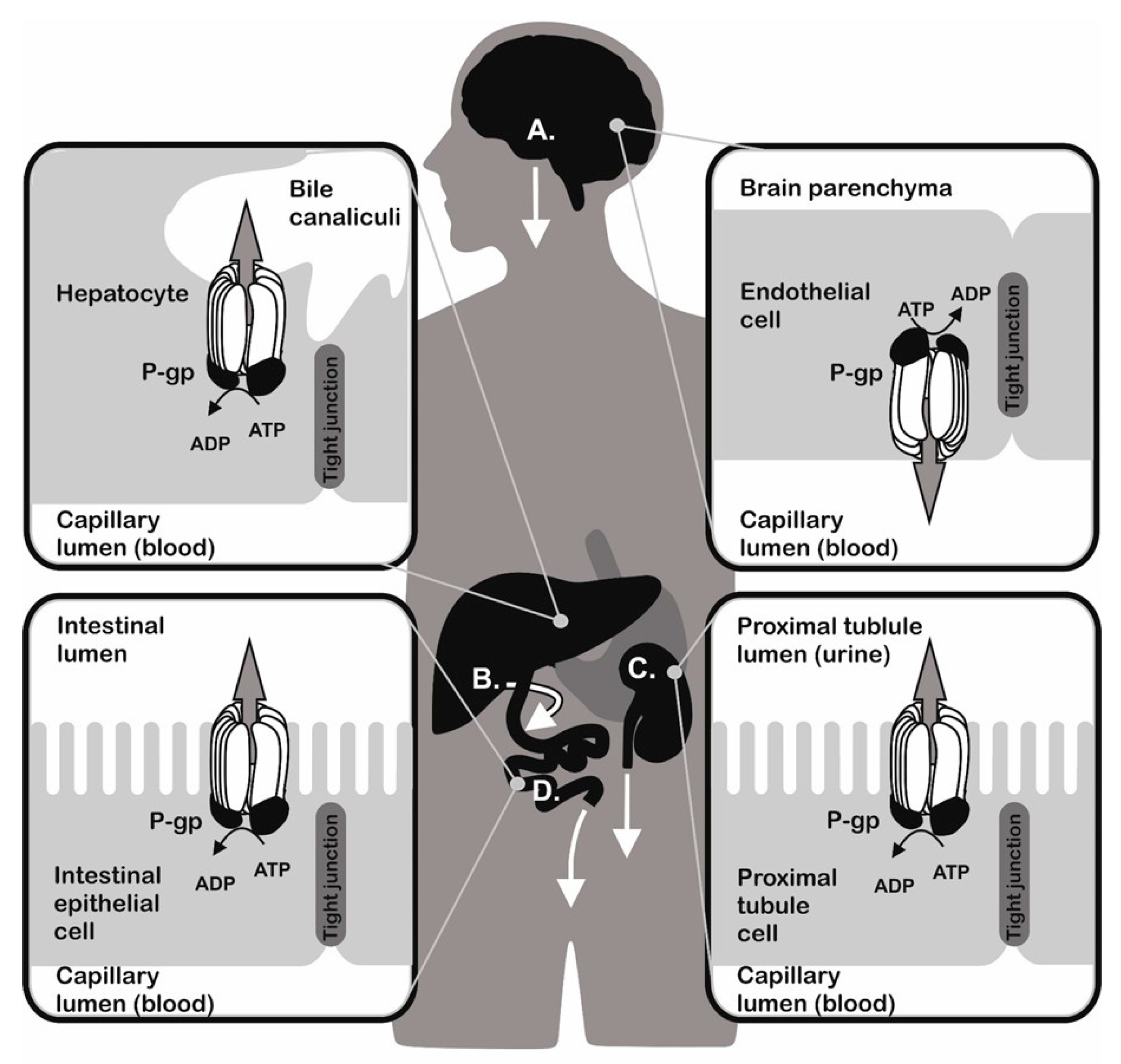{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. P-Glycoprotein and Multidrug Resistance (MDR)
3. Protein Quality Control in Endoplasmic Reticulum (ER)
4. ER Stress, Induction and Consequences
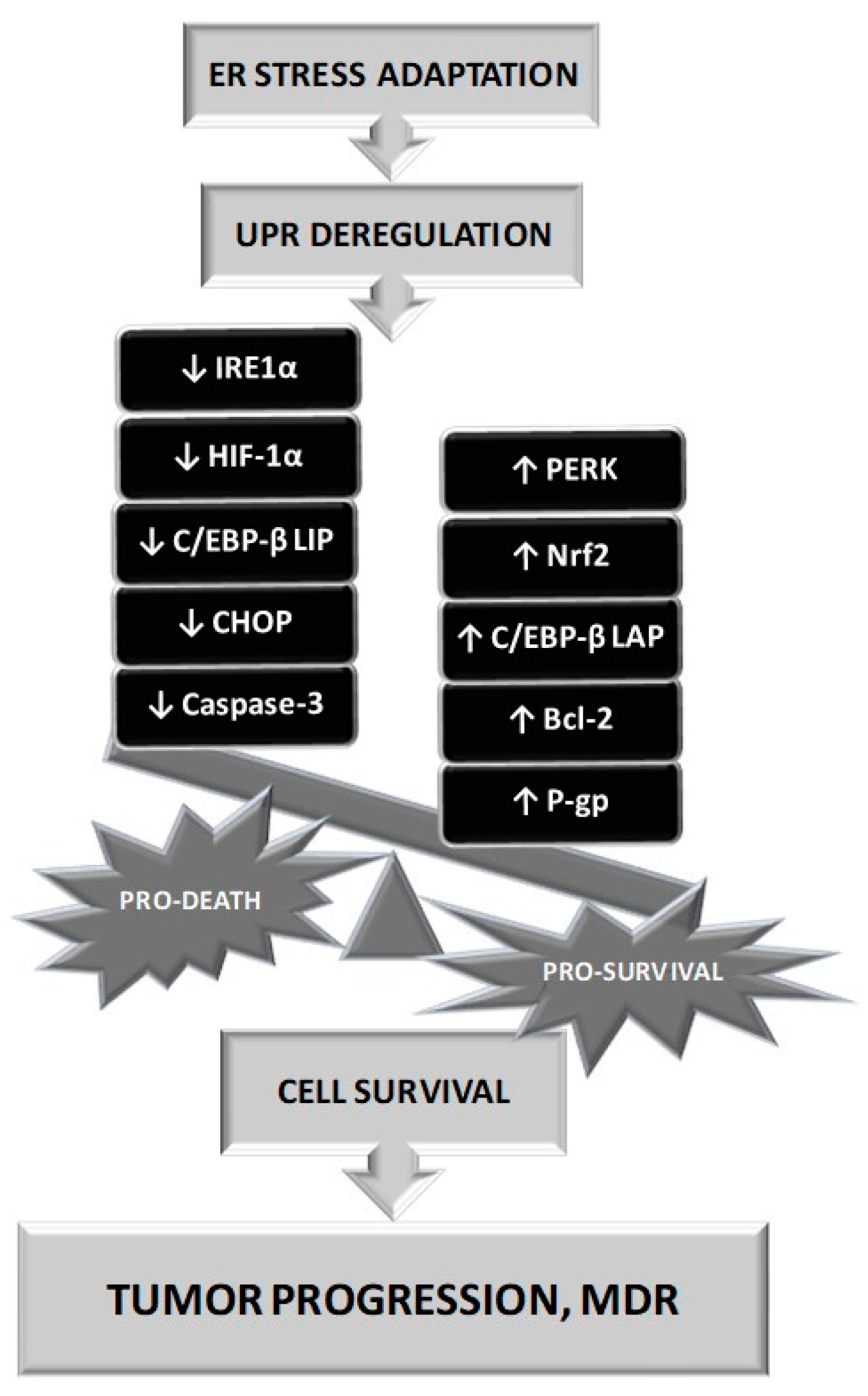
- The transcription factor X-box-binding protein (XBP1) regulates protein folding, trafficking and ERAD. After IRE1α induction of specific splicing of XBP1 mRNA, spliced variant sXBP1 enters the nucleus and regulates the expression of downstream products as UPR target genes, such as those encoding chaperones and components of ERAD [82,83,84]. The early pro survival response inhibits the expression of pro apoptotic C/EBP homologous protein (CHOP). However, prolonged ERS is associated with an increase of ROS levels and IRE-1α/XBP1 mediated activation of both CHOP and Bim and inhibition of Bcl-2, which leads to apoptosis [85,86,87,88,89].
- Activation of the PERK pathway leads to phosphorylation of eukaryotic initiation translation factor 2α (eIF2α), which triggers an antioxidant, pro survival response mediated by transcription factor 4 (ATF4). These phosphorylation leads to inhibition of translation [90]. In the case of sustained PERK stimulation, ATF4 signalling pathway induces dephosphorylation of eIF2α and promotes the expression of CHOP to mediate ERS-induced apoptosis [91,92].
- The ERS sensor ATF6 moves after activation from the ER to the GA, where it is cleaved by site 1 and site 2 proteases (S1P and S2P) to generate a cytosolic active transcription factor regulating protein folding and degradation [93].
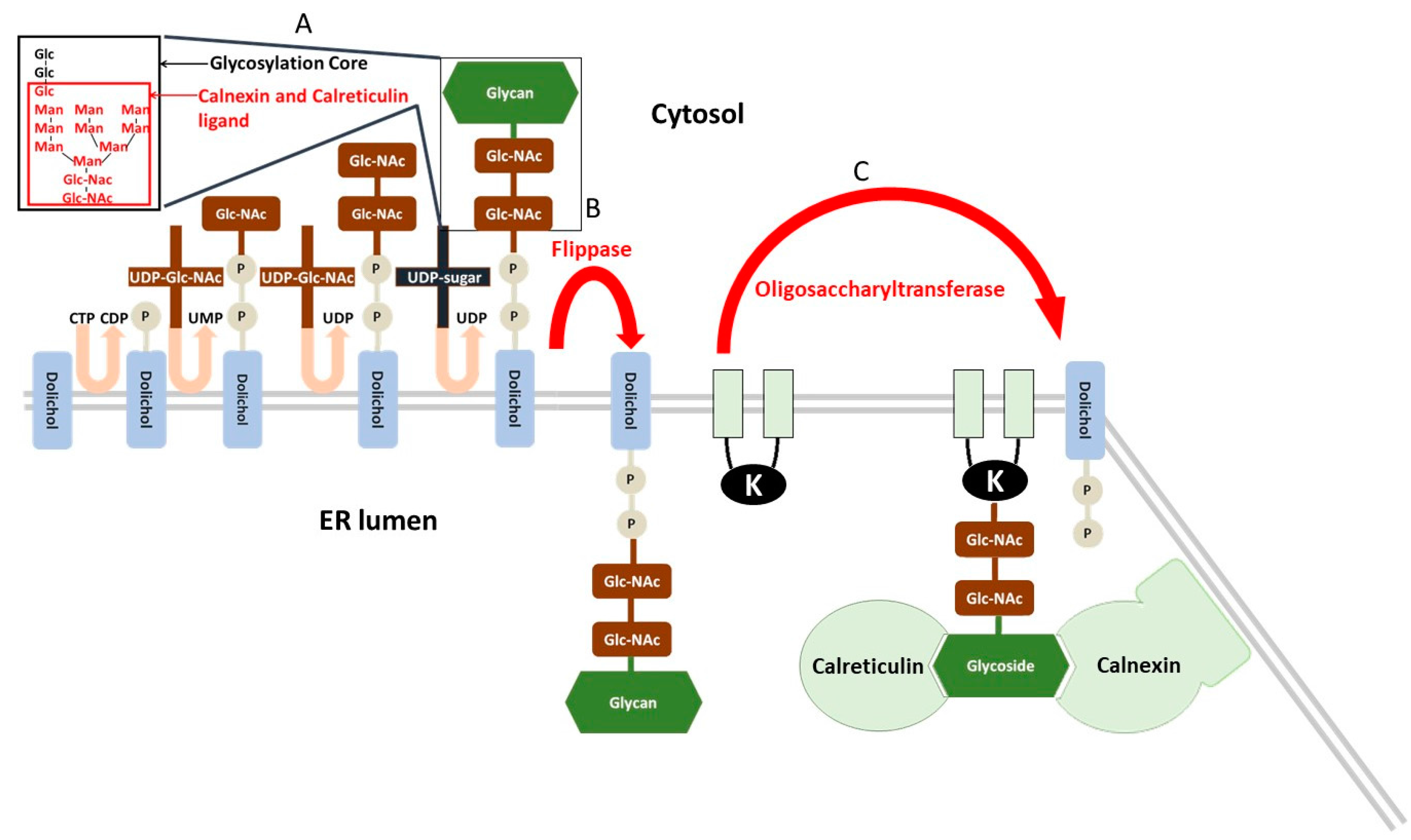
- Inhibitors of protein N-glycosylation, such as tunicamycin, an inhibitor of the UDP-N-acetylglucosamine-dolichol phosphate N-acetylglucosamine-1-phosphate transferase, that realize the first step of glycosylation core synthesis (Figure 2).
- Inducers of calcium depletion of ER, such as thapsigargin, that block the calcium pump of this organelle. Lack of calcium content in the ER eliminates proper function of calnexin and calreticulin and induces malfunction of protein quality control in the ER.
- Inhibitors of transport of proteins from the ER to the GA, such as brefeldin A, that additionally induce retrograde protein transport from the GA to the ER. This leads to the accumulation of unfolded proteins in the ER.
- Strong reducing agents, such as DTT, that block disulphide-bond formation and induce ERS within minutes.
- Proteasome inhibitors, such as, MG132 that block ERAD and cause misfolded protein accumulation in the ER.
5. ER Stress, Cancer and MDR
6. Therapeutic Approaches
7. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATF6 | activating transcription factor 6 |
| ABC | ATP-binding cassette |
| BiP | immunoglobulin binding protein |
| CHOP | C/EBP homologous protein |
| eIF2α | eukaryotic initiation translation factor 2α |
| ER | endoplasmic reticulum |
| ERAD | ER associated protein degradation |
| ERS | ER stress |
| GA | golgi apparatus |
| GRP78 | glucose regulated protein 78 |
| HSP | heat-shock protein |
| iASPP | inhibitor of apoptosis-stimulating protein p53 |
| IRE1α | inositol-requiring enzyme 1α |
| keap1 | Kelch-like ECH-associated protein 1 |
| MDR | multidrug resistance |
| mTOR | mechanistic target of rapamycin |
| Nrf2 | nuclear factor-erythroid 2-related factor 2 |
| OS9 | osteosarcoma amplified 9 |
| PERK | pancreatic ER kinase |
| P-gp | P-glycoprotein |
| ROS | reactive oxygen species |
| S1P and S2P | site 1 and site 2 proteases |
| UGGT | UDP-glucose:glycoprotein glycosyltransferase |
| URP | unfolded protein response |
| VEGF | vascular endothelial growth factor |
| XBP1 | X-box-binding protein |
| XTP3 | lectin of ER |
| XTP3-B | transactivated protein or ER lectin |
References
- Borst, P.; Schinkel, A.H. Genetic dissection of the function of mammalian P-glycoproteins. Trends Genet. 1997, 13, 217–222. [Google Scholar] [CrossRef]
- Efferth, T.; Volm, M. Multiple resistance to carcinogens and xenobiotics: P-glycoproteins as universal detoxifiers. Arch. Toxicol 2017, 91, 2515–2538. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.; Costa, J.; Reis-Henriques, M.A. ABC transporters in fish species: A review. Front. Physiol. 2014, 5, 266. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, R.M. ABC multidrug transporters in schistosomes and other parasitic flatworms. Parasitol. Int. 2013, 62, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; George, A.M. Multidrug resistance in parasites: ABC transporters, P-glycoproteins and molecular modelling. Int J. Parasitol. 2005, 35, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D.; Jones, P.M.; George, A.M. Multidrug efflux pumps: The structures of prokaryotic ATP-binding cassette transporter efflux pumps and implications for our understanding of eukaryotic P-glycoproteins and homologues. FEBS J. 2010, 277, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Merola, V.M.; Eubig, P.A. Toxicology of avermectins and milbemycins (macrocylic lactones) and the role of p-glycoprotein in dogs and cats. Vet. Clin. North Am. Small Anim. Pract. 2012, 42, 313–333. [Google Scholar] [CrossRef] [PubMed]
- Myllynen, P.; Kummu, M.; Sieppi, E. ABCB1 and ABCG2 expression in the placenta and fetus: An interspecies comparison. Expert Opin. Drug. Metab. Toxicol. 2010, 6, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.J. Too much of a good thing: How insects cope with excess ions or toxins in the diet. J. Exp. Biol. 2009, 212, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Goffeau, A. Yeast ATP-binding cassette transporters conferring multidrug resistance. Annu. Rev. Microbiol. 2012, 66, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47, 381–389. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef] [PubMed]
- Jolliet-Riant, P.; Tillement, J.P. Drug transfer across the blood-brain barrier and improvement of brain delivery. Fundam. Clin. Pharmacol. 1999, 13, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Molsa, M.; Heikkinen, T.; Hakkola, J.; Hakala, K.; Wallerman, O.; Wadelius, M.; Wadelius, C.; Laine, K. Functional role of P-glycoprotein in the human blood-placental barrier. Clin. Pharmacol. Ther. 2005, 78, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Mayer, U.; Wagenaar, E.; Mol, C.A.; van Deemter, L.; Smit, J.J.; van der Valk, M.A.; Voordouw, A.C.; Spits, H.; van Tellingen, O.; et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. USA 1997, 94, 4028–4033. [Google Scholar] [CrossRef] [PubMed]
- Kvackajova-Kisucka, J.; Barancik, M.; Breier, A. Drug transporters and their role in multidrug resistance of neoplastic cells. Gen. Physiol. Biophys. 2001, 20, 215–237. [Google Scholar] [PubMed]
- Thomas, H.; Coley, H.M. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003, 10, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, S. Efflux pump inhibitors: A strategy to combat P-glycoprotein and the nora multidrug resistance pump. ChemMedChem 2010, 5, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Ibraheem, Z.O.; Abd Majid, R.; Noor, S.M.; Sedik, H.M.; Basir, R. Role of different PFCRT and PFMDR-1 mutations in conferring resistance to antimalaria drugs in plasmodium falciparum. Malar. Res. Treat. 2014, 2014, 950424. [Google Scholar] [PubMed]
- Tomasova, L.; Konopelski, P.; Ufnal, M. Gut bacteria and hydrogen sulfide: The new old players in circulatory system homeostasis. Molecules 2016, 21, 1558. [Google Scholar] [CrossRef] [PubMed]
- Ufnal, M.; Pham, K. The gut-blood barrier permeability—A new marker in cardiovascular and metabolic diseases? Med. Hypotheses 2017, 98, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Barancik, M.; Sulova, Z.; Uhrik, B. P-glycoprotein—Implications of metabolism of neoplastic cells and cancer therapy. Curr. Cancer Drug. Targets 2005, 5, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into P-glycoprotein as a drug target. Anticancer. Agents Med. Chem. 2013, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Kahng, J.; Kim, M.; Lim, J.; Kim, Y.; Cho, B.; Kim, H.K.; Min, W.S.; Kim, C.C.; Lee, K.Y.; et al. Expression of functional markers in acute nonlymphoblastic leukemia. Acta Haematol. 2000, 104, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Imrichova, D.; Messingerova, L.; Seres, M.; Kavcova, H.; Pavlikova, L.; Coculova, M.; Breier, A.; Sulova, Z. Selection of resistant acute myeloid leukemia SKM-1 and MOLM-13 cells by vincristine-, mitoxantrone- and lenalidomide-induced upregulation of P-glycoprotein activity and downregulation of CD33 cell surface exposure. Eur. J. Pharm. Sci. 2015, 77, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Messingerova, L.; Imrichova, D.; Kavcova, H.; Seres, M.; Sulova, Z.; Breier, A. A decrease in cellular microRNA-27a content is involved in azacytidine-induced P-glycoprotein expression in SKM-1 cells. Toxicol. In Vitro 2016, 36, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Messingerova, L.; Imrichova, D.; Kavcova, H.; Turakova, K.; Breier, A.; Sulova, Z. Acute myeloid leukemia cells MOLM-13 and SKM-1 established for resistance by azacytidine are crossresistant to P-glycoprotein substrates. Toxicol. In Vitro 2015, 29, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Polekova, L.; Barancik, M.; Mrazova, T.; Pirker, R.; Wallner, J.; Sulova, Z.; Breier, A. Adaptation of mouse leukemia cells L1210 to vincristine. Evidence for expression of P-glycoprotein. Neoplasma 1992, 39, 73–77. [Google Scholar] [PubMed]
- Roninson, I.B.; Chin, J.E.; Choi, K.G.; Gros, P.; Housman, D.E.; Fojo, A.; Shen, D.W.; Gottesman, M.M.; Pastan, I. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 4538–4542. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Fojo, A.; Chin, J.E.; Roninson, I.B.; Richert, N.; Pastan, I.; Gottesman, M.M. Human multidrug-resistant cell lines: Increased MDR1 expression can precede gene amplification. Science 1986, 232, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.B.; Lind, M.J.; Cawkwell, L. Establishment of in-vitro models of chemotherapy resistance. Anticancer. Drugs 2007, 18, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Ling, V. Purification of p-glycoprotein from plasma membrane vesicles of chinese hamster ovary cell mutants with reduced colchicine permeability. J. Biol. Chem. 1979, 254, 12701–12705. [Google Scholar] [PubMed]
- Gottesman, M.M.; Ling, V. The molecular basis of multidrug resistance in cancer: The early years of P-glycoprotein research. FEBS Lett. 2006, 580, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Yang, Z.; Tang, S.; Han, Y.; Ma, J. Effects of P-glycoprotein and its inhibitors on apoptosis in K562 cells. Molecules 2014, 19, 13061–13075. [Google Scholar] [CrossRef] [PubMed]
- Bear, C.E. Drugs transported by P-glycoprotein inhibit a 40 PS outwardly rectifying chloride channel. Biochem. Biophys. Res. Commun. 1994, 200, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.M.; Wei, L.Y.; Roepe, P.D. Are altered pHi and membrane potential in hu MDR 1 transfectants sufficient to cause mdr protein-mediated multidrug resistance? J. Gen. Physiol. 1996, 108, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Idriss, H.T.; Hannun, Y.A.; Boulpaep, E.; Basavappa, S. Regulation of volume-activated chloride channels by p-glycoprotein: Phosphorylation has the final say! J. Physiol. 2000, 524, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Pallis, M.; Russell, N. P-glycoprotein plays a drug-efflux-independent role in augmenting cell survival in acute myeloblastic leukemia and is associated with modulation of a sphingomyelin-ceramide apoptotic pathway. Blood 2000, 95, 2897–2904. [Google Scholar] [PubMed]
- Ruefli, A.A.; Johnstone, R.W. A role for P-glycoprotein in regulating cell growth and survival. Clin. Appl. Immunol. Rev. 2003, 4, 31–47. [Google Scholar] [CrossRef]
- Tainton, K.M.; Smyth, M.J.; Jackson, J.T.; Tanner, J.E.; Cerruti, L.; Jane, S.M.; Darcy, P.K.; Johnstone, R.W. Mutational analysis of P-glycoprotein: Suppression of caspase activation in the absence of atp-dependent drug efflux. Cell Death Differ. 2004, 11, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Gibalova, L.; Sedlak, J.; Labudova, M.; Barancik, M.; Rehakova, A.; Breier, A.; Sulova, Z. Multidrug resistant P-glycoprotein positive L1210/VCR cells are also cross-resistant to cisplatin via a mechanism distinct from P-glycoprotein-mediated drug efflux activity. Gen. Physiol. Biophys. 2009, 28, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Gibalova, L.; Seres, M.; Rusnak, A.; Ditte, P.; Labudova, M.; Uhrik, B.; Pastorek, J.; Sedlak, J.; Breier, A.; Sulova, Z. P-glycoprotein depresses cisplatin sensitivity in l1210 cells by inhibiting cisplatin-induced caspase-3 activation. Toxicol. In Vitro 2012, 26, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F.; Gottesman, M.M. Is the multidrug transporter a flippase? Trends Biochem. Sci. 1992, 17, 18–21. [Google Scholar] [CrossRef]
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Greer, D.A.; Ivey, S. Distinct N-glycan glycosylation of P-glycoprotein isolated from the human uterine sarcoma cell line MES-SA/DX5. Biochim. Biophys. Acta 2007, 1770, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Thapsigargin or curcumin does not promote maturation of processing mutants of the abc transporters, CFTR, and P-glycoprotein. Biochem. Biophys. Res. Commun. 2004, 325, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Presley, J.F.; Cole, N.B.; Schroer, T.A.; Hirschberg, K.; Zaal, K.J.; Lippincott-Schwartz, J. ER-to-Golgi transport visualized in living cells. Nature 1997, 389, 81–85. [Google Scholar] [PubMed]
- Fu, D.; Roufogalis, B.D. Actin disruption inhibits endosomal traffic of P-glycoprotein-EGFP and resistance to daunorubicin accumulation. Am. J. Physiol.-Cell Physiol. 2007, 292, C1543–C1552. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, Y.; Dutt, P.; Lippincott-Schwartz, J.; Arias, I.M. Rab11a and myosin Vb are required for bile canalicular formation in WIF-B9 cells. Proc. Natl. Acad. Sci. USA 2005, 102, 15087–15092. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Groenendyk, J.; Michalak, M. Glycoprotein quality control and endoplasmic reticulum stress. Molecules 2015, 20, 13689–13704. [Google Scholar] [CrossRef] [PubMed]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Deprez, P.; Gautschi, M.; Helenius, A. More than one glycan is needed for er glucosidase ii to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol. Cell 2005, 19, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.Q.; Sharma, S.; Leister, K.J.; Joshi, L. A tight-knit group: Protein glycosylation, endoplasmic reticulum stress and the unfolded protein response. In Endoplasmic Reticulum Stress in Health and Disease; Agostinis, P., Afshin, S., Eds.; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar]
- Pelletier, M.F.; Marcil, A.; Sevigny, G.; Jakob, C.A.; Tessier, D.C.; Chevet, E.; Menard, R.; Bergeron, J.J.; Thomas, D.Y. The heterodimeric structure of glucosidase ii is required for its activity, solubility, and localization in vivo. Glycobiology 2000, 10, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Shailubhai, K.; Pukazhenthi, B.S.; Saxena, E.S.; Varma, G.M.; Vijay, I.K. Glucosidase I, a transmembrane endoplasmic reticular glycoprotein with a luminal catalytic domain. J. Biol. Chem. 1991, 266, 16587–16593. [Google Scholar] [PubMed]
- Sanyal, S.; Frank, C.G.; Menon, A.K. Distinct flippases translocate glycerophospholipids and oligosaccharide diphosphate dolichols across the endoplasmic reticulum. Biochemistry 2008, 47, 7937–7946. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.H.; Stanley, P.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Essentials of Glycobiology, 2nd ed.; CSH Laboratory Press: New York, NY, USA, 2009. [Google Scholar]
- Spiro, R.G.; Zhu, Q.; Bhoyroo, V.; Soling, H.D. Definition of the lectin-like properties of the molecular chaperone, calreticulin, and demonstration of its copurification with endomannosidase from rat liver golgi. J. Biol. Chem. 1996, 271, 11588–11594. [Google Scholar] [CrossRef] [PubMed]
- Ware, F.E.; Vassilakos, A.; Peterson, P.A.; Jackson, M.R.; Lehrman, M.A.; Williams, D.B. The molecular chaperone calnexin binds GLc1Man9GLcNAc2 oligosaccharide as an initial step in recognizing unfolded glycoproteins. J. Biol. Chem. 1995, 270, 4697–4704. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Forster, F. Structure of the mammalian oligosaccharyl-transferase complex in the native er protein translocon. Nat. Commun. 2014, 5, 3072. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.B. Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Tremblay, L.O.; You, Z.; Herscovics, A.; Wada, I.; Nagata, K. Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kong α1-antitrypsin by human er mannosidase I. J. Biol. Chem. 2003, 278, 26287–26294. [Google Scholar] [CrossRef] [PubMed]
- Ninagawa, S.; Okada, T.; Sumitomo, Y.; Kamiya, Y.; Kato, K.; Horimoto, S.; Ishikawa, T.; Takeda, S.; Sakuma, T.; Yamamoto, T.; et al. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 2014, 206, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.C.; Ferrero-Garcia, M.A.; Parodi, A.J. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:Glycoprotein glucosyltransferase. Biochemistry 1992, 31, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Bhattacharya, A.; Qi, L. Endoplasmic reticulum quality control in cancer: Friend or foe. Semin. Cancer Biol. 2015, 33, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Ye, Y.; Rapoport, T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 2002, 3, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14296–14301. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Standera, S.; Buerger, E.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Koning, F.; Kloetzel, P.M.; Seeger, M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J. Mol. Biol. 2005, 354, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Rapoport, T.A. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14132–14138. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Yao, J.; Zeng, W.; Mizuno, Y.; Kamm, K.E.; Stull, J.T.; Harding, H.P.; Ron, D.; Muallem, S. ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from PERK-knockout mice. J. Cell Sci. 2006, 119, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Panayi, G.S.; Corrigall, V.M. Immunoglobulin heavy-chain-binding protein (BiP): A stress protein that has the potential to be a novel therapy for rheumatoid arthritis. Biochem. Soc. Trans. 2014, 42, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Vandewynckel, Y.P.; Laukens, D.; Geerts, A.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Janssens, S.; Heindryckx, F.; Van Vlierberghe, H. The paradox of the unfolded protein response in cancer. Anticancer. Res. 2013, 33, 4683–4694. [Google Scholar] [PubMed]
- Back, S.H.; Schroder, M.; Lee, K.; Zhang, K.; Kaufman, R.J. ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 2005, 35, 395–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Ahmed, H.; Yang, P.; Czinn, S.J.; Blanchard, T.G. Endoplasmic reticulum stress and IRE-1 signaling cause apoptosis in colon cancer cells in response to andrographolide treatment. Oncotarget 2016, 7, 41432–41444. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Banerjee, V.; Czinn, S.; Blanchard, T. Increased reactive oxygen species levels cause er stress and cytotoxicity in andrographolide treated colon cancer cells. Oncotarget 2017, 8, 26142–26153. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Hubbard, S.R. How IRE1 reacts to er stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Zeriouh, W.; Nani, A.; Belarbi, M.; Dumont, A.; de Rosny, C.; Aboura, I.; Ghanemi, F.Z.; Murtaza, B.; Patoli, D.; Thomas, C.; et al. Phenolic extract from oleaster (Olea europaea var. Sylvestris) leaves reduces colon cancer growth and induces caspase-dependent apoptosis in colon cancer cells via the mitochondrial apoptotic pathway. PLoS ONE 2017, 12, e0170823. [Google Scholar]
- Knutsen, J.H.; Rodland, G.E.; Boe, C.A.; Haland, T.W.; Sunnerhagen, P.; Grallert, B.; Boye, E. Stress-induced inhibition of translation independently of eIF2α phosphorylation. J. Cell Sci. 2015, 128, 4420–4427. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eiF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The role of the PERK/eiF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process srebps. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Pagliarini, V.; Giglio, P.; Bernardoni, P.; De Zio, D.; Fimia, G.M.; Piacentini, M.; Corazzari, M. Downregulation of E2F1 during ER stress is required to induce apoptosis. J. Cell Sci. 2015, 128, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring er stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [PubMed]
- Shen, K.; Johnson, D.W.; Vesey, D.A.; McGuckin, M.A.; Gobe, G.C. Role of the unfolded protein response in determining the fate of tumor cells and the promise of multi-targeted therapies. Cell Stress Chaperones 2017, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef] [PubMed]
- Drogat, B.; Auguste, P.; Nguyen, D.T.; Bouchecareilh, M.; Pineau, R.; Nalbantoglu, J.; Kaufman, R.J.; Chevet, E.; Bikfalvi, A.; Moenner, M. IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-A expression and contributes to angiogenesis and tumor growth in vivo. Cancer Res. 2007, 67, 6700–6707. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Lipson, K.L.; Sargent, K.E.; Mercurio, A.M.; Hunt, J.S.; Ron, D.; Urano, F. Transcriptional regulation of VEGF-a by the unfolded protein response pathway. PLoS ONE 2010, 5, e9575. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qi, X.; Chen, Z.; Shaw, L.; Cai, J.; Smith, L.H.; Grant, M.B.; Boulton, M.E. Targeting the IRE1A/XBP1 and ATF6 arms of the unfolded protein response enhances VEGF blockade to prevent retinal and choroidal neovascularization. Am. J. Pathol. 2013, 182, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Romero-Ramirez, L.; Cao, H.; Regalado, M.P.; Kambham, N.; Siemann, D.; Kim, J.J.; Le, Q.T.; Koong, A.C. X box-binding protein 1 regulates angiogenesis in human pancreatic adenocarcinomas. Transl. Oncol. 2009, 2, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Zhao, K.; Wang, X.; Li, H.; Yu, M.; He, M.; Xue, X.; Zhu, Y.; Zhang, C.; Cheng, Y.; et al. iASPP is an antioxidative factor and drives cancer growth and drug resistance by competing with Nrf2 for keap1 binding. Cancer Cell 2017, 32, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. Perk promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Han, H.; Wu, L.; Pan, B.; Dong, B.; Yin, C.C.; Tian, Z.; Liu, X.; Yang, Y.; Zhang, H.; et al. iASPP facilitates tumor growth by promoting mTOR-dependent autophagy in human non-small-cell lung cancer. Cell Death Dis. 2017, 8, e3150. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Liu, Y.; Liu, H.; Chen, X.; Liu, M.; Che, H.; Guo, F.; Wang, C.; Zhang, D.; Wu, J.; et al. PERK silence inhibits glioma cell growth under low glucose stress by blockage of p-AKT and subsequent HK2’s mitochondria translocation. Sci. Rep. 2015, 5, 9065. [Google Scholar] [CrossRef] [PubMed]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Banh, A.; Papandreou, I.; Cao, H.; Galvez, M.G.; Gurtner, G.C.; Denko, N.C.; Le, Q.T.; Koong, A.C. Imaging the unfolded protein response in primary tumors reveals microenvironments with metabolic variations that predict tumor growth. Cancer Res. 2010, 70, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Dalton, L.E.; Clarke, H.J.; Knight, J.; Lawson, M.H.; Wason, J.; Lomas, D.A.; Howat, W.J.; Rintoul, R.C.; Rassl, D.M.; Marciniak, S.J. The endoplasmic reticulum stress marker chop predicts survival in malignant mesothelioma. Br. J. Cancer 2013, 108, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.P.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Onda, M.; Nagai, H.; Nagahata, T.; Ogawa, K.; Emi, M. Upregulation and overexpression of human X-box binding protein 1 (hXBP-1) gene in primary breast cancers. Breast Cancer 2003, 10, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Yoshimatsu, K.; Watanabe, K.; Yokomizo, H.; Otani, T.; Matsumoto, A.; Osawa, G.; Onda, M.; Ogawa, K. Overexpression of human X-box binding protein 1 (XBP-1) in colorectal adenomas and adenocarcinomas. Anticancer Res. 2007, 27, 127–131. [Google Scholar] [PubMed]
- Kim, K.M.; Yu, T.K.; Chu, H.H.; Park, H.S.; Jang, K.Y.; Moon, W.S.; Kang, M.J.; Lee, D.G.; Kim, M.H.; Lee, J.H.; et al. Expression of er stress and autophagy-related molecules in human non-small cell lung cancer and premalignant lesions. Int. J. Cancer 2012, 131, E362–E370. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and GRP78 genes in human hepatocellular carcinoma: A possible involvement of the er stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Thorpe, J.A.; Schwarze, S.R. IRE1α controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell Stress Chaperones 2010, 15, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Bagratuni, T.; Wu, P.; Gonzalez de Castro, D.; Davenport, E.L.; Dickens, N.J.; Walker, B.A.; Boyd, K.; Johnson, D.C.; Gregory, W.; Morgan, G.J.; et al. XBP1S levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood 2010, 116, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.R.; Sukhdeo, K.; Protopopova, M.; Sinha, R.; Enos, M.; Carrasco, D.E.; Zheng, M.; Mani, M.; Henderson, J.; Pinkus, G.S.; et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007, 11, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Schardt, J.A.; Weber, D.; Eyholzer, M.; Mueller, B.U.; Pabst, T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin. Cancer Res. 2009, 15, 3834–3841. [Google Scholar] [CrossRef] [PubMed]
- Donze, O.; Jagus, R.; Koromilas, A.E.; Hershey, J.W.; Sonenberg, N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J. 1995, 14, 3828–3834. [Google Scholar] [PubMed]
- Sequeira, S.J.; Ranganathan, A.C.; Adam, A.P.; Iglesias, B.V.; Farias, E.F.; Aguirre-Ghiso, J.A. Inhibition of proliferation by perk regulates mammary acinar morphogenesis and tumor formation. PLoS ONE 2007, 2, e615. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Green, W.; Finzi, G.; Sessa, F.; Nouraie, M.; Lee, E.L.; Morgano, A.; Moschetta, A.; Cattaneo, M.; Mariani-Costantini, R.; et al. Sel1l, an upr response protein, a potential marker of colonic cell transformation. Dig. Dis. Sci. 2012, 57, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Fontanella, E.; Canton, C.; Delia, D.; Biunno, I. SEL1L affects human pancreatic cancer cell cycle and invasiveness through modulation of PTEN and genes related to cell-matrix interactions. Neoplasia 2005, 7, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Granelli, P.; Cattaneo, M.; Ferrero, S.; Bottiglieri, L.; Bosari, S.; Fichera, G.; Biunno, I. SEL1L and squamous cell carcinoma of the esophagus. Clin. Cancer Res. 2004, 10, 5857–5861. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, J.; Mai, B.; Amos, C.; Killary, A.M.; Sen, S.; Wei, C.; Frazier, M.L. A single-nucleotide polymorphism in tumor suppressor gene SEL1L as a predictive and prognostic marker for pancreatic ductal adenocarcinoma in caucasians. Mol. Carcinog. 2012, 51, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Mellai, M.; Cattaneo, M.; Storaci, A.M.; Annovazzi, L.; Cassoni, P.; Melcarne, A.; De Blasio, P.; Schiffer, D.; Biunno, I. SEL1L SNP rs12435998, a predictor of glioblastoma survival and response to radio-chemotherapy. Oncotarget 2015, 6, 12452–12467. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Mahon, P.C.; Oh, J.; Kelly, B.; Krishnamachary, B.; Pearson, M.; Chan, D.A.; Giaccia, A.J.; Semenza, G.L. OS-9 interacts with hypoxia-inducible factor 1α and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1α. Mol. Cell 2005, 17, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K.; Konishi, H.; Arima, C.; Tomida, S.; Takeuchi, T.; Shimada, Y.; Yatabe, Y.; Mitsudomi, T.; Osada, H.; Takahashi, T. Novel metastasis-related gene cim functions in the regulation of multiple cellular stress-response pathways. Cancer Res. 2010, 70, 9949–9958. [Google Scholar] [CrossRef] [PubMed]
- Sha, H.; Sun, S.; Francisco, A.B.; Ehrhardt, N.; Xue, Z.; Liu, L.; Lawrence, P.; Mattijssen, F.; Guber, R.D.; Panhwar, M.S.; et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab. 2014, 20, 458–470. [Google Scholar] [CrossRef] [PubMed]
- DeLaBarre, B.; Christianson, J.C.; Kopito, R.R.; Brunger, A.T. Central pore residues mediate the p97/VCP activity required for erad. Mol. Cell 2006, 22, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, M.E.; Rico, C.; Tsoi, M.; Vivancos, M.; Filimon, S.; Paquet, M.; Boerboom, D. Pharmacological targeting of valosin containing protein (VCP) induces DNA damage and selectively kills canine lymphoma cells. BMC Cancer 2015, 15, 479. [Google Scholar] [CrossRef] [PubMed]
- Pasetto, M.; Antignani, A.; Ormanoglu, P.; Buehler, E.; Guha, R.; Pastan, I.; Martin, S.E.; FitzGerald, D.J. Whole-genome RNAI screen highlights components of the endoplasmic reticulum/Golgi as a source of resistance to immunotoxin-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, E1135–E1142. [Google Scholar] [CrossRef] [PubMed]
- Valle, C.W.; Min, T.; Bodas, M.; Mazur, S.; Begum, S.; Tang, D.; Vij, N. Critical role of VCP/p97 in the pathogenesis and progression of non-small cell lung carcinoma. PLoS ONE 2011, 6, e29073. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Beverly, L.J. Regulation of VCP/p97 demonstrates the critical balance between cell death and epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 2015, 6, 17725–17737. [Google Scholar] [CrossRef] [PubMed]
- Cagnetta, A.; Caffa, I.; Acharya, C.; Soncini, D.; Acharya, P.; Adamia, S.; Pierri, I.; Bergamaschi, M.; Garuti, A.; Fraternali, G.; et al. APO866 increases antitumor activity of cyclosporin-a by inducing mitochondrial and endoplasmic reticulum stress in leukemia cells. Clin. Cancer Res. 2015, 21, 3934–3945. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, L.; Li, J.; Cheng, Y.; Chen, J.; Shen, M.; Zhang, S.; Wei, H. Insulin resistance contributes to multidrug resistance in HepG2 cells via activation of the PERK signaling pathway and upregulation of Bcl-2 and P-gp. Oncol. Rep. 2016, 35, 3018–3024. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.; Cao, Y.; Rodriguez, P.C. Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity: A promising opportunity for cancer immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Kopecka, J.; Panada, E.; Barak, S.; Rubinstein, M. The role of C/EBP-β lip in multidrug resistance. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. Perk induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef] [PubMed]
- Dadey, D.Y.; Kapoor, V.; Khudanyan, A.; Urano, F.; Kim, A.H.; Thotala, D.; Hallahan, D.E. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 2016, 7, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [PubMed]
- Hamada, A.; Miyano, H.; Watanabe, H.; Saito, H. Interaction of imatinib mesilate with human P-glycoprotein. J. Pharmacol. Exp. Ther. 2003, 307, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Kusio-Kobialka, M.; Podszywalow-Bartnicka, P.; Peidis, P.; Glodkowska-Mrowka, E.; Wolanin, K.; Leszak, G.; Seferynska, I.; Stoklosa, T.; Koromilas, A.E.; Piwocka, K. The PERK-eIF2α phosphorylation arm is a pro-survival pathway of BCR-ABL signaling and confers resistance to imatinib treatment in chronic myeloid leukemia cells. Cell Cycle 2012, 11, 4069–4078. [Google Scholar] [CrossRef] [PubMed]
- Maytin, E.V.; Habener, J.F. Transcription factors C/EBPα, C/EBPβ, and CHOP (Gadd153) expressed during the differentiation program of keratinocytes in vitro and in vivo. J. Investig. Dermatol. 1998, 110, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Meir, O.; Dvash, E.; Werman, A.; Rubinstein, M. C/EBP-β regulates endoplasmic reticulum stress-triggered cell death in mouse and human models. PLoS ONE 2010, 5, e9516. [Google Scholar] [CrossRef]
- Li, X.; Li, J.P.; Yuan, H.Y.; Gao, X.; Qu, X.J.; Xu, W.F.; Tang, W. Recent advances in P-glycoprotein-mediated multidrug resistance reversal mechanisms. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Globisch, C.; Pajeva, I.K.; Wiese, M. Structure-activity relationships of a series of tariquidar analogs as multidrug resistance modulators. Bioorg. Med. Chem. 2006, 14, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Weidner, L.D.; Fung, K.L.; Kannan, P.; Moen, J.K.; Kumar, J.S.; Mulder, J.; Innis, R.B.; Gottesman, M.M.; Hall, M.D. Tariquidar is an inhibitor and not a substrate of human and mouse P-glycoprotein. Drug Metab. Dispos. 2016, 44, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Widemann, B.C.; Pastakia, D.; Chen, C.C.; Yang, S.X.; Cole, D.; Balis, F.M. Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P-glycoprotein inhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.R.; Chang, S.Y.; Hong, E.H.; Kwon, B.E.; Kim, H.M.; Kim, Y.J.; Lee, J.; Cho, H.J.; Cheon, J.H.; Ko, H.J. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget 2014, 5, 12331–12345. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Lang, J.Y.; Hung, M.C.; Sengupta, K.; Banerjee, S.K.; Baksi, K.; Banerjee, D.K. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J. Biol. Chem. 2011, 286, 29127–29138. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel perk kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Auf, G.; Jabouille, A.; Guerit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1α is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Romeril, S.P.; Shu, A.; Ralph, J.; Medina, J.R.; Feng, Y.; Li, W.H.; Grant, S.W.; Heerding, D.A.; Minthorn, E.; et al. Discovery of GSK2656157: An optimized perk inhibitor selected for preclinical development. ACS Med. Chem. Lett. 2013, 4, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Naczki, C.; Koritzinsky, M.; Fels, D.; Blais, J.; Hu, N.; Harding, H.; Novoa, I.; Varia, M.; Raleigh, J.; et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005, 24, 3470–3481. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.; Ding, L.W.; Sun, Q.Y.; Torres-Fernandez, L.A.; Tan, S.Z.; Xiao, J.; Lim, S.L.; Garg, M.; Lee, K.L.; Kitajima, S.; et al. Selective inhibition of unfolded protein response induces apoptosis in pancreatic cancer cells. Oncotarget 2014, 5, 4881–4894. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1α rnase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Kebache, S.; Fazel, A.; Wong, H.N.; Jenna, S.; Emadali, A.; Lee, E.H.; Bergeron, J.J.; Kaufman, R.J.; Larose, L.; et al. Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol. Biol. Cell 2004, 15, 4248–4260. [Google Scholar] [CrossRef] [PubMed]
- Niederreiter, L.; Fritz, T.M.; Adolph, T.E.; Krismer, A.M.; Offner, F.A.; Tschurtschenthaler, M.; Flak, M.B.; Hosomi, S.; Tomczak, M.F.; Kaneider, N.C.; et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J. Exp. Med. 2013, 210, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Ranatunga, S.; Kriss, C.L.; Cubitt, C.L.; Tao, J.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.C. Inhibition of er stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Investig. 2014, 124, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mrna splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/EIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013, 15, R2. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an ire1α endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of er stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C.; et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wei, L.; Fan, F.; Ji, S.; Zhang, S.; Geng, J.; Hong, L.; Fan, X.; Chen, Q.; Tian, J.; et al. Integration of hippo signalling and the unfolded protein response to restrain liver overgrowth and tumorigenesis. Nat. Commun. 2015, 6, 6239. [Google Scholar] [CrossRef] [PubMed]
- Morrow, K.; Hernandez, C.P.; Raber, P.; Del Valle, L.; Wilk, A.M.; Majumdar, S.; Wyczechowska, D.; Reiss, K.; Rodriguez, P.C. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia 2013, 27, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Dunner, K., Jr.; Boise, L.H.; Chiao, P.J.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005, 65, 11510–11519. [Google Scholar] [CrossRef] [PubMed]
- Auner, H.W.; Moody, A.M.; Ward, T.H.; Kraus, M.; Milan, E.; May, P.; Chaidos, A.; Driessen, C.; Cenci, S.; Dazzi, F.; et al. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS ONE 2013, 8, e74415. [Google Scholar] [CrossRef] [PubMed]
- Brem, G.J.; Mylonas, I.; Bruning, A. Eeyarestatin causes cervical cancer cell sensitization to bortezomib treatment by augmenting ER stress and CHOP expression. Gynecol. Oncol. 2013, 128, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Gronberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-tumor interactome screen reveals er stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Baronchelli, S.; Schiffer, D.; Mellai, M.; Caldera, V.; Saccani, G.J.; Dalpra, L.; Daga, A.; Orlandi, R.; DeBlasio, P.; et al. Down-modulation of SEL1L, an unfolded protein response and endoplasmic reticulum-associated degradation protein, sensitizes glioma stem cells to the cytotoxic effect of valproic acid. J. Biol. Chem. 2014, 289, 2826–2838. [Google Scholar] [CrossRef] [PubMed]
- Zha, W.; Wang, G.; Xu, W.; Liu, X.; Wang, Y.; Zha, B.S.; Shi, J.; Zhao, Q.; Gerk, P.M.; Studer, E.; et al. Inhibition of P-glycoprotein by hiv protease inhibitors increases intracellular accumulation of berberine in murine and human macrophages. PLoS ONE 2013, 8, e54349. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, G.; Wei, H.; Sun, J.; Chen, J.; Xie, B.; Wang, B.; Gu, J.; Li, C.; Tian, B.; et al. The endoplasmic reticulum stress response is associated with insulin resistance-mediated drug resistance in HEPG2 cells. Neoplasma 2015, 62, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, G.; Mathur, A.; Mallade, P.; Gerlach, S.; Willis, J.; Datta, A.; Srivastav, S.; Abdel-Mageed, A.B.; Mondal, D. Nelfinavir targets multiple drug resistance mechanisms to increase the efficacy of doxorubicin in MCF-7/DOX breast cancer cells. Biochimie 2016, 124, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Asano, T.; Okubo, K.; Isono, M.; Asano, T. Nelfinavir and ritonavir kill bladder cancer cells synergistically by inducing endoplasmic reticulum stress. Oncol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.C.; Yang, J.S.; Lin, M.W.; Hsu, S.C.; Lin, J.J.; Lin, H.J.; Hsia, T.C.; Liao, C.L.; Yang, M.D.; Fan, M.J.; et al. Emodin has cytotoxic and protective effects in rat C6 glioma cells: Roles of Mdr1a and nuclear factor kB in cell survival. J. Pharmacol. Exp. Ther. 2009, 330, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Seres, M.; Ditte, P.; Breier, A.; Sulova, Z. Effect of thapsigargin on P-glycoprotein-negative and P-glycoprotein-positive L1210 mouse leukaemia cells. Gen. Physiol. Biophys. 2010, 29, 396–401. [Google Scholar] [CrossRef] [PubMed]
- El-Khattouti, A.; Sheehan, N.T.; Monico, J.; Drummond, H.A.; Haikel, Y.; Brodell, R.T.; Megahed, M.; Hassan, M. CD133(+) melanoma subpopulation acquired resistance to caffeic acid phenethyl ester-induced apoptosis is attributed to the elevated expression of ABCB5: Significance for melanoma treatment. Cancer Lett. 2015, 357, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Liang, F.; Zhang, Z.; Liu, Y.; Sheng, H.; Xie, M. S1 kills MCF-7/ADR cells more than MCF-7 cells: A protective mechanism of endoplasmic reticulum stress. Biomed. Pharmacother. 2013, 67, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Chiang, P.C.; Guh, J.H. Tunicamycin induces resistance to camptothecin and etoposide in human hepatocellular carcinoma cells: Role of cell-cycle arrest and GRP78. Naunyn-Schmiedebergs Arch. Pharmacol. 2009, 380, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaichitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-d-Glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Yoshimura, A.; Furukawa, T.; Sumizawa, T.; Nakazima, Y.; Akiyama, S. Glycosylation of P-glycoprotein in a multidrug-resistant kb cell line, and in the human tissues. Biochim. Biophys. Acta 1991, 1073, 309–315. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, J.Y.; Hait, W.N.; Yang, J.M. Regulation of the stability of P-glycoprotein by ubiquitination. Mol. Pharmacol. 2004, 66, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.; Weber, T.K.; Arceci, R.; Ramchurren, N.; Kastrinakis, W.V.; Steele, G., Jr.; Summerhayes, I.C. Inhibition of N-linked glycosylation of P-glycoprotein by tunicamycin results in a reduced multidrug resistance phenotype. Br. J. Cancer 1995, 71, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Pavlikova, L.; Seres, M.; Hano, M.; Bohacova, V.; Sevcikova, I.; Kyca, T.; Breier, A.; Sulova, Z. L1210 cells overexpressing ABCB1 drug transporters are resistant to inhibitors of the N- and O-glycosylation of proteins. Molecules 2017, 22, 1104. [Google Scholar] [CrossRef] [PubMed]
- Pavlikova, L.; Seres, M.; Imrichova, D.; Hano, M.; Rusnak, A.; Zamorova, M.; Katrlik, J.; Breier, A.; Sulova, Z. The expression of P-gp in leukemia cells is associated with cross-resistance to protein N-glycosylation inhibitor tunicamycin. Gen. Physiol. Biophys. 2016, 35, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Seres, M.; Cholujova, D.; Bubencikova, T.; Breier, A.; Sulova, Z. Tunicamycin depresses P-glycoprotein glycosylation without an effect on its membrane localization and drug efflux activity in L1210 cells. Int. J. Mol. Sci. 2011, 12, 7772–7784. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Jaffrey, S.R. Proteomic identification of protein ubiquitination events. Biotechnol. Genet. Eng. Rev. 2013, 29, 73–109. [Google Scholar] [CrossRef] [PubMed]
- Ronai, Z.A. Monoubiquitination in proteasomal degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 8894–8896. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Chen, Z.J. Diversity of polyubiquitin chains. Dev. Cell 2009, 16, 485–486. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, W.; Wu, W. Micrornas change the landscape of cancer resistance. Methods Mol. Biol. 2018, 1699, 83–89. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hano, M.; Tomášová, L.; Šereš, M.; Pavlíková, L.; Breier, A.; Sulová, Z. Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors. Molecules 2018, 23, 337. https://doi.org/10.3390/molecules23020337
Hano M, Tomášová L, Šereš M, Pavlíková L, Breier A, Sulová Z. Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors. Molecules. 2018; 23(2):337. https://doi.org/10.3390/molecules23020337
Chicago/Turabian StyleHano, Milan, Lenka Tomášová, Mário Šereš, Lucia Pavlíková, Albert Breier, and Zdena Sulová. 2018. "Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors" Molecules 23, no. 2: 337. https://doi.org/10.3390/molecules23020337
APA StyleHano, M., Tomášová, L., Šereš, M., Pavlíková, L., Breier, A., & Sulová, Z. (2018). Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors. Molecules, 23(2), 337. https://doi.org/10.3390/molecules23020337