Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
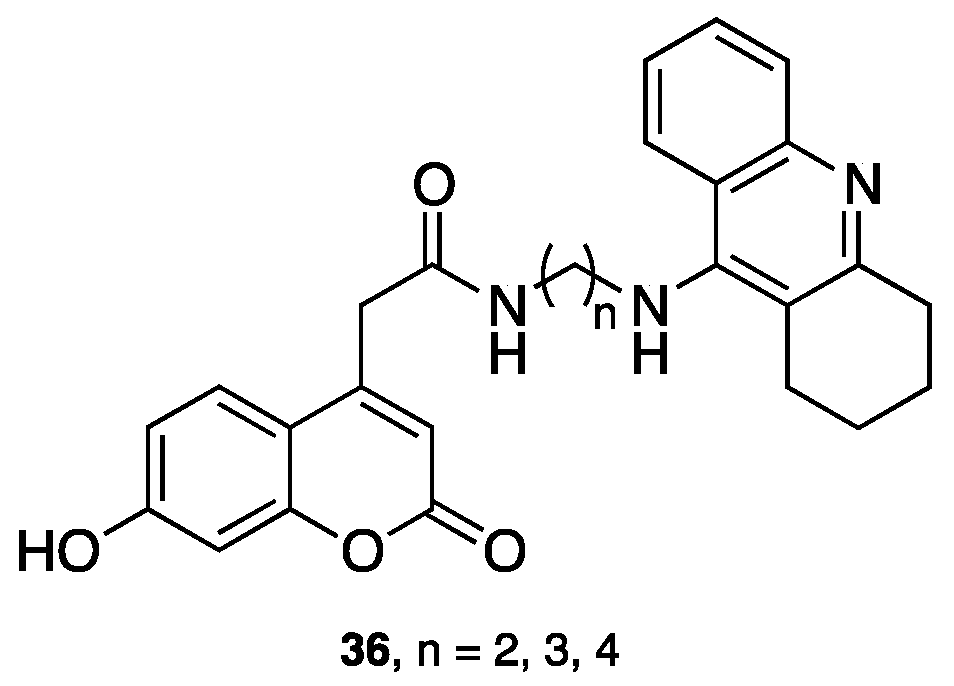{kind=link}
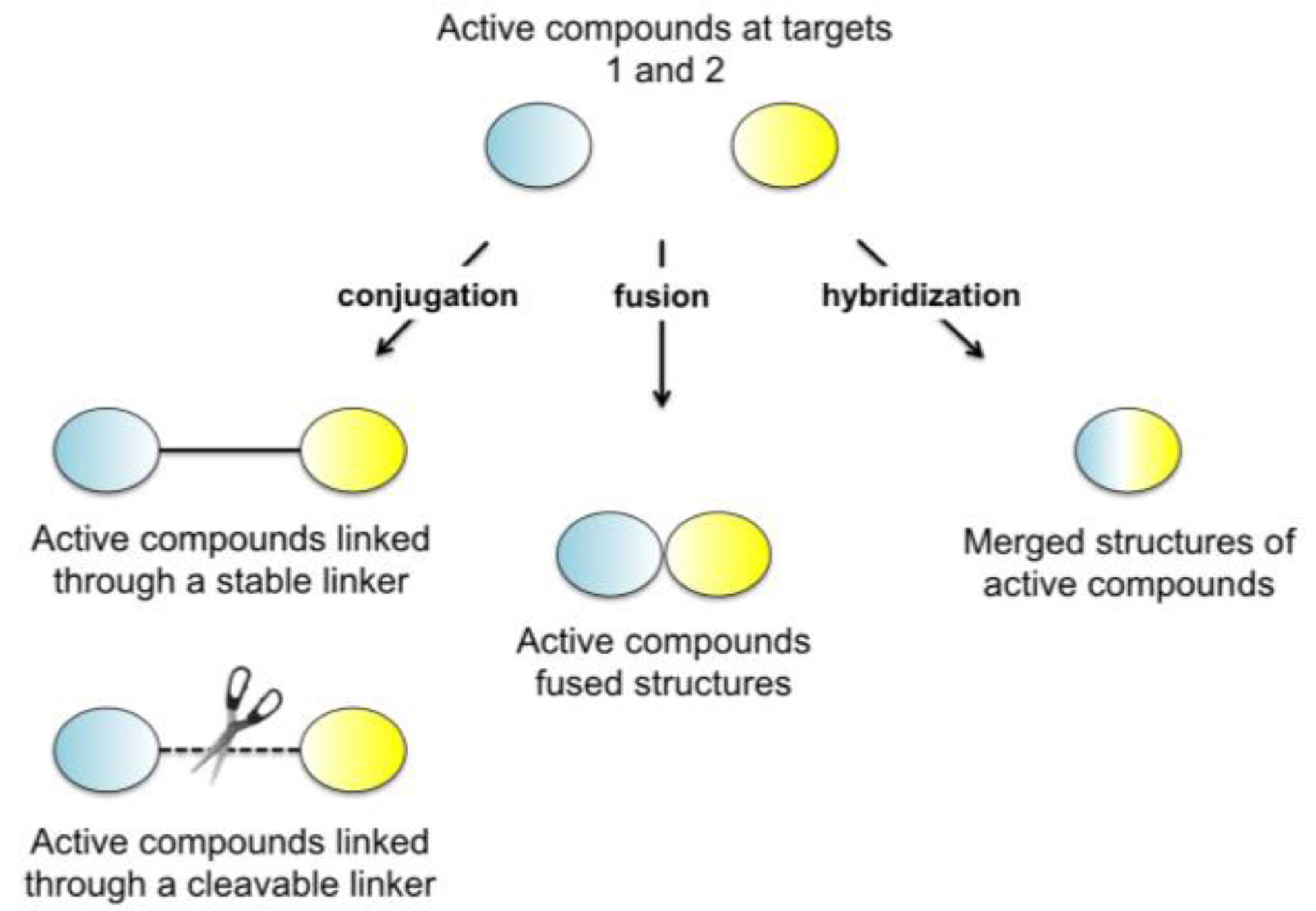{kind=link}
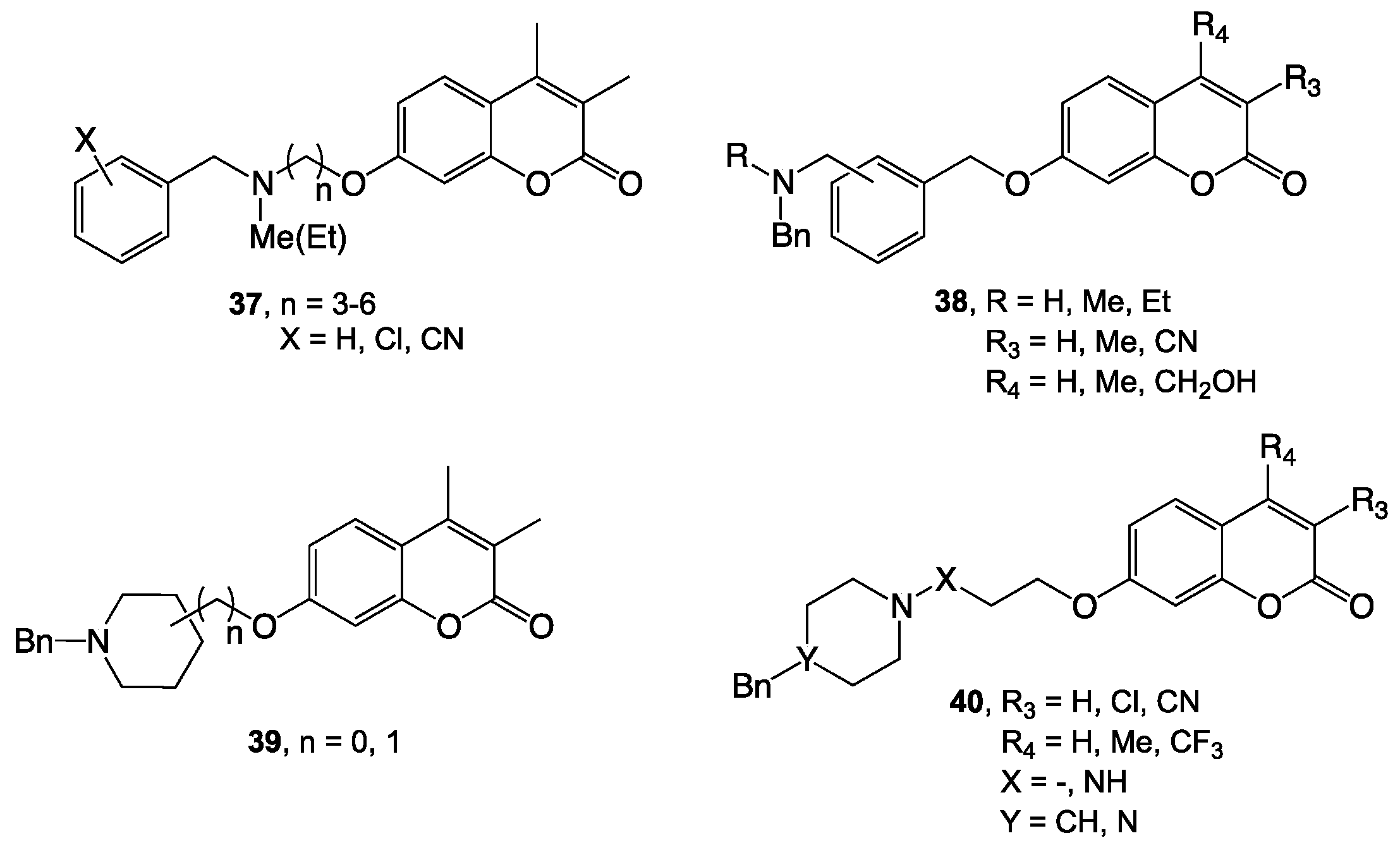{kind=link}
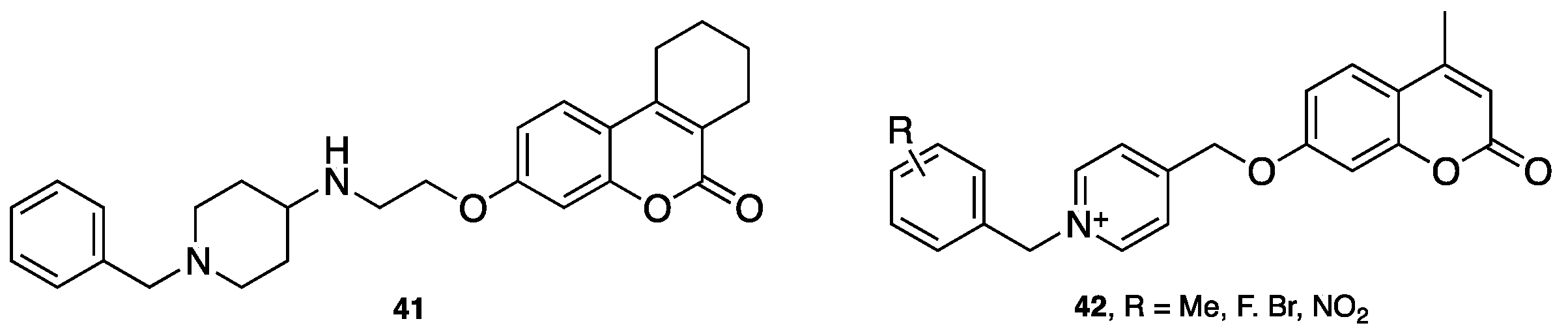{kind=link}
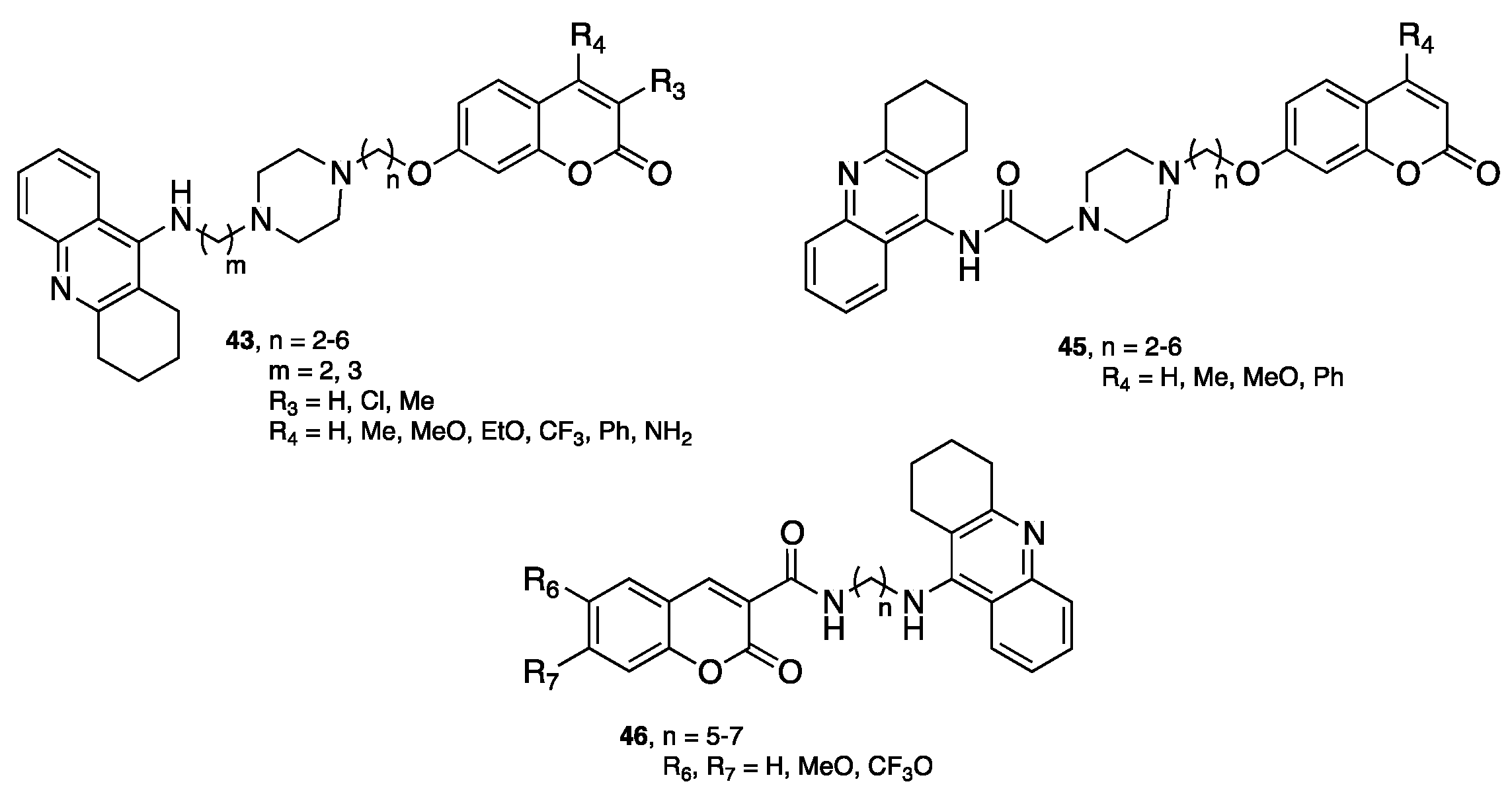{kind=link}
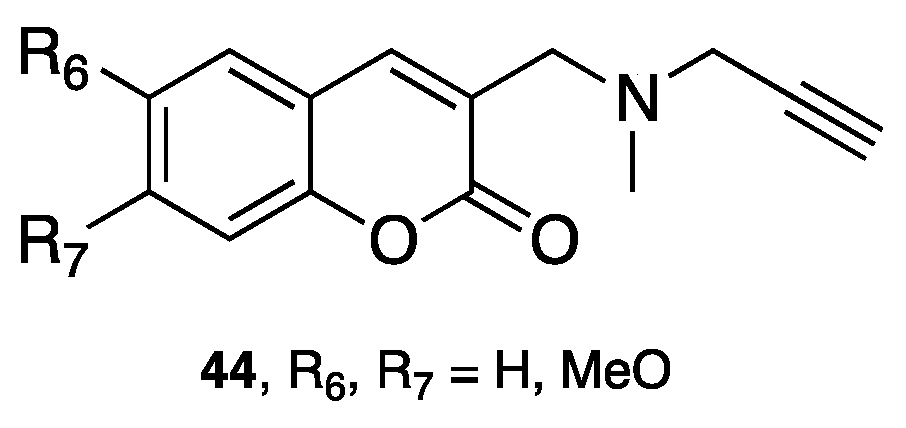{kind=link}
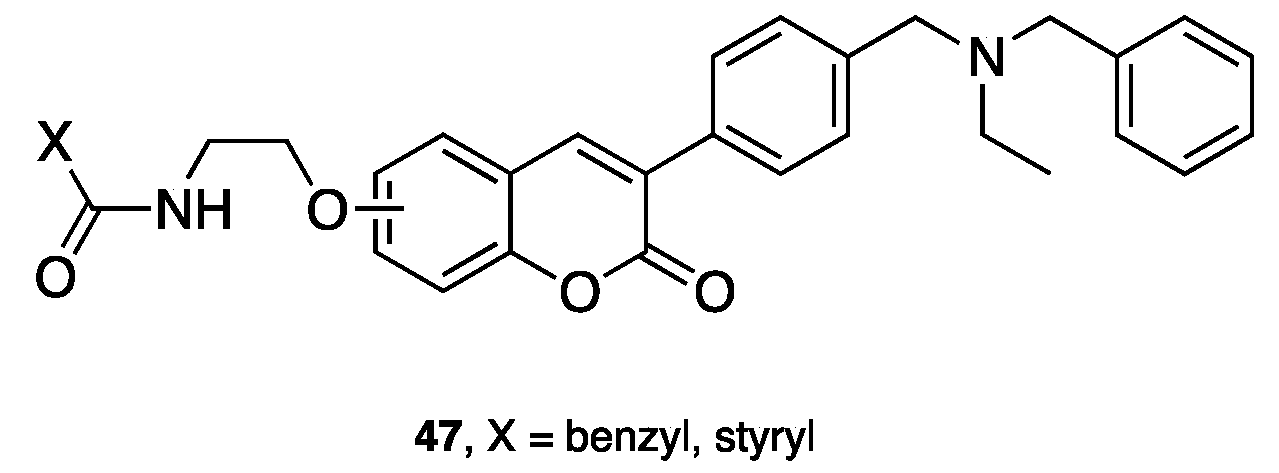{kind=link}
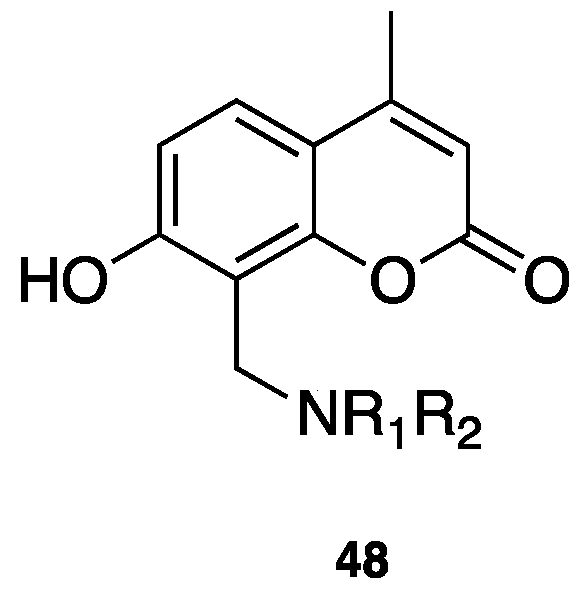{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
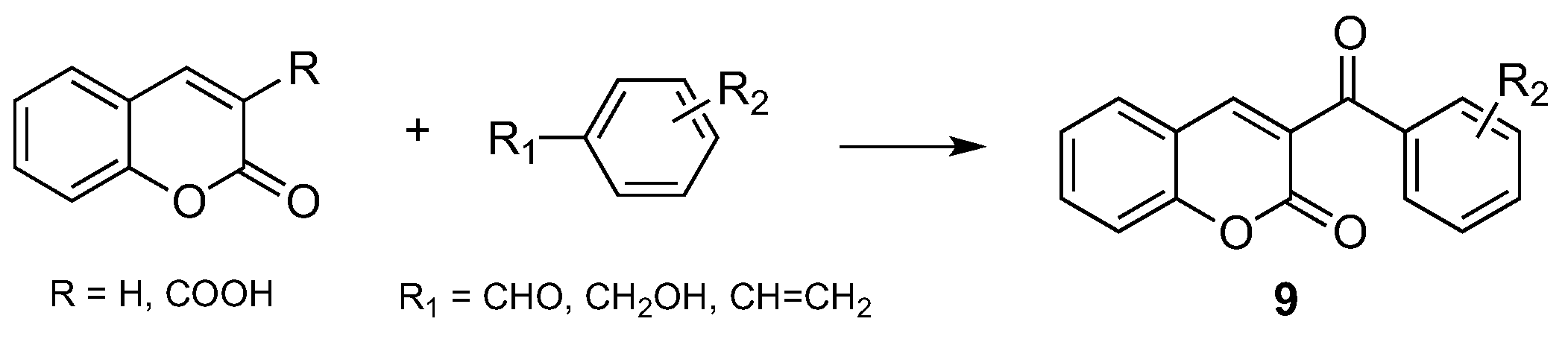{kind=link}
{kind=link}
{kind=link}
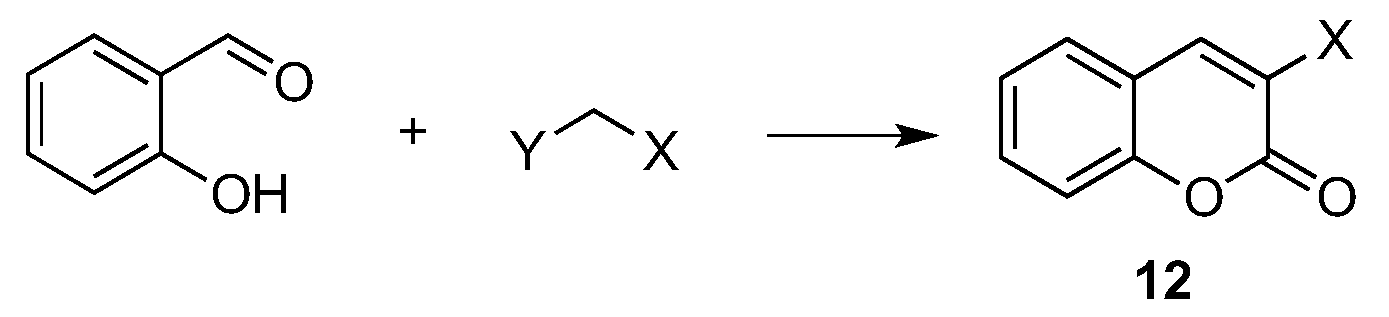{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
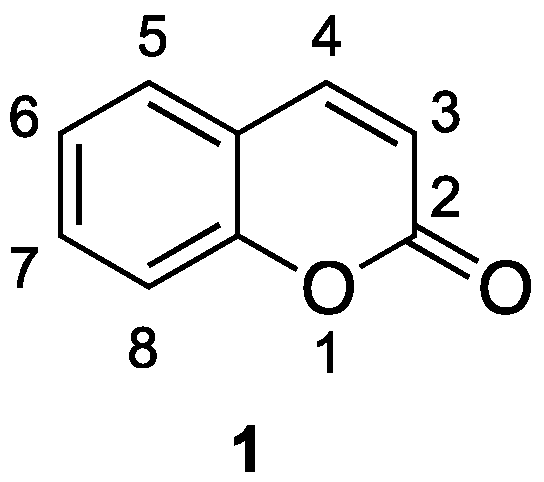
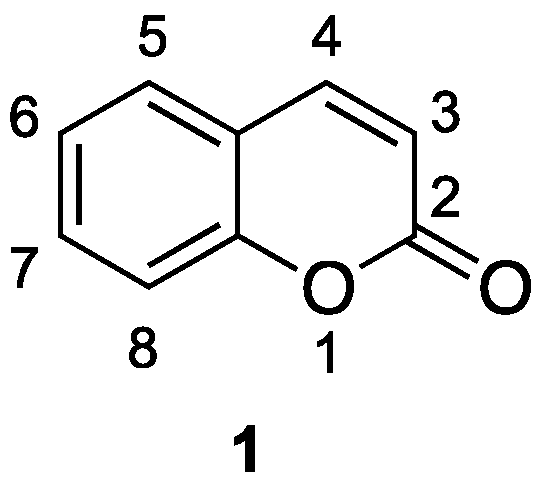
1.1. General Overview on Coumarins
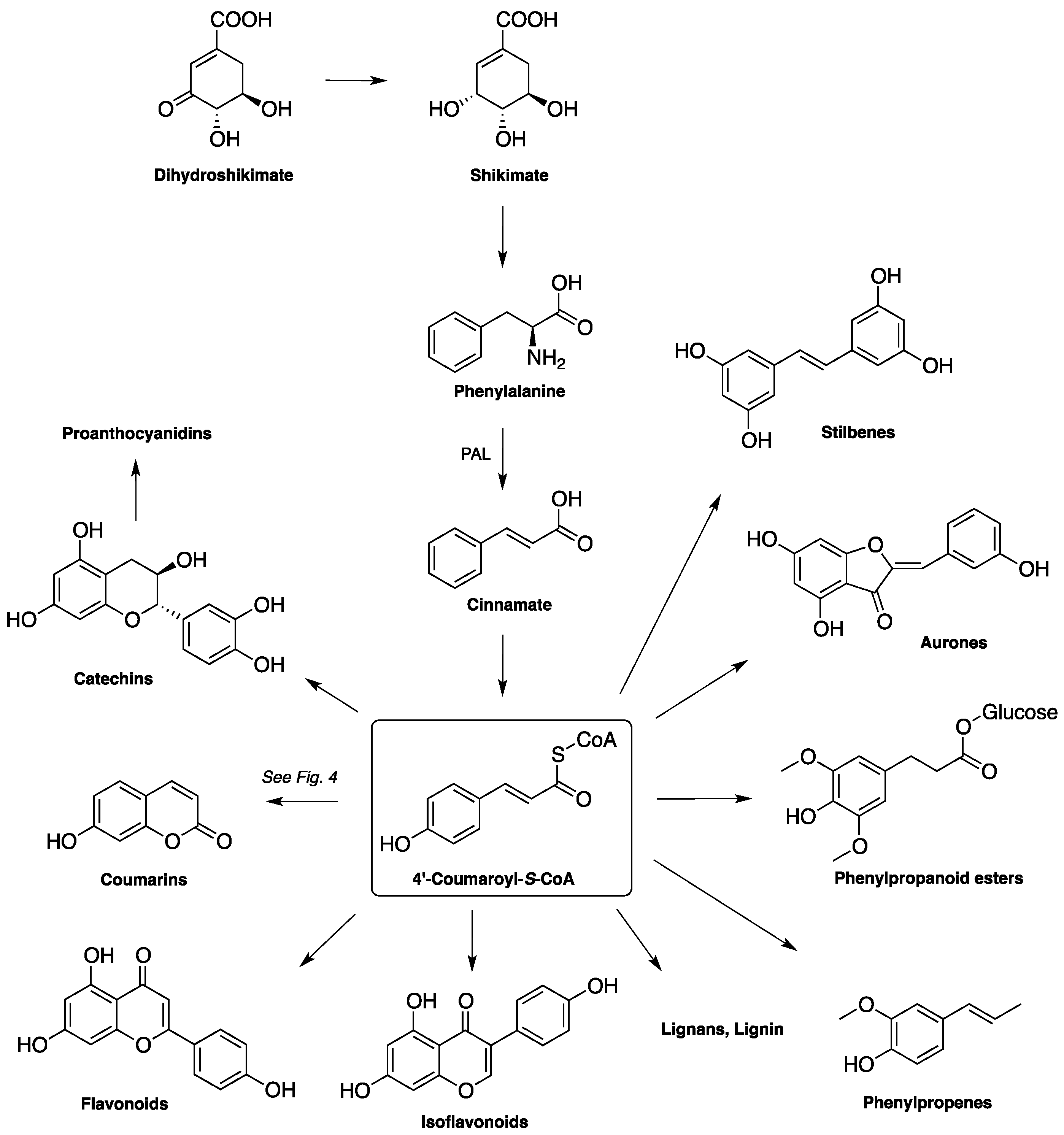
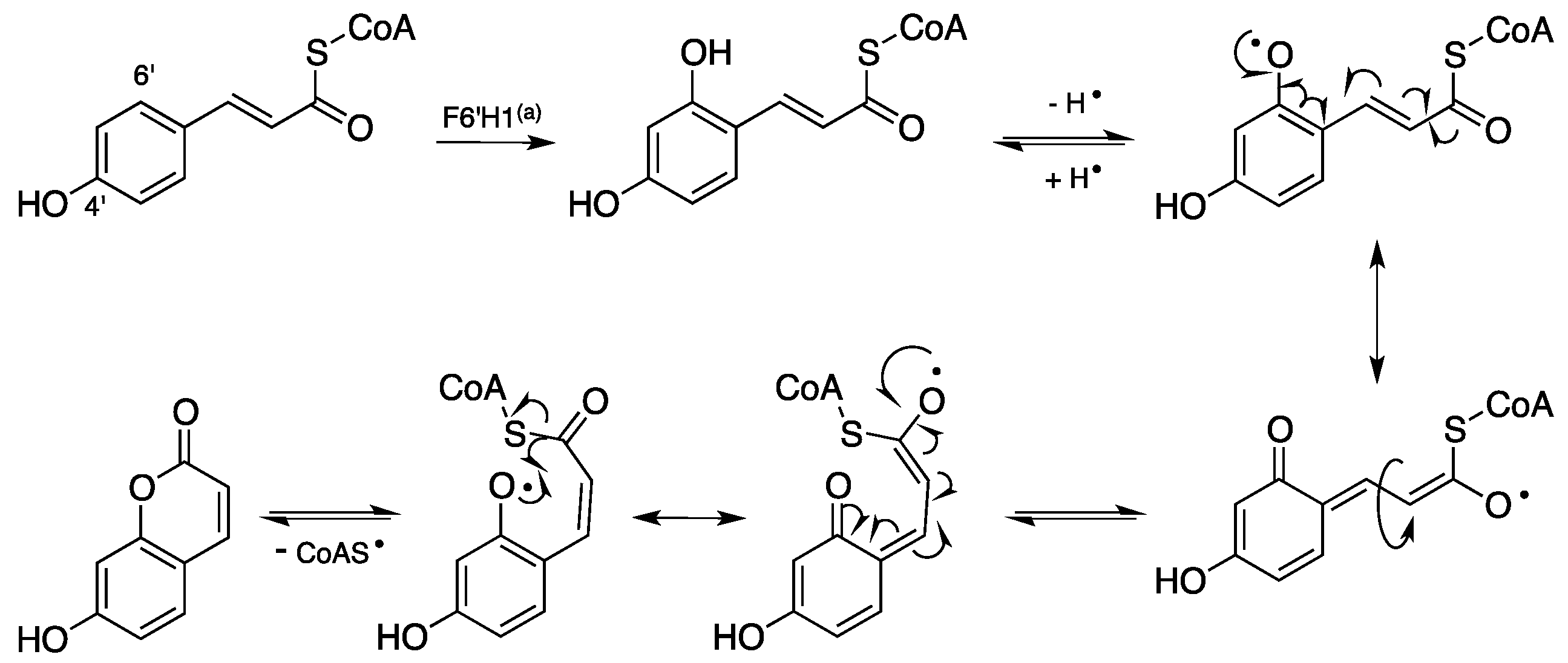
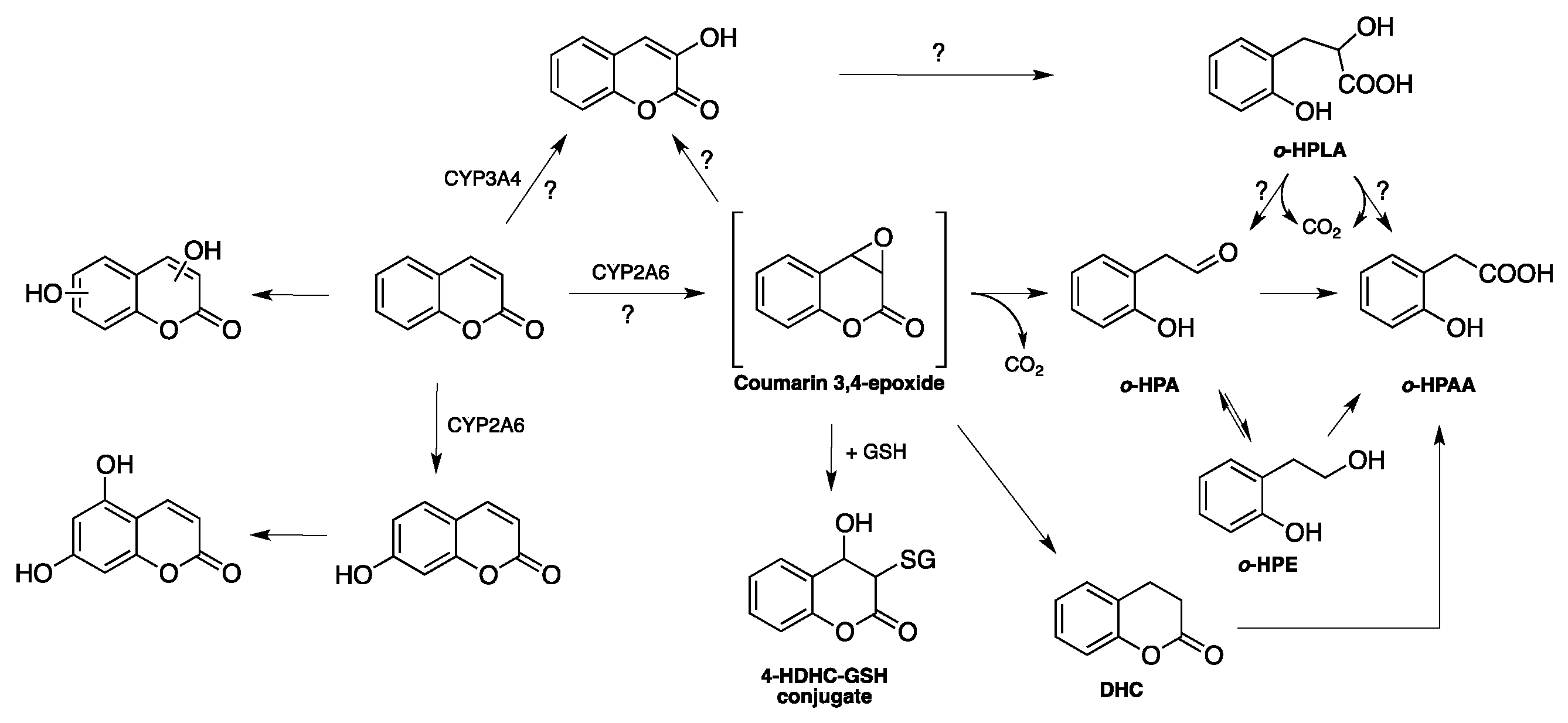
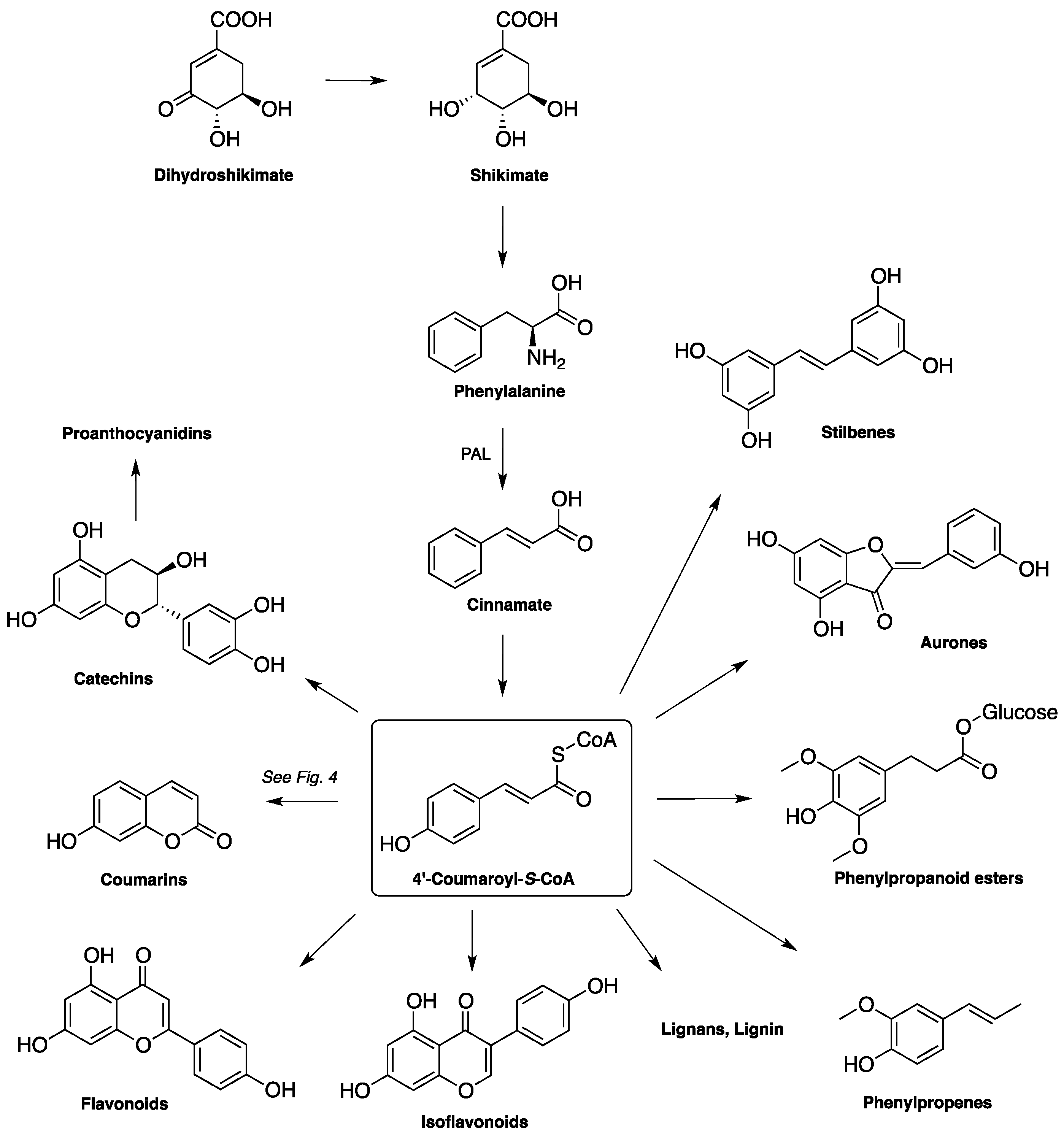
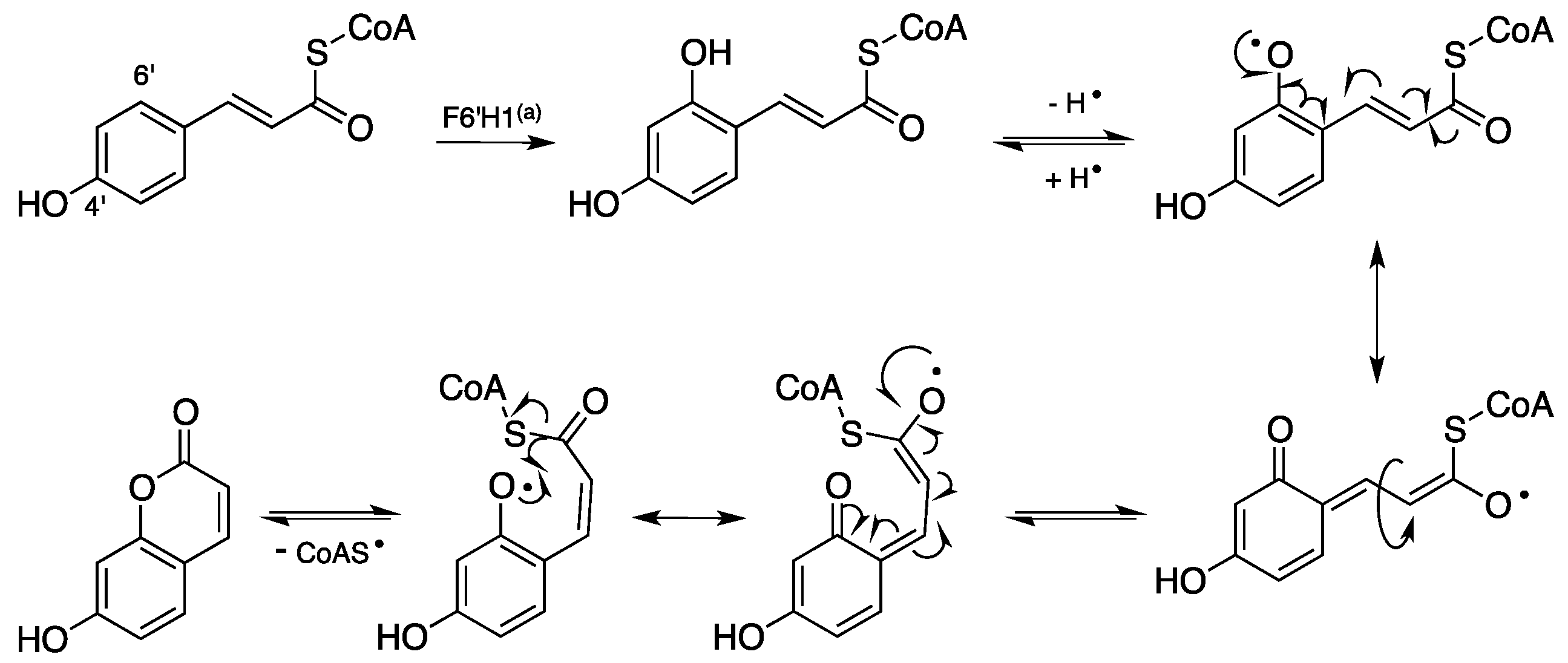
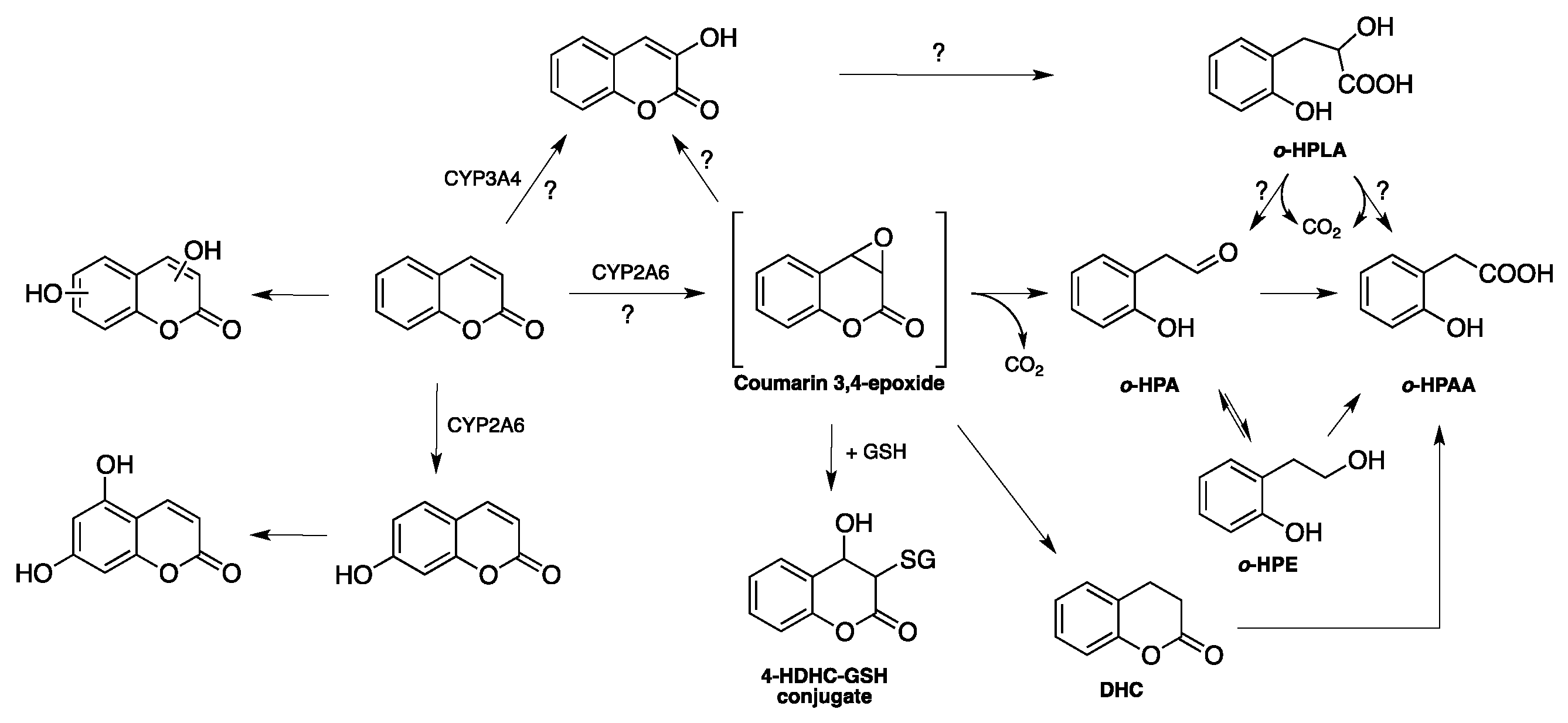
1.2. Biosynthesis and Metabolism of Coumarins
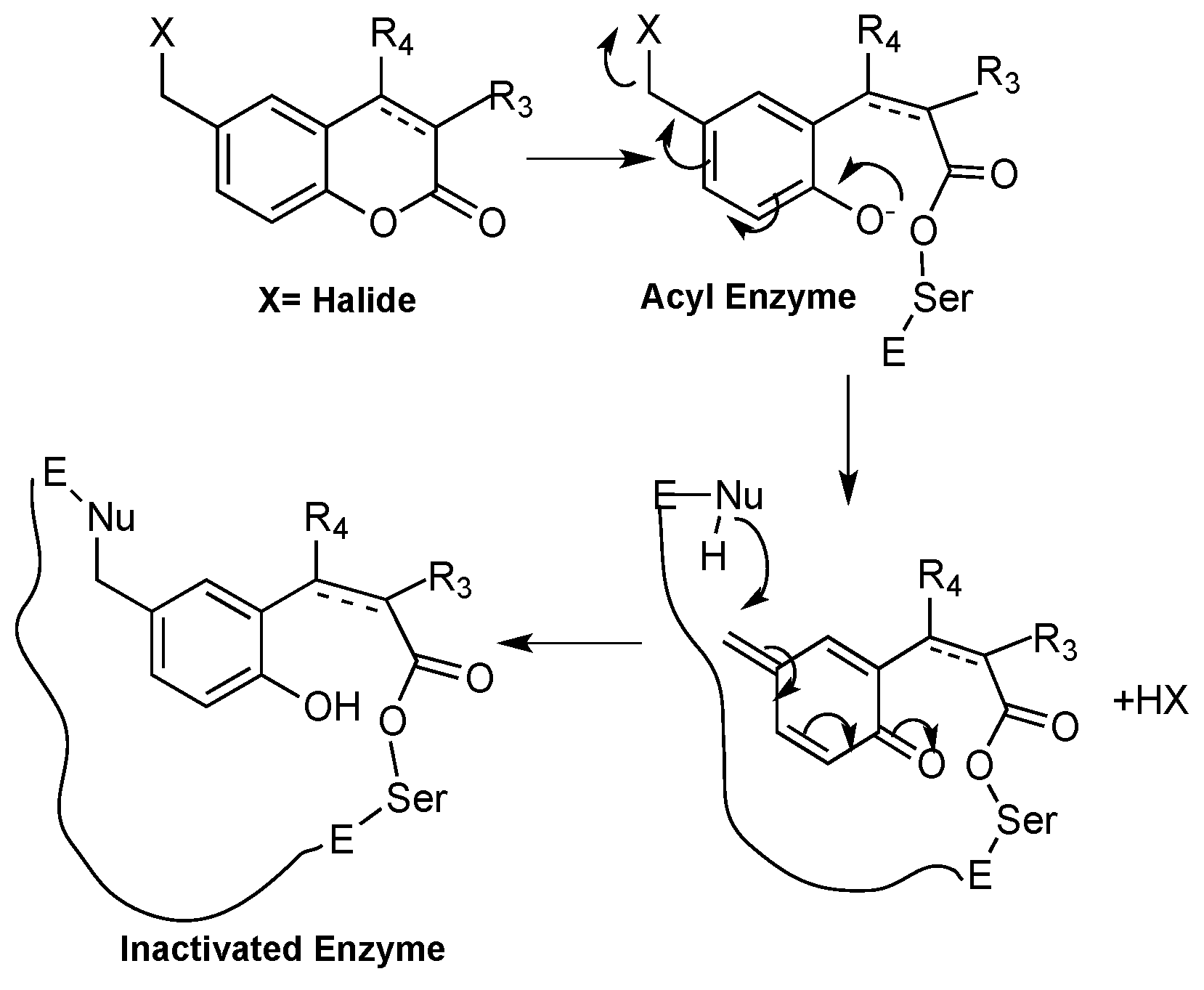
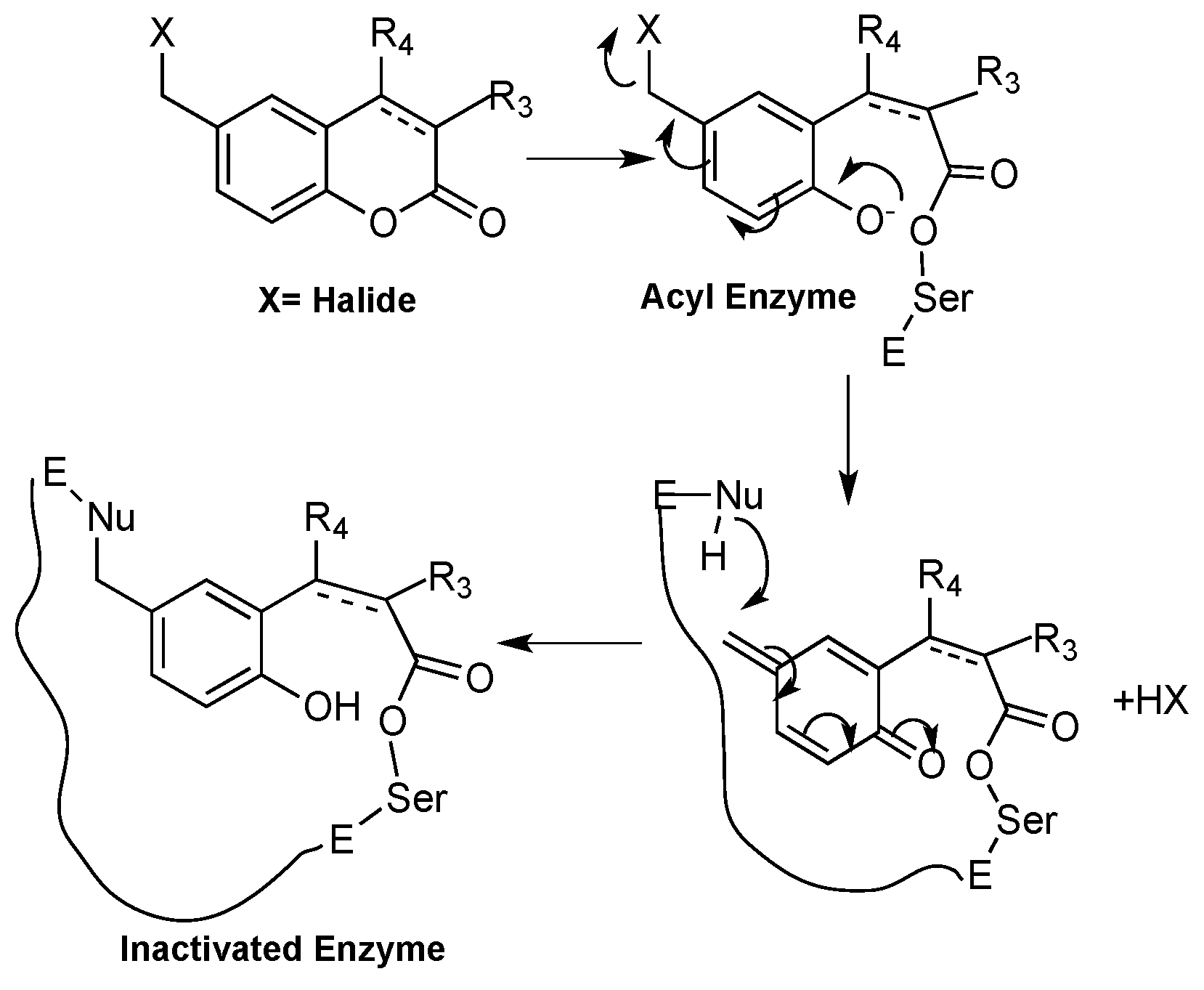
1.3. Toxicity of Coumarins
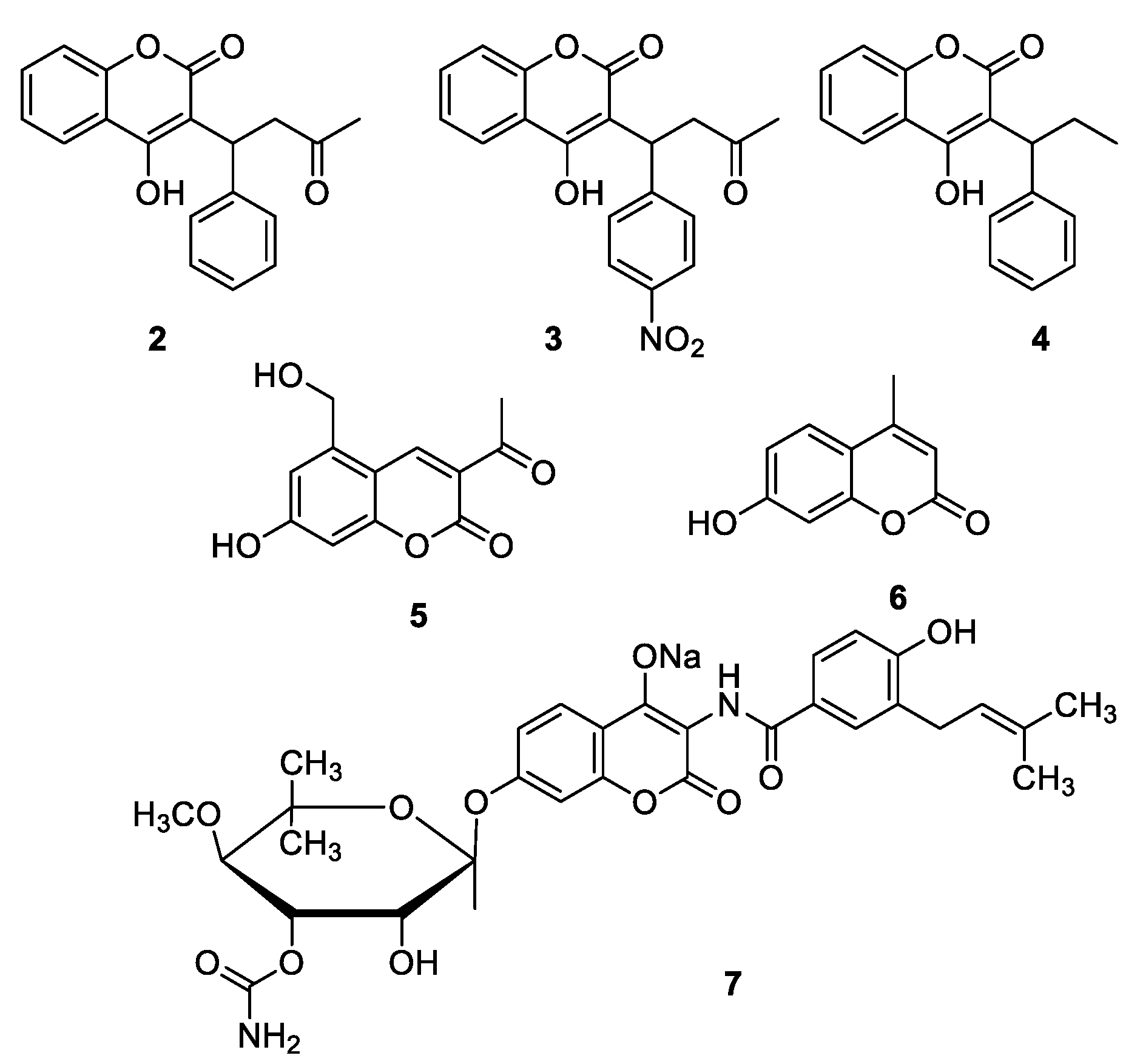
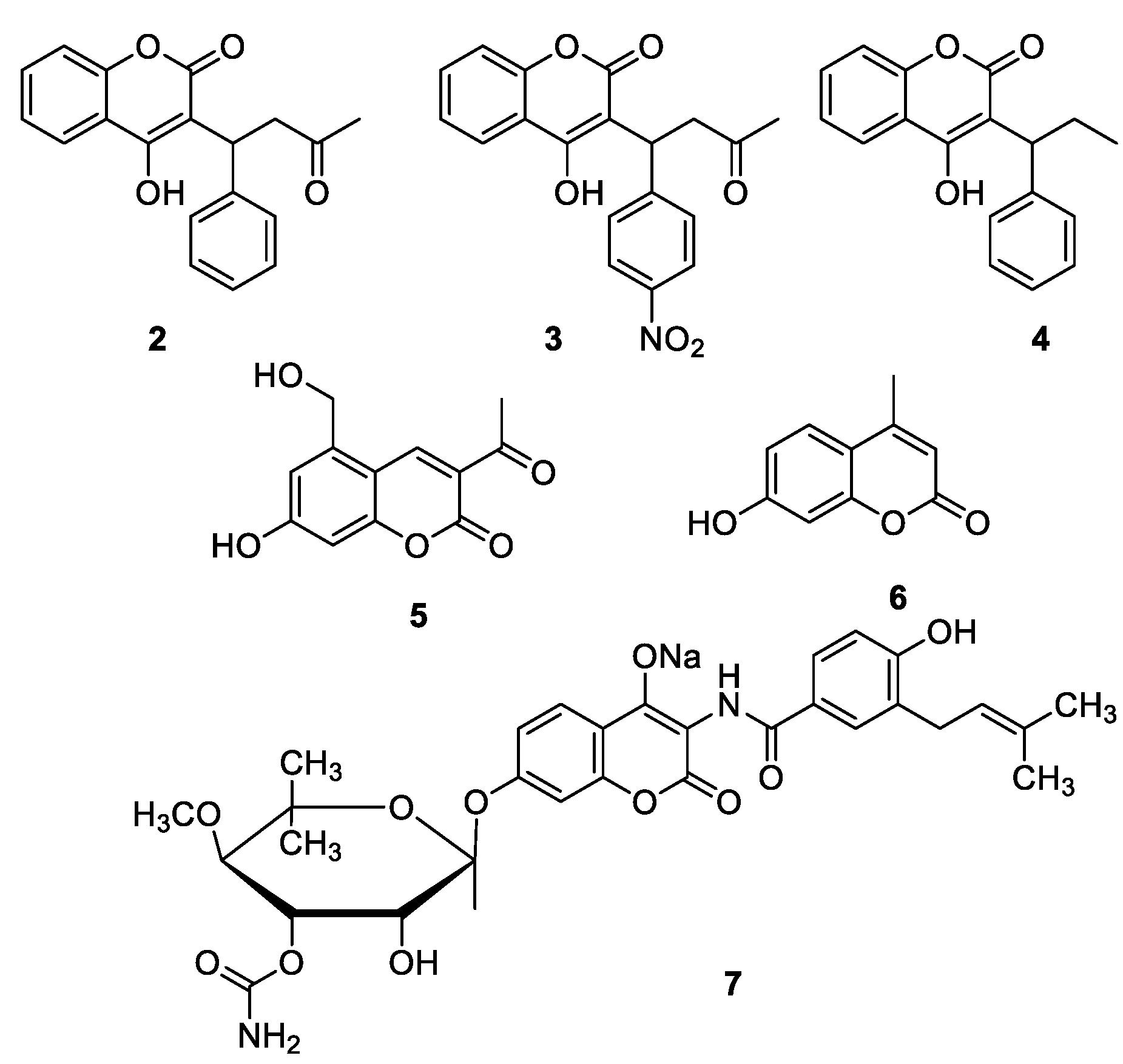
1.4. Pharmacological Activities of Coumarins
2. Synthetic Strategies for the Preparation of Coumarins
2.1. Regioselective Synthesis of 3-Substituted Coumarins
2.2. Synthesis of Coumarins by the Pechmann Reaction
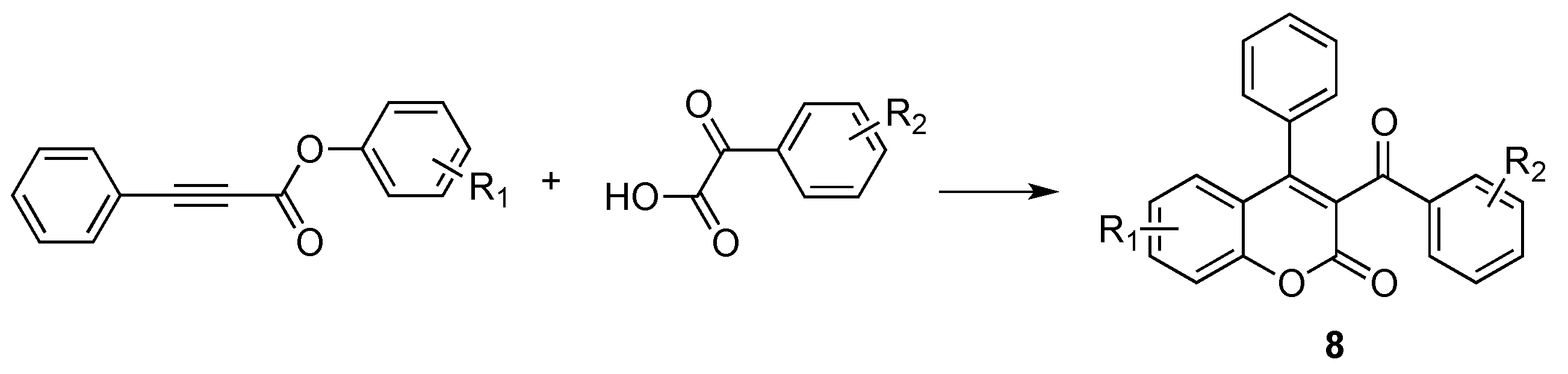
2.3. Miscellaneous Reactions for the Synthesis of Coumarins
3. Coumarins as Enzyme Inhibitors
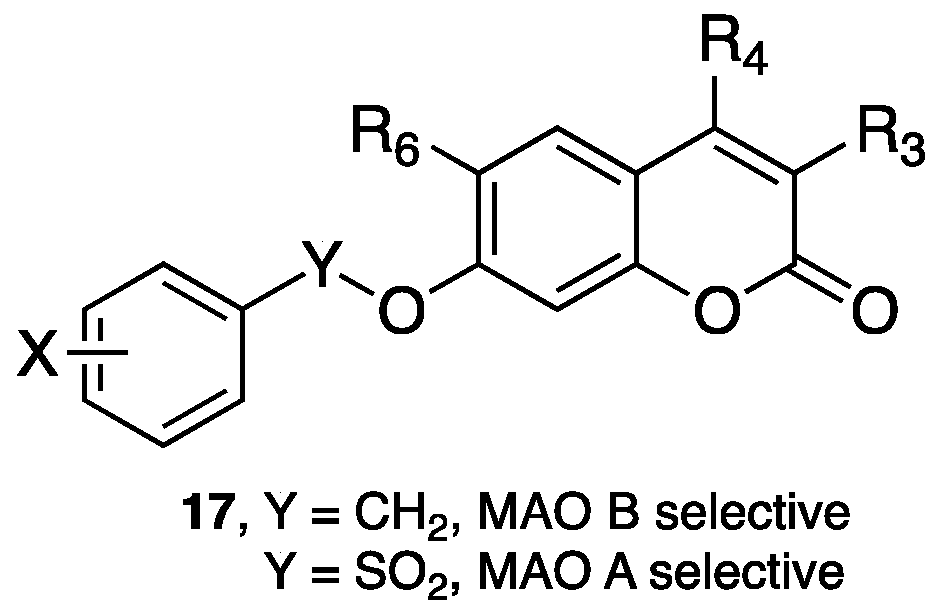
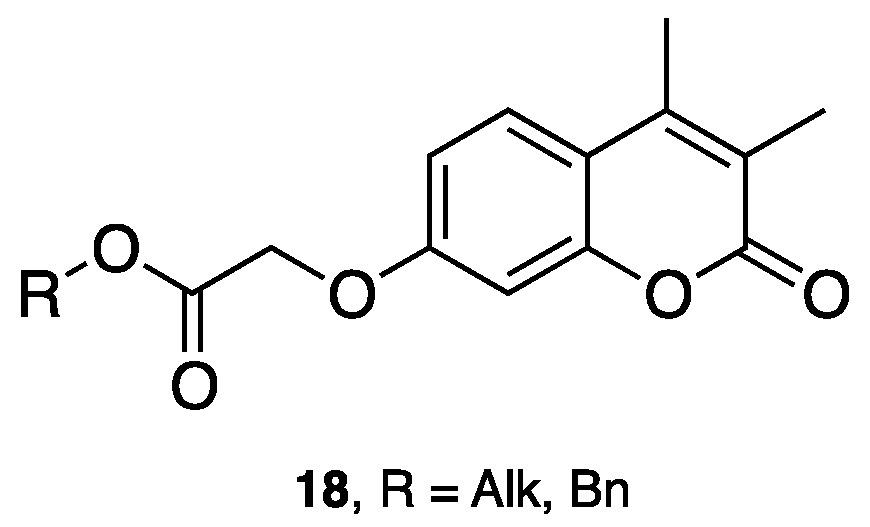
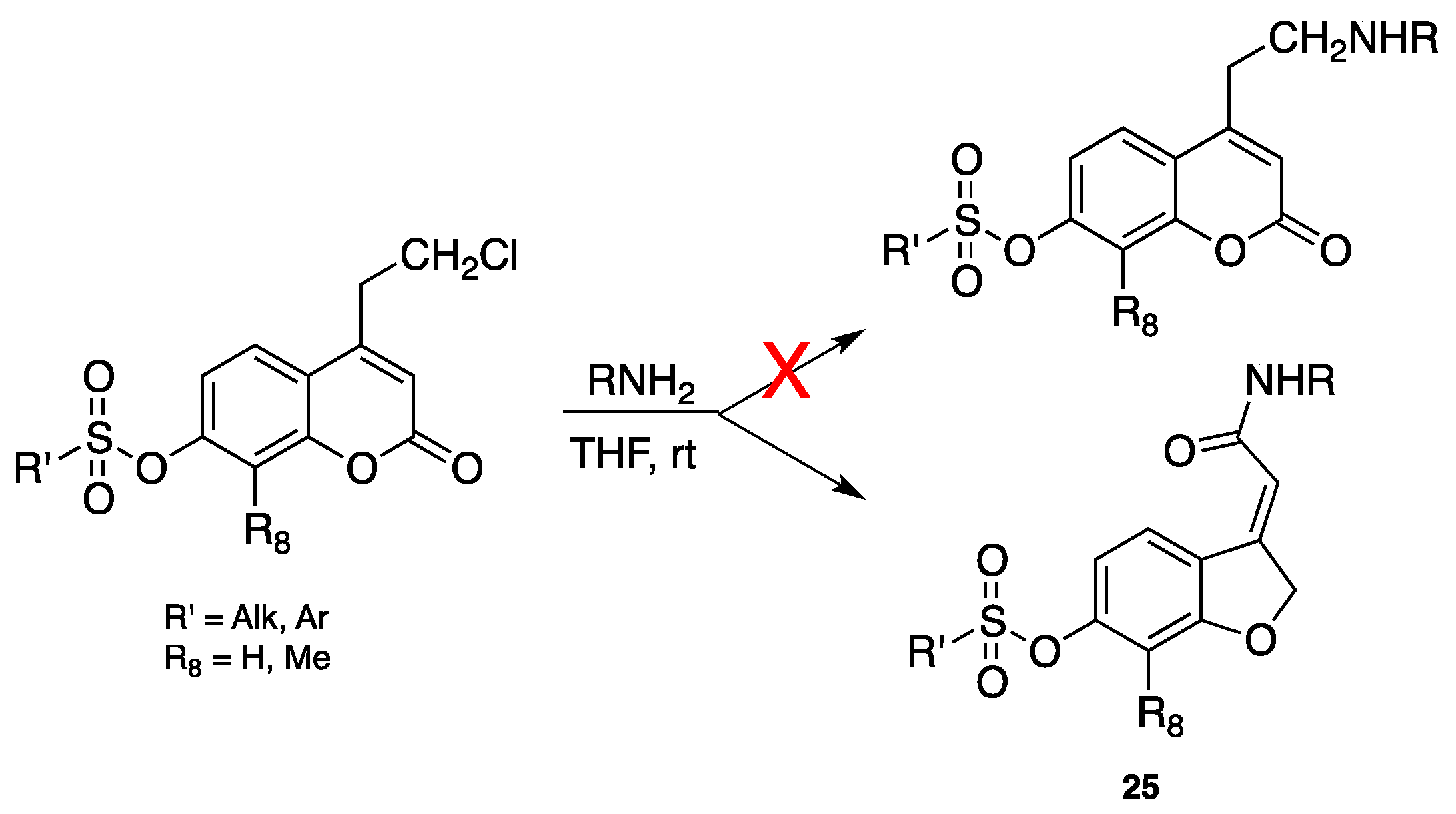
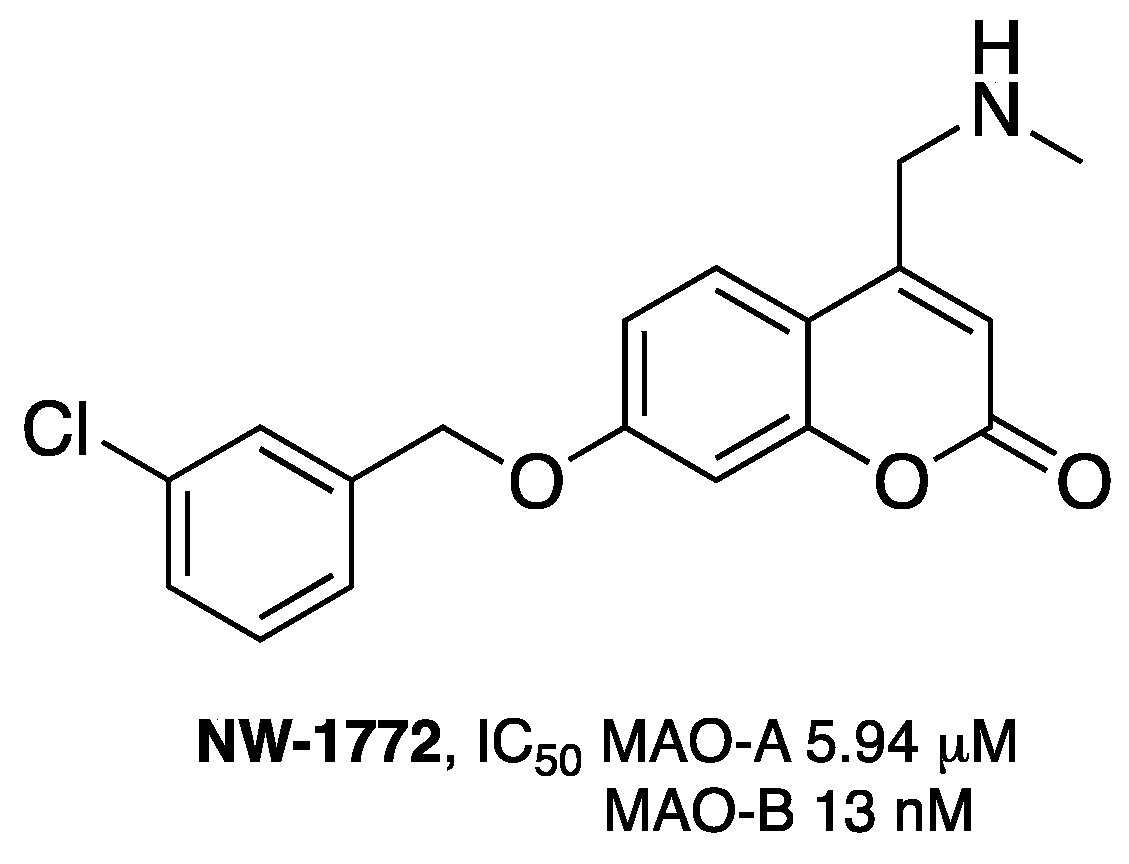
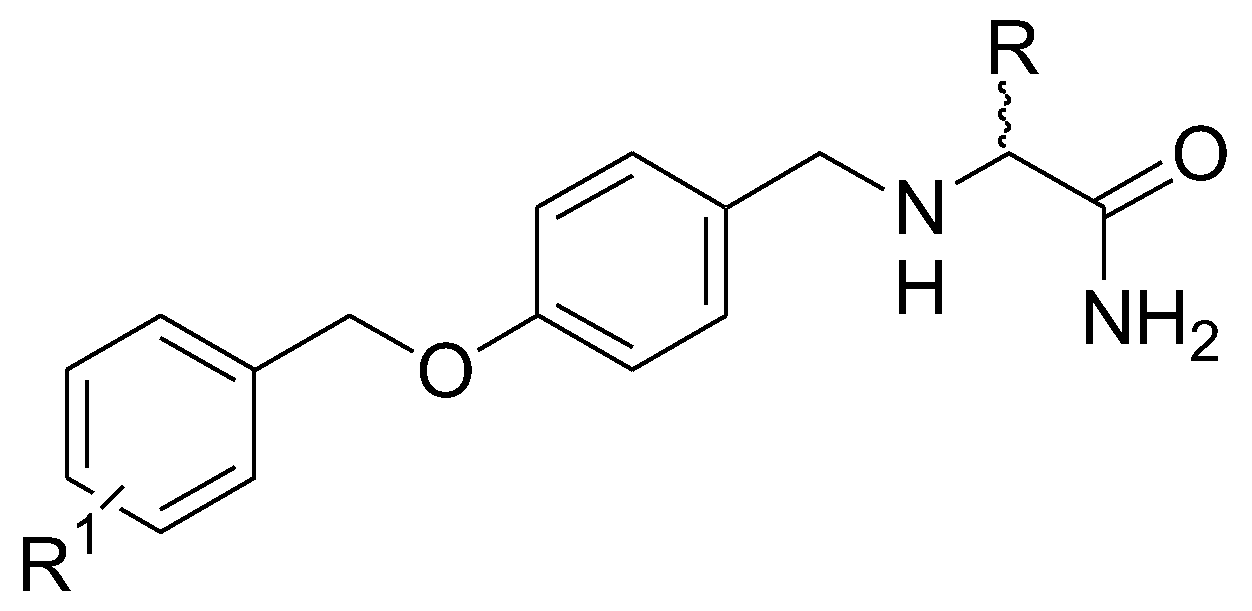
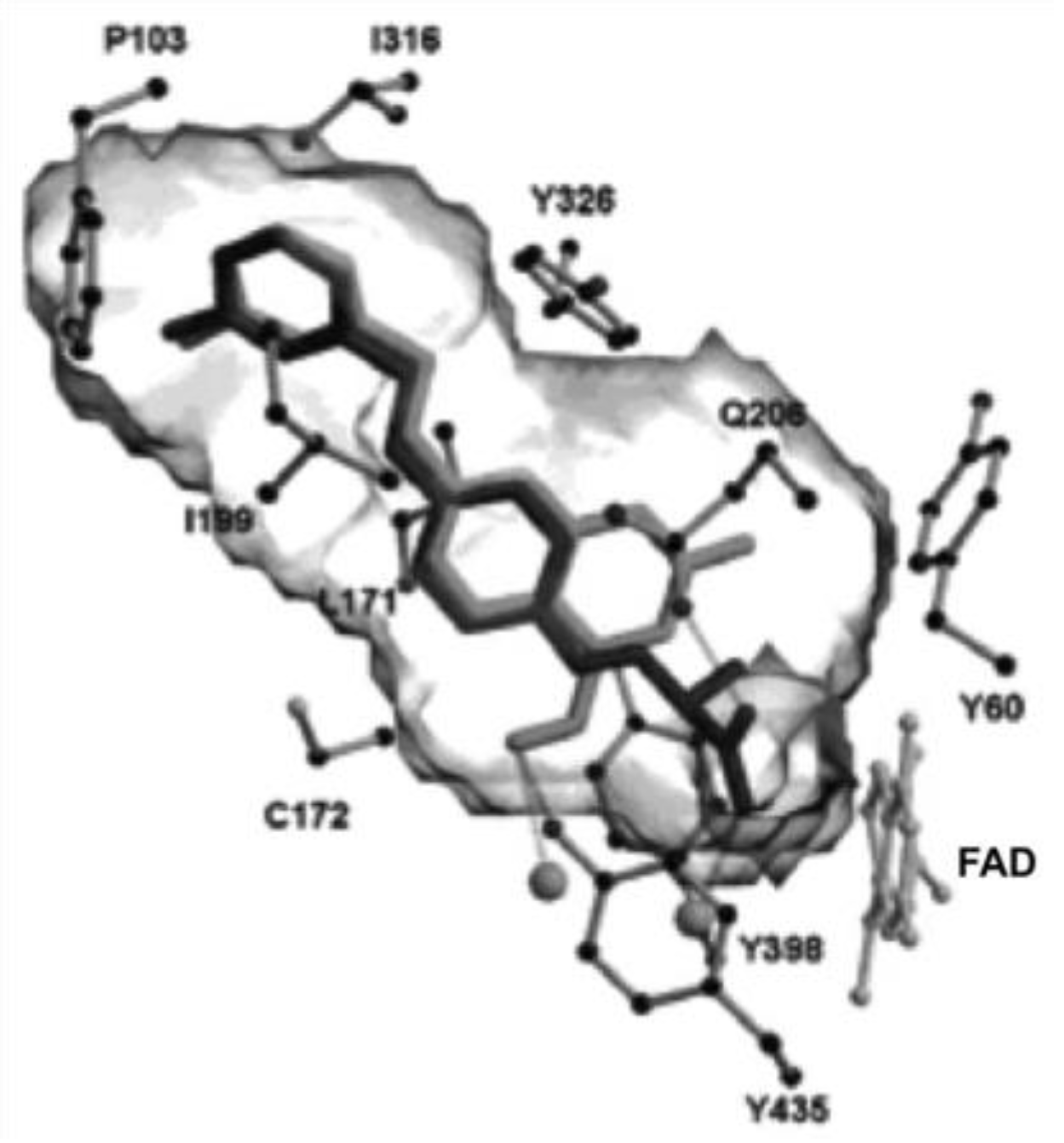
3.1. Coumarins as MAO Inhibitors
3.2. Coumarins as ChE Inhibitors
3.3. Coumarins as Multitarget MAO–ChE Inhibitors
3.4. Coumarins as Pleiotropic Agents in NDs
3.5. Coumarins Acting on Other Targets
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Penta, S. (Ed.) Advances in Structure and Activity Relationship of Coumarin Derivatives; Academic Press-Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Detsi, A.; Kontogiorgis, C.; Hadjipavlou-Litina, D. Coumarin derivatives: An updated patent review (2015–2016). Expert Opin. Ther. Pat. 2017, 27, 1201–1227. [Google Scholar] [CrossRef] [PubMed]
- Barot, K.P.; Jain, S.V.; Kremer, L.; Singh, S.; Ghate, M.D. Recent advances and therapeutic journey of coumarins: Current status and perspectives. Med. Chem. Res. 2015, 24, 2771–2798. [Google Scholar] [CrossRef]
- Asif, M. Pharmacologically potentials of different substituted coumarin derivatives. Chem. Int. 2015, 1, 1–11. [Google Scholar]
- Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriarte, E. Simple coumarins and analogues in medicinal chemistry: Occurrence, synthesis and biological activity. Curr. Med. Chem. 2005, 12, 887–916. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015, 101, 475–495. [Google Scholar] [CrossRef] [PubMed]
- Klenkar, J.; Molnar, M. Natural and synthetic coumarins as potential anticancer agents. J. Chem. Pharm. Res. 2015, 7, 1223–1238. [Google Scholar]
- Hassan, M.Z.; Osman, H.; Ali, M.A.; Ahsan, M.J. Therapeutic potential of coumarins as antiviral agents. Eur. J. Med. Chem. 2016, 123, 236–255. [Google Scholar] [CrossRef] [PubMed]
- Garro, H.A.; Pungitore, C.R. Coumarins as potential inhibitors of DNA polymerases and reverse transcriptases. Searching new antiretroviral and antitumoral drugs. Curr. Drug Discov. Technol. 2015, 12, 66–79. [Google Scholar] [CrossRef] [PubMed]
- De Souza, L.G.; Rennã, M.N.; Figueroa-Villar, J.D. Coumarins as cholinesterase inhibitors: A review. Chem.-Biol. Interact. 2016, 254, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.D.; Nahar, L. Progress in the chemistry of naturally occurring coumarins. Prog. Chem. Org. Nat. Prod. 2017, 7, CD012744. [Google Scholar]
- Keri, R.S.; Sasidhar, B.S.; Nagaraja, B.M.; Santos, M.A. Recent progress in the drug development of coumarin derivatives as potent antituberculosis agents. Eur. J. Med. Chem. 2015, 100, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, G.; Abdelwahab, A.B.; Chaimbault, P. Natural and synthetic coumarins with effects on inflammation. Molecules 2016, 21, 1322. [Google Scholar] [CrossRef] [PubMed]
- Revankar, H.M.; Bukhari, S.N.A.; Kumar, G.B.; Qin, H.-L. Coumarins scaffolds as COX inhibitors. Bioorg. Chem. 2017, 71, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Trenor, S.R.; Shultz, A.R.; Love, B.J.; Long, T.E. Coumarins in Polymers: From Light Harvesting to Photo-Cross-Linkable Tissue Scaffolds. Chem. Rev. 2004, 104, 3059–3077. [Google Scholar] [CrossRef] [PubMed]
- Tasior, M.; Kim, D.; Singha, S.; Krzeszewski, M.; Ahn, K.H.; Gryko, D.T. π-Expanded coumarins: Synthesis, optical properties and applications. J. Mater. Chem. 2015, 3, 1421–1446. [Google Scholar] [CrossRef]
- Vogel, A. Darstellung von Benzoesaure aus der Tonka-Boline und aus den MeliIoten-oder Steinklee-Blumen. Ann. Phys. 1820, 64, 161–166. [Google Scholar] [CrossRef]
- Dean, F.M. Naturally occurring coumarins. Fortschr. Chem. Org. Naturst. IX 1952, 9, 225–291. [Google Scholar]
- Murray, R.D.H.; Mendez, J.; Brown, S.A. The Natural Coumarins: Occurrence, Chemistry and Biochemistry; John Wiley & Sons: New York, NY, USA, 1982. [Google Scholar]
- Murray, R.D.H. Naturally occurring plant coumarins. Fortschr. Chem. Org. Naturst. 1991, 58, 84–322. [Google Scholar]
- O’Kennedy, R.; Thornes, R.D. (Eds.) Coumarins: Biology, Applications, and Mode of Action; John Wiley & Sons: New York, NY, USA, 1997. [Google Scholar]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; Mizutani, M.; Kawamura, N.; Yamamoto, R.; Tamai, M.; Yamaguchi, H.; Skata, K.; Shimizu, B. Scopoletin is biosynthesized via ortho-hydroxylation of feruloyl CoA by a 2-oxolutarate-dependent dioxygenase in Arabidopsis thaliana. Plant J. 2008, 55, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.G. Coumarin Metabolism, Toxicity and Carcinogenicity: Relevance for Human Risk Assessment. Food Chem. Toxicol. 1999, 37, 423–453. [Google Scholar] [CrossRef]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Danielson, P.B. The Cytochrome P450 Superfamily: Biochemistry, Evolution and Drug Metabolism in Humans. Curr. Drug Metab. 2002, 3, 561–597. [Google Scholar] [CrossRef] [PubMed]
- Raunio, H.; Rahnasto-Rilla, M. CYP2A6: Genetics, structure, regulation, and function. Drug Metab. Drug Interact. 2012, 27, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, O.; Rautio, A.; Raunio, H.; Pasanen, M. CYP2A6: A human coumarin 7-hydroxylase. Toxicology 2000, 144, 139–147. [Google Scholar] [CrossRef]
- Farinola, N.; Piller, N.B. CYP2A6 polymorphisms: Is there a role for pharmacogenomics in preventing coumarin-induced hepatotoxicity in lymphedema patients? Pharmacogenomics 2007, 8, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Born, S.L.; Caudill, D.; Smith, B.J.; Lehman-McKeeman, L.D. In vitro kinetics of coumarin 3,4-epoxidation: Application to species differences in toxicity and carcinogenicity. Toxicol. Sci. 2000, 58, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Lacy, A.; O’Kennedy, R. Studies on coumarines and coumarin related compounds to determine their therapeutic role in the treatment of cancer. Curr. Pharm. Des. 2004, 10, 3797–3811. [Google Scholar] [CrossRef] [PubMed]
- Fentem, J.H.; Hammond, A.H.; Garle, M.J.; Fry, M.J. Toxicity of coumarin and various methyl derivatives in cultures of rat hepatocytes and V79 cells. Toxicol. In Vitro 1992, 6, 21–25. [Google Scholar] [CrossRef]
- Tanaka, Y.; Fujii, W.; Hori, H.; Kitagawa, Y.; Ozaki, K. Relationship between coumarin-induced hepatocellular toxicity and mitochondrial function in rats. Food Chem. Toxicol. 2016, 90, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Loprinzi, C.L.; Sloan, J.; Kugler, J. Coumarin-induced hepatotoxicity. J. Clin. Oncol. 1997, 15, 3167–3168. [Google Scholar] [CrossRef] [PubMed]
- Ratanasavanh, D.; Lamiable, D.; Biour, M.; Guédès, Y.; Gersberg, M.; Leutenegger, E.; Riché, C. Metabolism and toxicity of coumarin on cultured human, rat, mouse and rabbit hepatocytes. Fundam. Clin. Pharmacol. 1996, 10, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Weigta, S.; Hueblera, N.; Streckerb, R.; Braunbeckb, T.; Broscharda, T.H. Developmental effects of coumarin and the anticoagulant coumarin derivative warfarin on zebrafish (Danio rerio) embryos. Reprod. Toxicol. 2012, 33, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Fort, D.J.; Stover, E.L.; Propst, T.; Hull, M.A.; Bantle, J.A. Evaluation of the developmental toxicities of coumarin, 4-hydroxycoumarin, and 7-hydroxycoumarin using FETAX. Drug Chem. Toxicol. 1998, 21, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Abraham, K.; Woehrlin, F.; Lindtner, O.; Heinemeyer, G.; Lampen, A. Toxicology and risk assessment of coumarin: Focus on human data. Mol. Nutr. Food Res. 2010, 54, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Saify, Z.S.; Khan, M.Z.; Zia-Ullah, M.I.; Atta-Ur-Rahman, C.; Perveen, S.; Chohan, Z.H.; Supuran, C.T. Synthesis of coumarin derivatives with cytotoxic, antibacterial and antifungal activity. J. Enzyme Inhib. Med. Chem. 2004, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Neyts, J.; Cleucq, E.; Singha, R.; Chang, Y.H.; Das, A.R.; Chakraborty, S.K.; Hong, S.C.; Tsay, S.C.; Hsu, M.H.; Hwu, J.R. Structure-activity relationship of new antihepatitis C virus agents: Heterobicycle-coumarin conjugates. J. Med. Chem. 2009, 52, 1486–1490. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Takahashi, K.; Kanayama, S.; Nakano, Y.; Imai, H.; Kibi, M.; Imahori, D.; Hasei, T.; Watanabe, T. Structures of antimutagenic constituents in the peels of Citrus limon. J. Nat. Med. 2017, 71, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Bubols, G.B.; Vianna, D.R.; Medina-Remon, A.; von Poser, G.; Lamuela-Raventos, R.M.; Eifler-Lima, V.L.; Garcia, S.C. The antioxidant activity of coumarins and flavonoids. Mini-Rev. Med. Chem. 2013, 13, 318–334. [Google Scholar] [PubMed]
- Al-Majedy, Y.K.; Al-Amiery, A.A.; Kadhum, A.A.H.; Mohamad, A.B. Antioxidant Activities of 4-Methylumbelliferone Derivatives. PLoS ONE 2016, 11, e0156625. [Google Scholar] [CrossRef] [PubMed]
- Kontogiorgis, C.A.; Hadjipavlou-Litina, D.J. Synthesis and anti-inflammatory activity of coumarin derivatives. J. Med. Chem. 2005, 48, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Surin, W.R.; Misra, A.; Prakash, P.; Singh, V.; Khanna, V.; Kumar, S.; Siddiqui, H.H.; Raj, K.; Barthwal, M.K.; et al. Antithrombotic activity of a newly synthesized coumarin derivative 3-(5-hydroxy-2,2-dimethyl-chroman-6-yl)-N-{2-[3-(5-hydroxy-2,2-dimethyl-chroman-6-yl)-propionylamino]-ethyl}-propionamide. Chem. Biol. Drug Des. 2013, 81, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Kumar, A.; Kumar, M.; Sarkar, J.; Sinha, S. Synthesis and in vitro evaluation of novel coumarin-chalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 7205–7211. [Google Scholar] [CrossRef] [PubMed]
- Abdelhafez, O.M.; Amin, K.M.; Batran, R.Z.; Maher, T.J.; Nada, S.A.; Sethumadhavan, S. Synthesis, anticoagulant and PIVKA-II induced by new 4-hydroxycoumarin derivatives. Bioorg. Med. Chem. 2010, 18, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Nargotra, S.; Sharma, S.; Alam, M.I.; Ahmed, Z.; Bhagat, A.; Taneja, S.C.; Qazi, G.N.; Koul, S. In silico identification of viper phospholipaseA2 inhibitors: Validation by in vitro, in vivo studies. J. Mol. Model. 2011, 17, 3063–3073. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.S.; Choi, J.S.; Islam, M.N.; Kim, Y.S.; Kim, H.P. Inhibition of 5-lipoxygenase and skin inflammation by the aerial parts of Artemisia capillaris and its constituents. Arch. Pharm. Res. 2011, 34, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Patil, P.O.; Bari, S.B.; Firke, S.D.; Deshmukh, P.K.; Donda, S.T.; Patil, D.A. A comprehensive review on synthesis and designing aspects of coumarin derivatives as monoamine oxidase inhibitors for depression and Alzheimer’s disease. Bioorg. Med. Chem. 2013, 21, 2434–2450. [Google Scholar] [CrossRef] [PubMed]
- Najmanová, I.; Doseděl, M.; Hrdina, R.; Anzenbacher, P.; Filipský, T.; Říha, M.; Mladěnka, P. Cardiovascular effects of coumarins besides their antioxidant activity. Curr. Top. Med. Chem. 2015, 15, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Kostova, I. Synthetic and natural coumarins as cytotoxic agents. Curr. Med. Chem. Anti-Cancer Agents 2005, 5, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Vullo, D.; Maresca, A.; Supuran, C.; Poulsen, T. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1539–1542. [Google Scholar] [CrossRef] [PubMed]
- Pochet, L.; Frédérick, R.; Masereel, B. Coumarin and isocoumarin as serine protease inhibitors. Curr. Pharm. Des. 2004, 10, 3781–3796. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Soualmia, F.; Furio, L.; Renard, J.F.; Kempen, I.; Qin, L.; Pagano, M.; Pirotte, B.; El Amri, C.; Hovnanian, A.; et al. Toward the first class of suicide inhibitors of kallikreins involved in skin diseases. J. Med. Chem. 2015, 58, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.K.; Katiyar, D. Recent Advances in Transition-Metal-Catalyzed Synthesis of Coumarins. Synthesis 2016, 48, 2303–2322. [Google Scholar]
- Calcio, G.; Tagliapietra, M.; Palmisano, C. Recent advances and perspectives in the synthesis of bioactive coumarins. RSC Adv. 2016, 6, 46394–46405. [Google Scholar] [CrossRef]
- Heravi, M.M.; Khaghaninejad, S.; Mostofi, M. Pechmann reaction in the synthesis of coumarin derivatives. Adv. Heterocycl. Chem. 2014, 112, 1–50. [Google Scholar]
- Lanman, B.A. Pechmann coumarin synthesis. In Name Reactions in Heterocyclic Chemistry II; Li, J.J., Ed.; John Wiley & Sons: New York, NY, USA, 2011. [Google Scholar]
- Vekariya, R.H.; Patel, H.D. Synthesis of α-bromocarbonyl compounds: recent advances. Tetrahedron 2014, 70, 3949–3961. [Google Scholar] [CrossRef]
- Yan, K.; Yang, D.; Wei, W.; Wang, F.; Shuai, Y.; Li, Q.; Wang, H. Silver-mediated radical cyclization of alkynoates and α-keto acids leading to coumarins via cascade double C-C bond formation. J. Org. Chem. 2015, 80, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
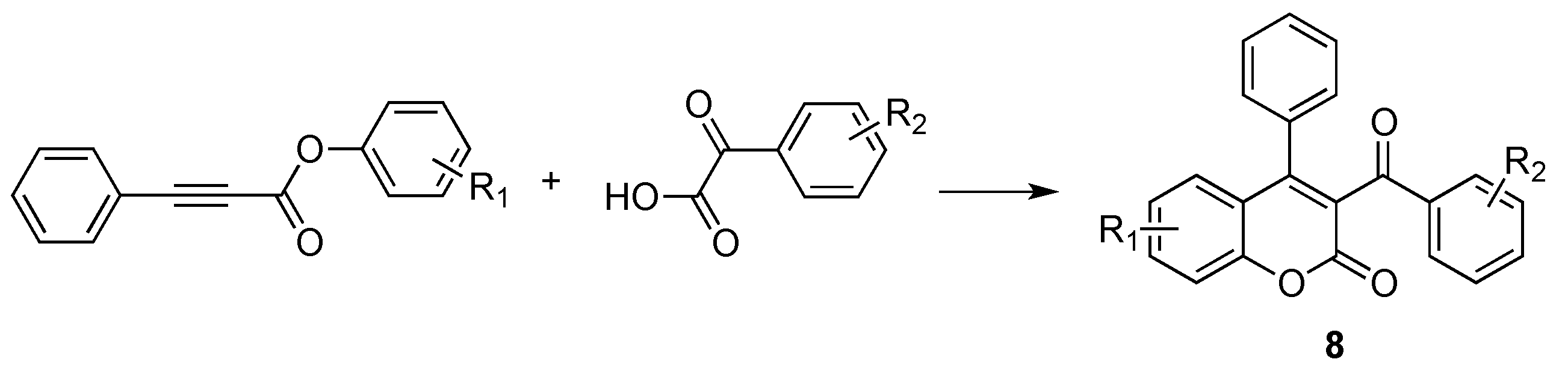
- Jafarpour, F.; Abbasnia, M. A Regioselective Metal-Free Construction of 3-Aroyl Coumarins by Csp2-H Functionalization. J. Org. Chem. 2016, 81, 11982–11986. [Google Scholar] [CrossRef] [PubMed]
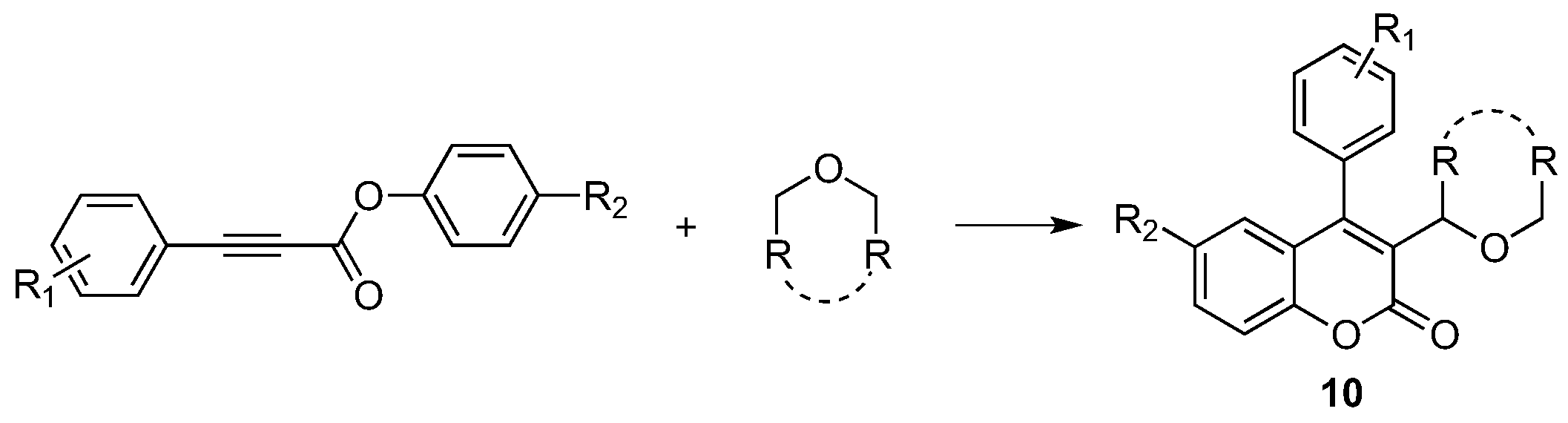
- Feng, S.; Xie, X.; Zhang, W.; Liu, L.; Zhong, Z.; Xu, D.; She, X. Visible-Light-Promoted Dual C-C Bond Formations of Alkynoates via a Domino Radical Addition/Cyclization Reaction: A Synthesis of Coumarins. Org. Lett. 2016, 18, 3846–3849. [Google Scholar] [CrossRef] [PubMed]
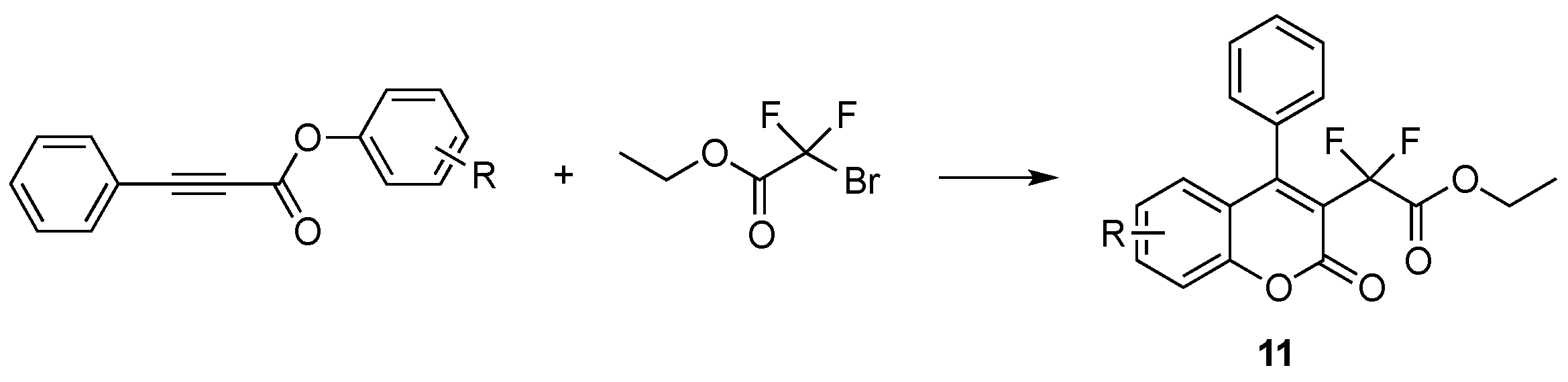
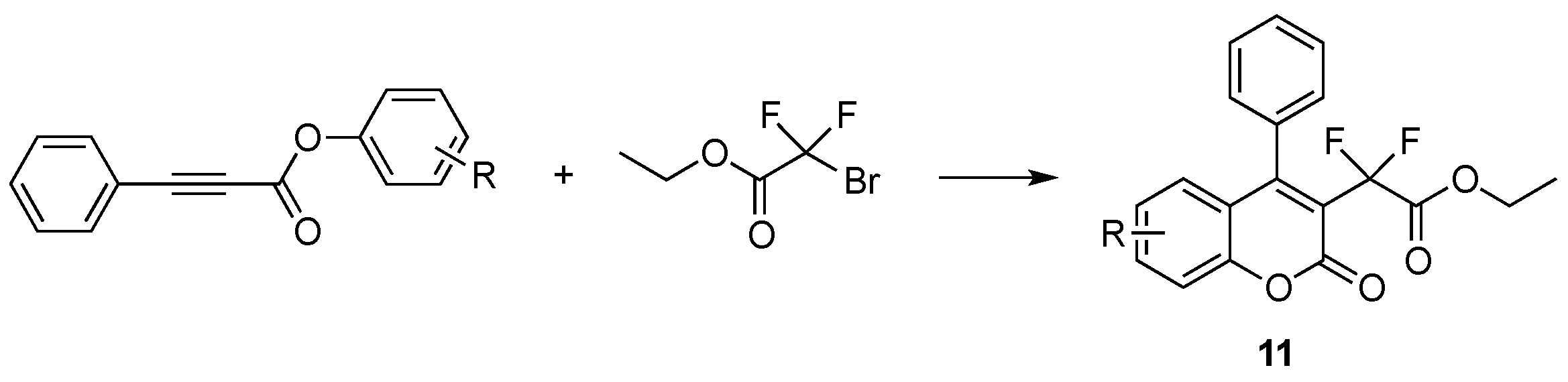
- Fu, W.; Zhu, M.; Zou, G.; Xu, C.; Wang, Z.; Ji, B. Visible-light-mediated radical aryldifluoroacetylation of alkynes with ethyl bromodifluoroacetate for the synthesis of 3-difluoroacetylated coumarins. J. Org. Chem. 2015, 80, 4766–4770. [Google Scholar] [CrossRef] [PubMed]
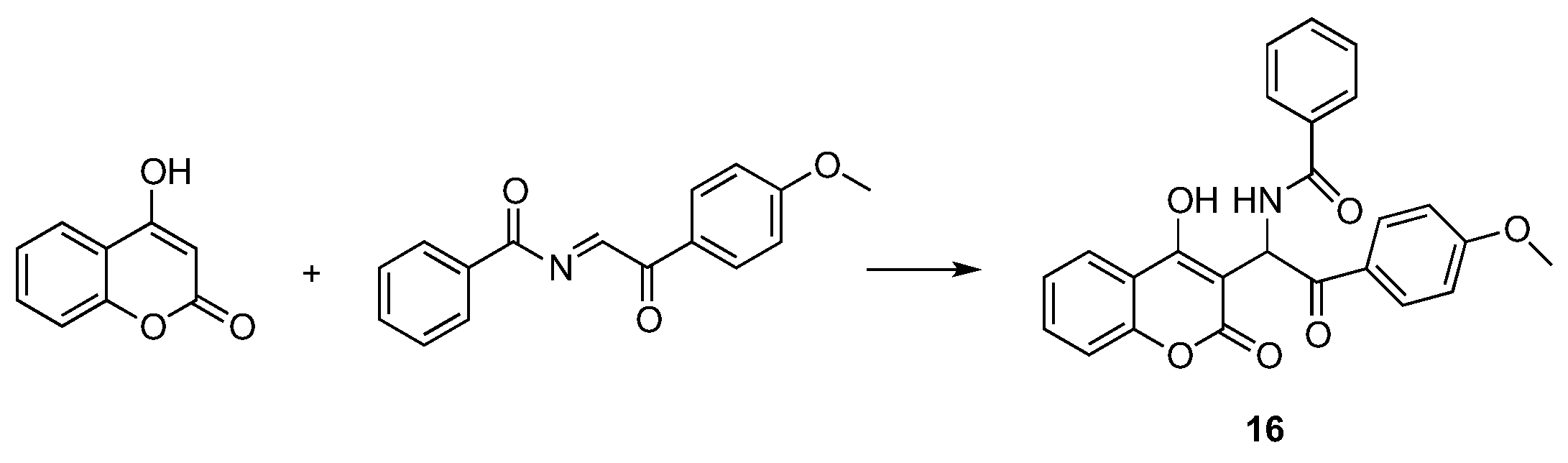
- Vekariya, R.H.; Patel, H.D. Recent Advances in the Synthesis of Coumarin Derivatives via Knoevenagel Condensation: A Review. Synth. Commun. 2014, 44, 2756–2788. [Google Scholar] [CrossRef]
- Kiyani, H.; Daroonkala, M.D. A cost-effective and green aqueous synthesis of 3-substituted coumarins catalyzed by potassium phthalimide. Bull. Chem. Soc. Ethiopia 2015, 2, 449–456. [Google Scholar] [CrossRef]
- Ghomi, J.S.; Akbarzadeh, Z. Ultrasonic accelerated Knoevenagel condensation by magnetically recoverable MgFe2O4 nanocatalyst: A rapid and green synthesis of coumarins under solvent-free conditions. Ultrasonics Sonochem. 2018, 40, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, Z.; Rezayati, S.; Bagheri, M.; Hajinasiri, R. Preparation of a novel, efficient, and recyclable magnetic catalyst, γ-Fe2O3@HAp-Ag nanoparticles, and a solvent- and halogen-free protocol for the synthesis of coumarin derivatives. Chin. Chem. Lett. 2017, 28, 75–82. [Google Scholar] [CrossRef]
- Prateeptongkum, S.; Duangdee, N.; Thongyoo, P. Facile iron(III) chloride hexahydrate catalyzed synthesis of coumarins. Arkivoc 2015, 15, 248–258. [Google Scholar]
- Moradi, L.; Belali, F. Solvent-free one-pot synthesis of coumarins using molybdate sulfuric acid as highly efficient catalyst. J. Iran. Chem. Soc. 2015, 12, 1927–1934. [Google Scholar] [CrossRef]
- Sun, R.; Gao, Y.; Ma, Y.; Yang, G.; Li, Y. SnCl4 grafted on silica gel: An efficient catalyst for solvent-free synthesis of coumarins via the Pechmann condensation. J. Iran. Chem. Soc. 2017, 14, 737–742. [Google Scholar] [CrossRef]
- Kour, M.; Paul, S. A green and convenient approach for the one-pot solvent-free synthesis of coumarins and β-amino carbonyl compounds using Lewis acid grafted sulfonated carbon@titania composite. Mon. Chem. 2017, 148, 327–337. [Google Scholar] [CrossRef]
- Tahanpesar, S. Synthesis of substituted coumarins catalyzed by sawdust-SO3H. An efficient and environmentally benign solid acid catalyst under solvent-free conditions. Russ. J. Gen. Chem. 2015, 85, 2135–2140. [Google Scholar] [CrossRef]
- Dengale, R.A.; Thorat, N.M.; Thopate, S.R. L-ascorbic acid: A green and competent promoter for solvent-free synthesis of flavones and coumarins under conventional as well as microwave heating. Lett. Org. Chem. 2016, 10, 734–741. [Google Scholar]
- Shirini, F.; Yahyazadeh, A.; Mohammadi, K. A solvent-free synthesis of coumarins using 1,3-disulfonic acid imidazolium hydrogen sulfate as a reusable and effective ionic liquid catalyst. Res. Chem. Intermed. 2015, 4, 6207–6218. [Google Scholar] [CrossRef]
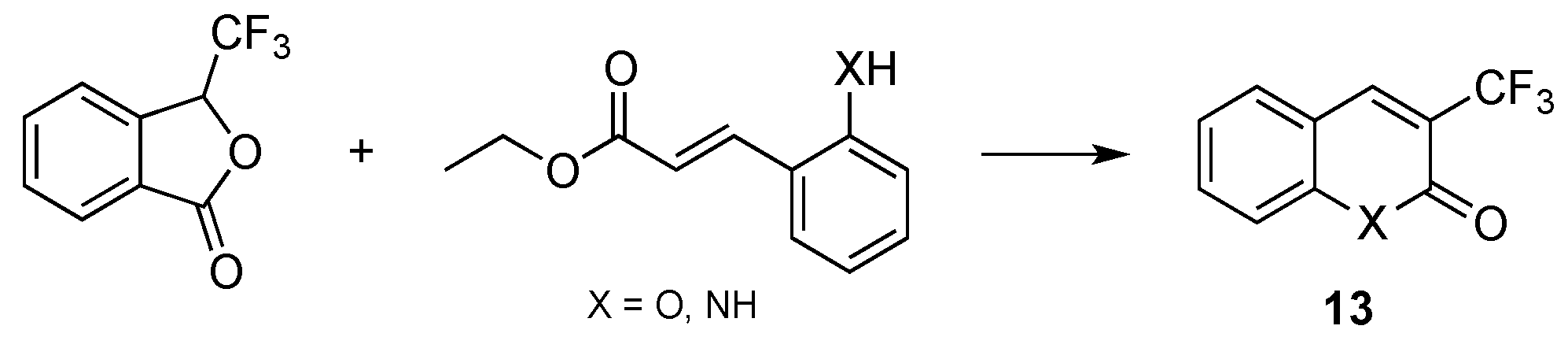
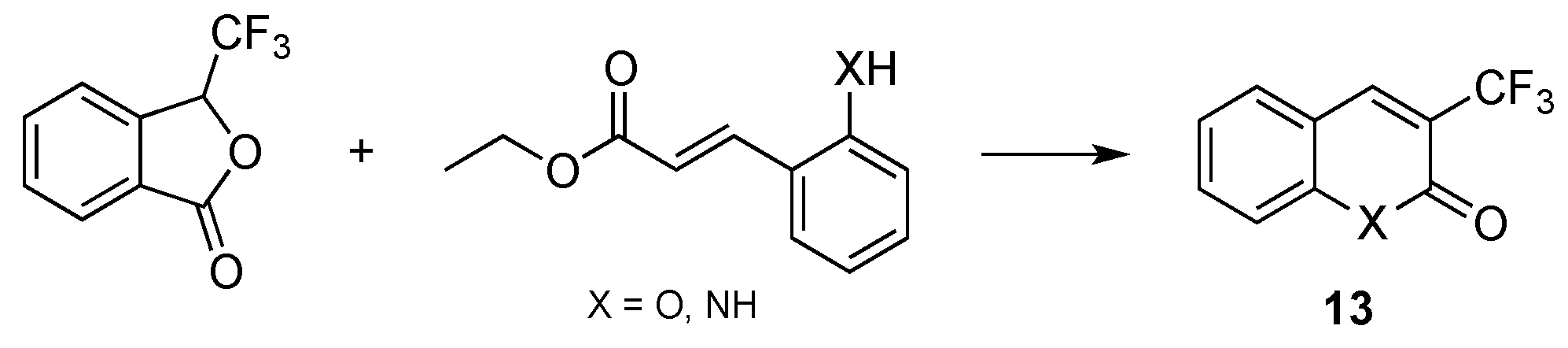
- Chaabouni, S.; Simonet, F.; François, A.; Abid, S.; Galaup, C.; Chassaing, S. 3-Trifluoromethylated Coumarins and Carbostyrils by Radical Trifluoromethylation of ortho-Functionalized Cinnamic Esters. Eur. J. Org. Chem. 2017, 4, 271–277. [Google Scholar] [CrossRef]
- Da Silveira Pinto, L.S.; De Souza, M.V.N. Sonochemistry as a General Procedure for the Synthesis of Coumarins, Including Multigram Synthesis. Synthesis 2017, 49, 2677–2682. [Google Scholar]
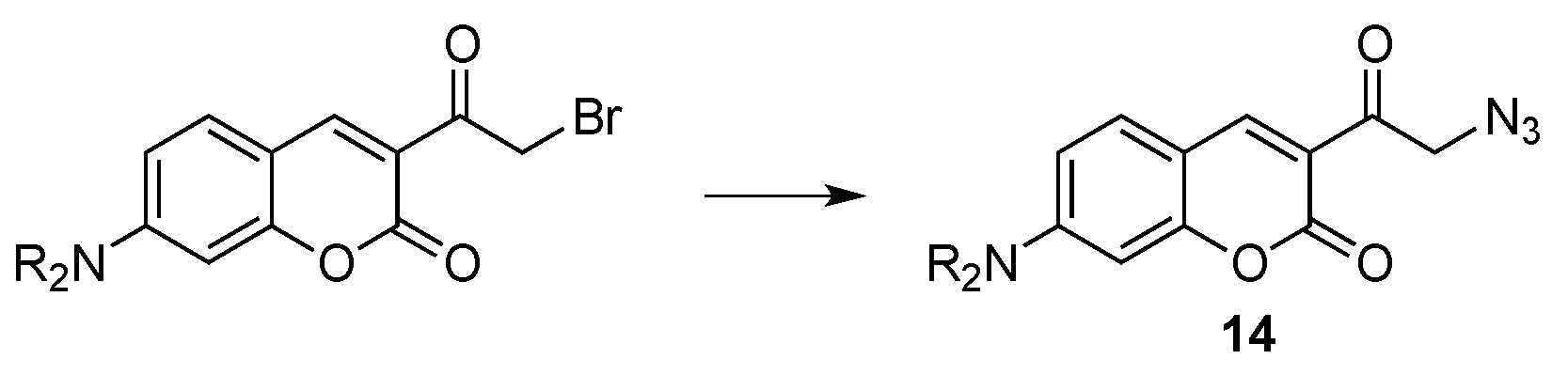
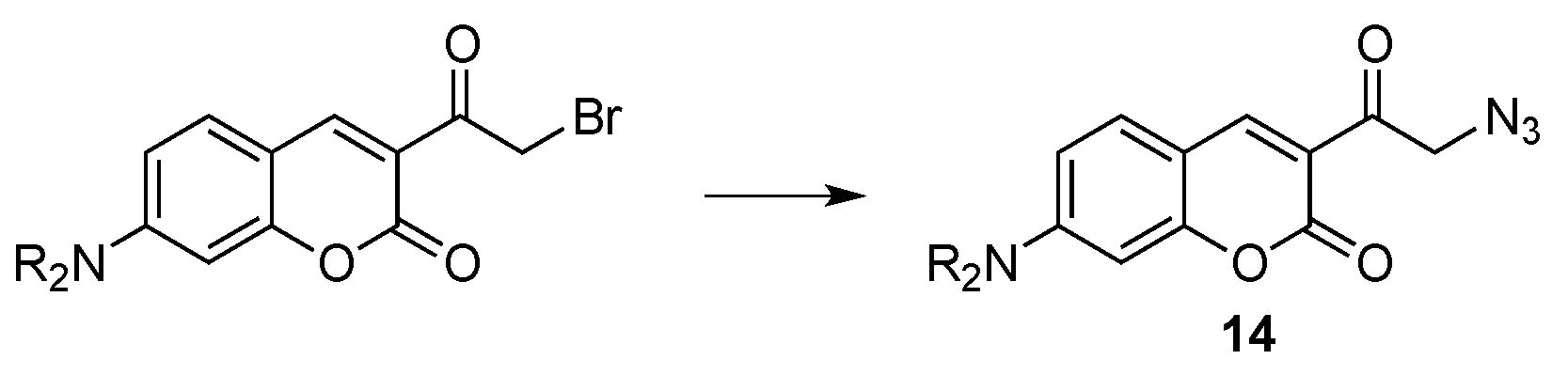
- Evans, V.; Duncanson, P.; Motevalli, M.; Watkinson, M. An investigation into the synthesis of azido-functionalised coumarins for application in 1,3-dipolar “click” cycloaddition reactions. Dyes Pigment. 2016, 135, 36–40. [Google Scholar] [CrossRef]
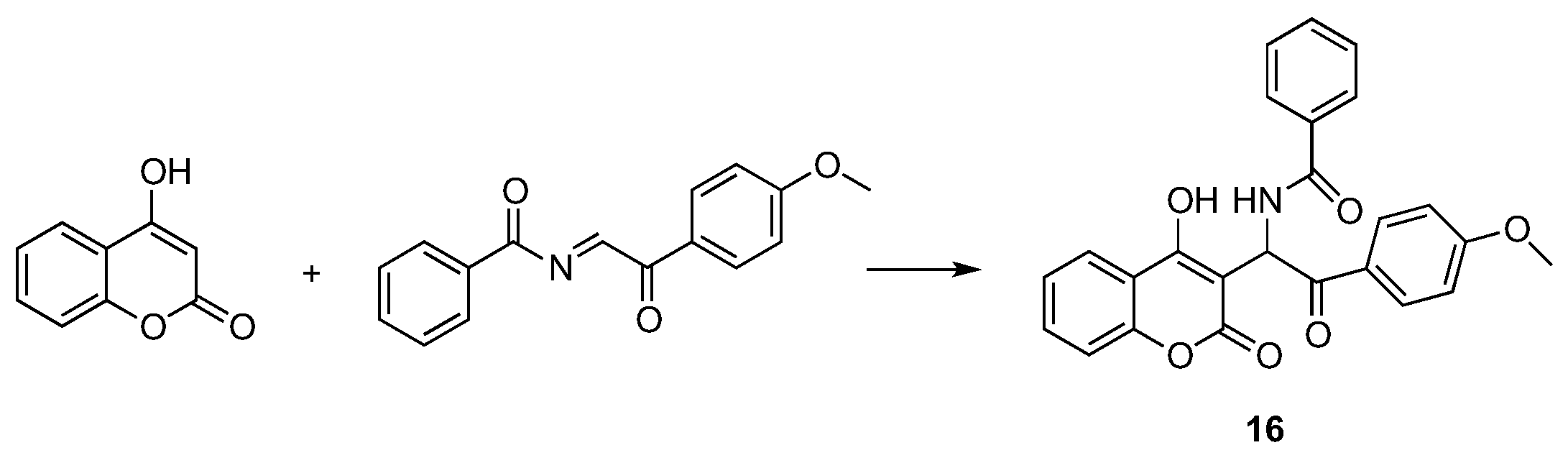
- Sivakumar, K.; Xie, F.; Cash, B.M.; Long, S.; Barnhill, H.N.; Wang, Q. A fluorogenic 1,3-dipolar cycloaddition reaction of 3-azidocoumarins and acetylenes. Org. Lett. 2004, 24, 4603–4606. [Google Scholar] [CrossRef] [PubMed]
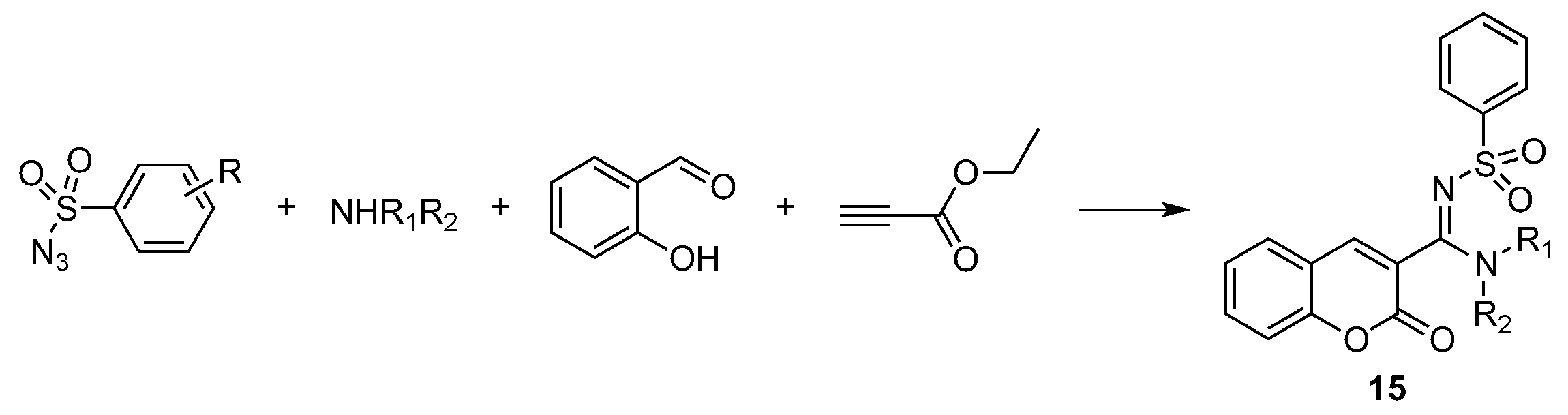
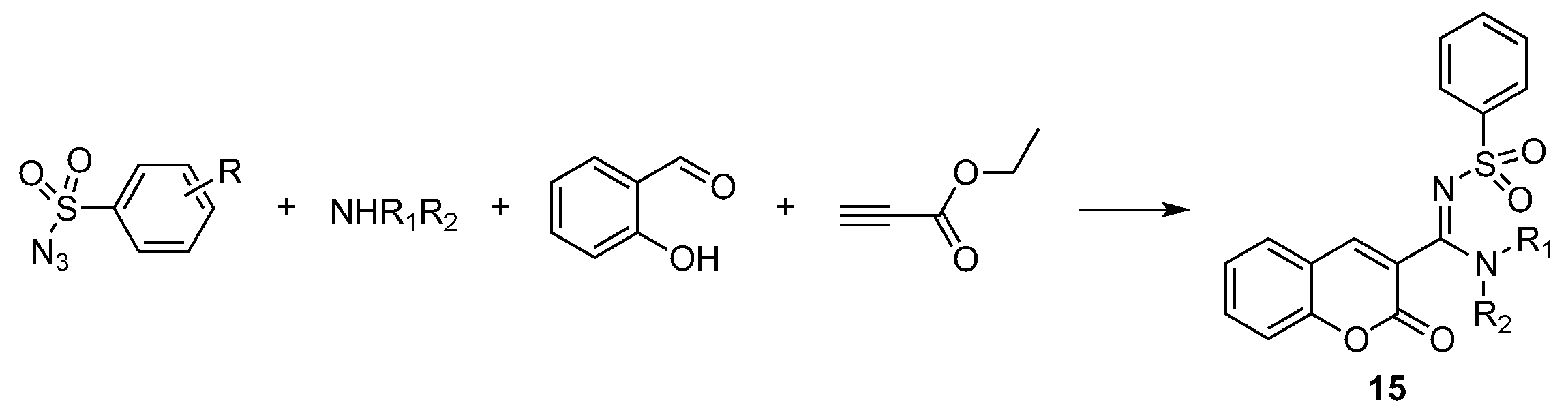
- Murugavel, G.; Punniyamurthy, T. Microwave-Assisted Copper-Catalyzed Four-Component Tandem Synthesis of 3- N -Sulfonylamidine Coumarins. J. Org. Chem. 2015, 80, 6291–6299. [Google Scholar] [CrossRef] [PubMed]
- Khodabakhshi, S.; Karami, B.; Eskandari, K. Molybdate sulfuric acid-catalyzed one-pot synthesis of substituted coumarins under solvent-free conditions. Res. Chem. Intermed. 2015, 41, 7263–7267. [Google Scholar] [CrossRef]
- Yang, S.-M.; Shim, G.Y.; Kim, B.-G.; Ahn, J.-H. Biological synthesis of coumarins in Escherichia coli. Microb. Cell Factor. 2015, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sun, X.; Yuan, Q.; Yan, Y. Combinatorial biosynthesis of plant-specific coumarins in bacteria. Metab. Eng. 2013, 18, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.M.; Tavares, L.B.B.; de Oliveira, D. Fungi as a source of natural coumarins production. Appl. Microbiol. Biotechnol. 2016, 100, 6571–6584. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.E.; Mattevi, A.; Binda, C.; Li, M.; Hubalek, F. Structure and mechanism of monoamine oxidases. Curr. Med. Chem. 2004, 11, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Wimbiscus, M.; Kostenko, O.; Malone, D. MAO inhibitors: Risks, benefits, and lore. Clevel. Clin. J. Med. 2010, 77, 859–882. [Google Scholar] [CrossRef] [PubMed]
- Rendenbach, B.; Weifenbach, H.; Teschendorf, H.J. Arylalkoxycumarine. Verfahren zu Ihre Herstellung und Diese Enthalende Therapeutische Mittel. Patent DE 3834861 A1; 1990 BASF AG, 19 April 1990. [Google Scholar]
- Schlecker, R.; Schmidt, P.; Thieme, P.C.; Lenke, D.; Teschendorf, H.J.; Traut, M.; Mueller, C.D.; Hofmann, J.P.; Kreiskott, H. Neue Sulfonsaureester von Hydroxycumarinen, ihre Herstellung und sie Enthaltende Arzneimittel. Patent DE 3243158 A1; 1984 BASF AG, 24 May 1984. [Google Scholar]
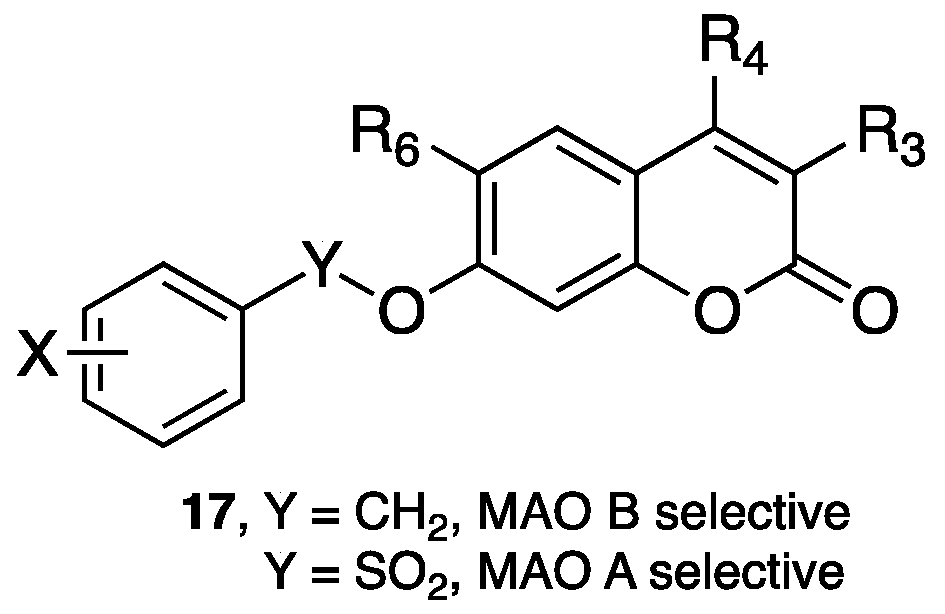
- Rendenbach-Mueller, B.; Schlecker, R.; Traut, M.; Weifenbach, H. Synthesis of coumarins as subtype-selective inhibitors of monoamine oxidase. Bioorg. Med. Chem. Lett. 1994, 4, 1195–1198. [Google Scholar] [CrossRef]
- Gnerre, C.; Catto, M.; Leonetti, F.; Weber, P.; Carrupt, P.A.; Altomare, C.; Carotti, A.; Testa, B. Inhibition of monoamine oxidases by functionalized coumarin derivatives: Biological activities, QSARs, and 3D-QSARs. J. Med. Chem. 2000, 43, 4747–4758. [Google Scholar] [CrossRef] [PubMed]
- Catto, M.; Nicolotti, O.; Leonetti, F.; Carotti, A.; Favia, A.D.; Soto-Otero, R.; Mendez-Alvarez, E.; Carotti, A. Structural insights into monoamine oxidase inhibitory potency and selectivity of 7-substituted coumarins from ligand- and target-based approaches. J. Med. Chem. 2006, 49, 4912–4925. [Google Scholar] [CrossRef] [PubMed]
- Novaroli, L.; Reist, M.; Favre, E.; Carotti, A.; Catto, M.; Carrupt, P.-A. Human recombinant monoamine oxidase B as reliable and efficient enzyme source for inhibitor screening. Bioorg. Med. Chem. 2005, 13, 6212–6217. [Google Scholar] [CrossRef] [PubMed]
- Novaroli, L.; Daina, A.; Favre, E.; Bravo, J.; Carotti, A.; Leonetti, F.; Catto, M.; Carrupt, P.-A.; Reist, M. Impact of species-dependent differences on screening, design, and development of MAO B inhibitors. J. Med. Chem. 2006, 49, 6264–6272. [Google Scholar] [CrossRef] [PubMed]
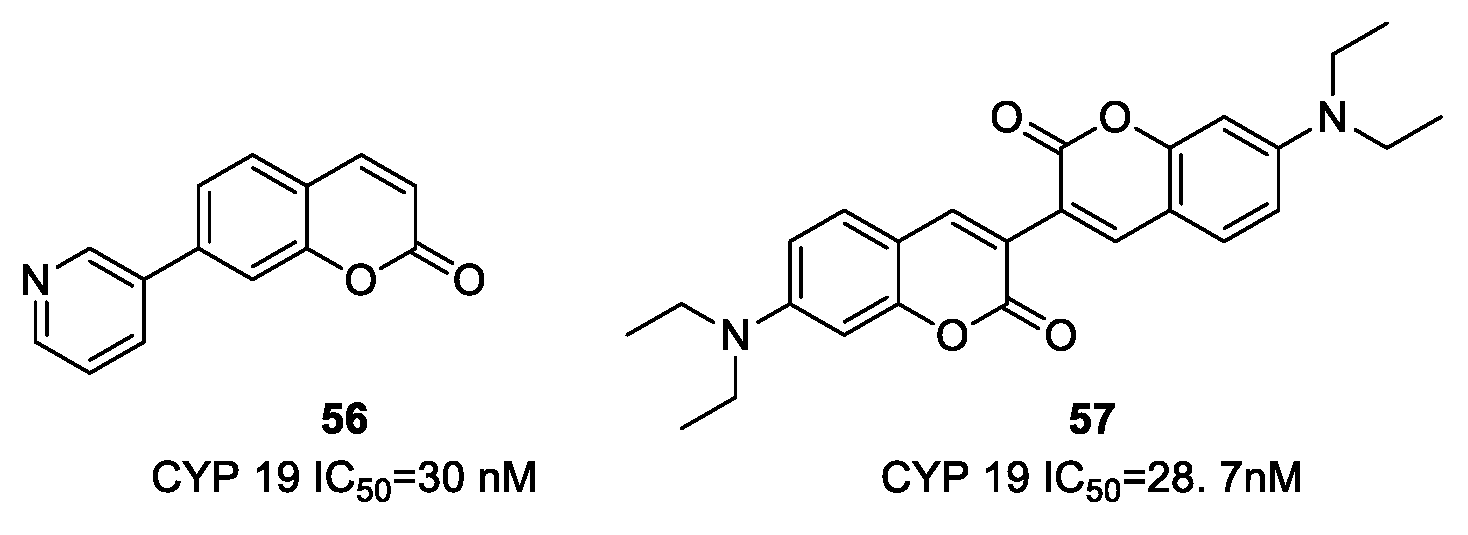
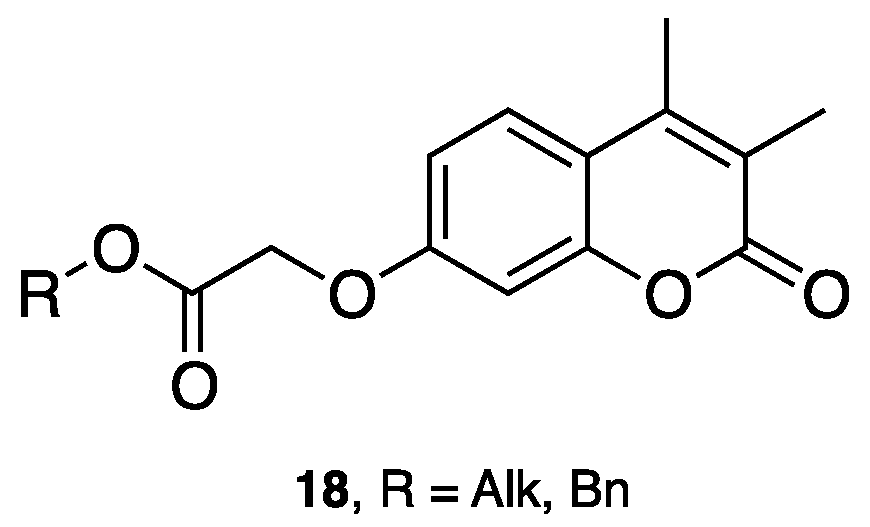
- Carotti, A.; Altomare, C.; Catto, M.; Gnerre, C.; Summo, L.; De Marco, A.; Rose, S.; Jenner, P.; Testa, B. Lipophilicity plays a major role in modulating monoamine oxidase B (MAO-B). Inhibition by 7-substituted coumarins. Chem. Biodivers. 2006, 3, 134–144. [Google Scholar] [CrossRef] [PubMed]
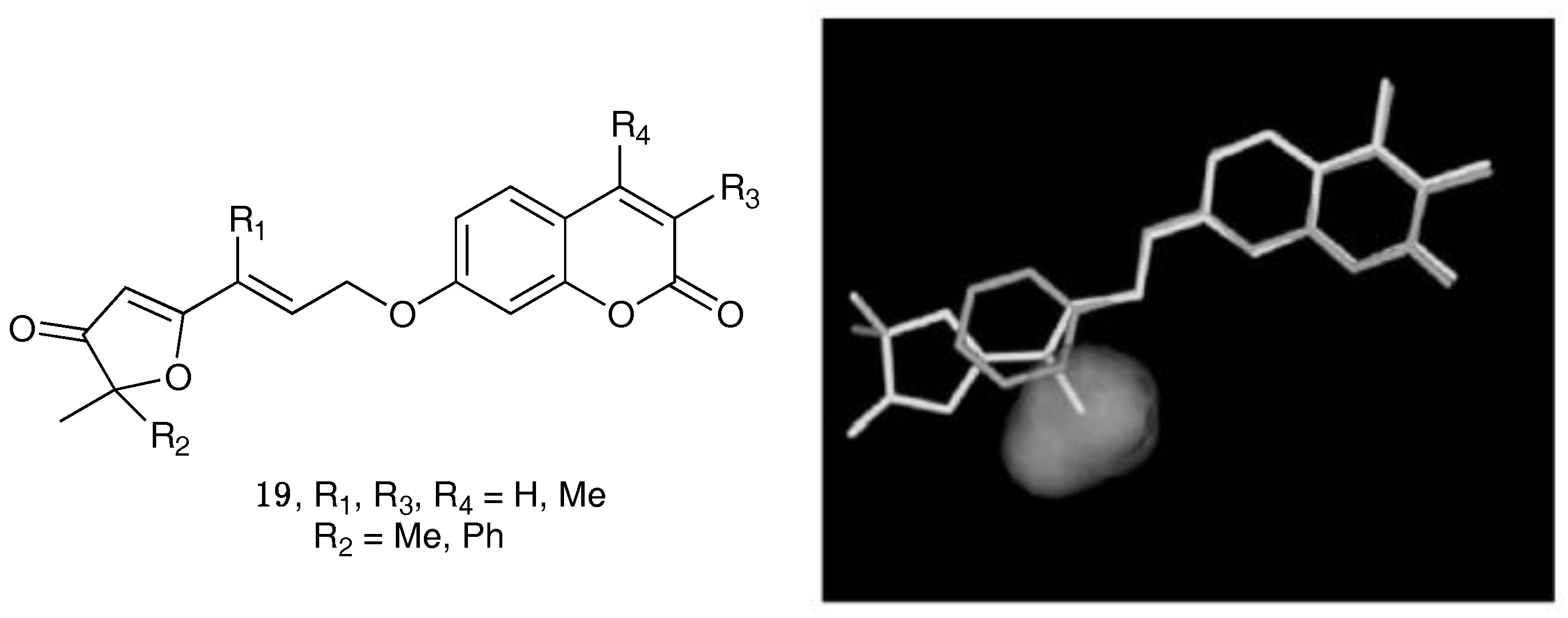
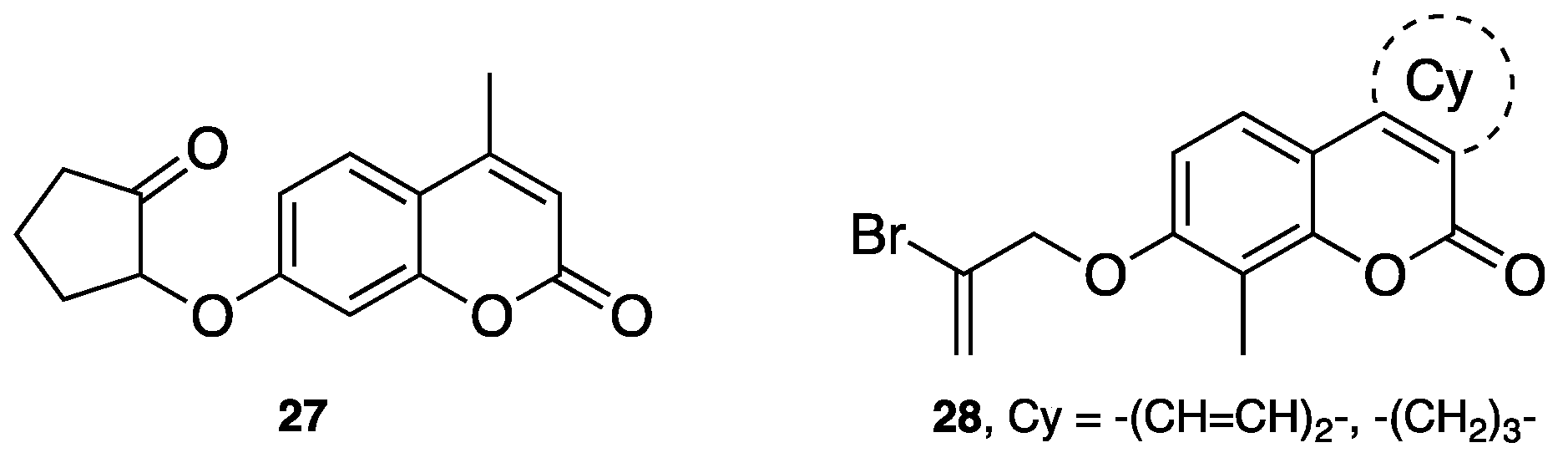
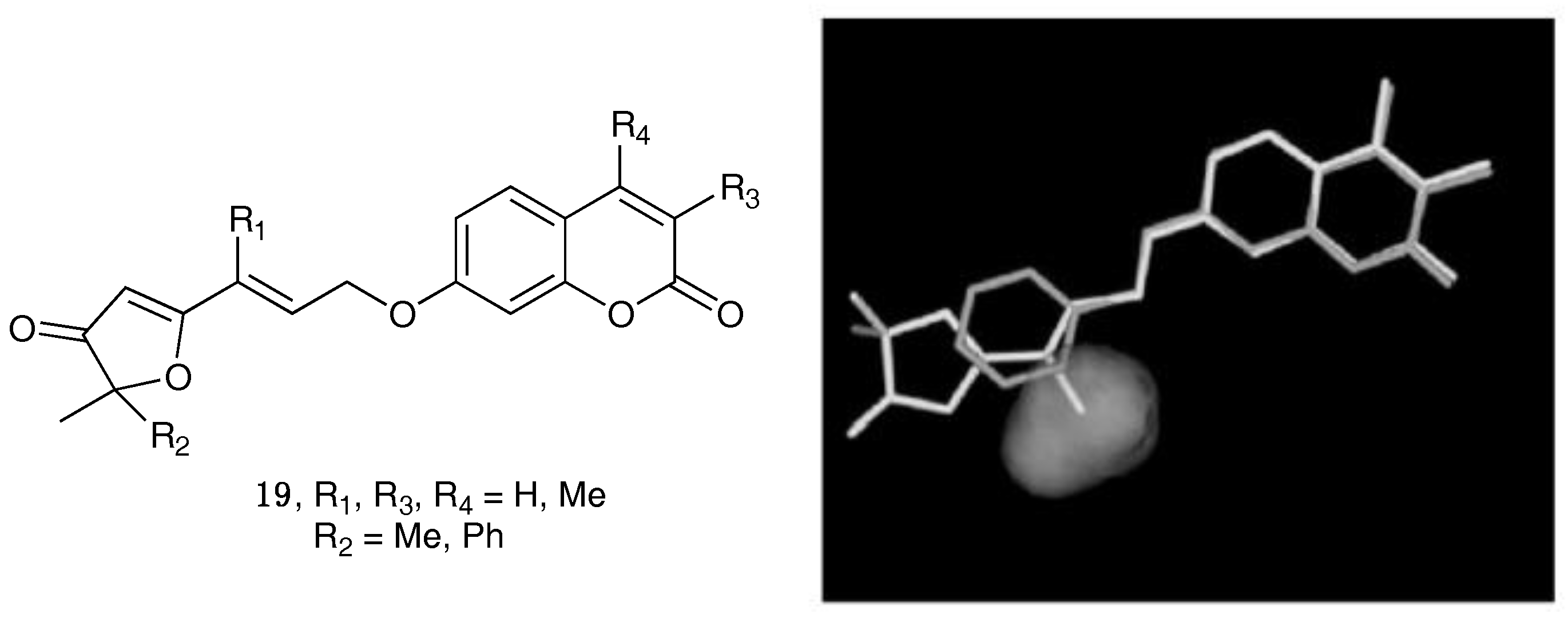
- Chimichi, S.; Boccalini, M.; Salvador, A.; Dall’Acqua, F.; Basso, G.; Viola, G. Synthesis and biological evaluation of new geiparvarin derivatives. ChemMedChem 2009, 4, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Carotti, A.; Carrieri, A.; Chimichi, S.; Boccalini, M.; Cosimelli, B.; Gnerre, C.; Carotti, A.; Carrupt, P.-A.; Testa, B. Natural and synthetic geiparvarins are strong and selective MAO-B inhibitors. Synthesis and SAR studies. Bioorg. Med. Chem. Lett. 2002, 12, 3551–3555. [Google Scholar] [CrossRef]
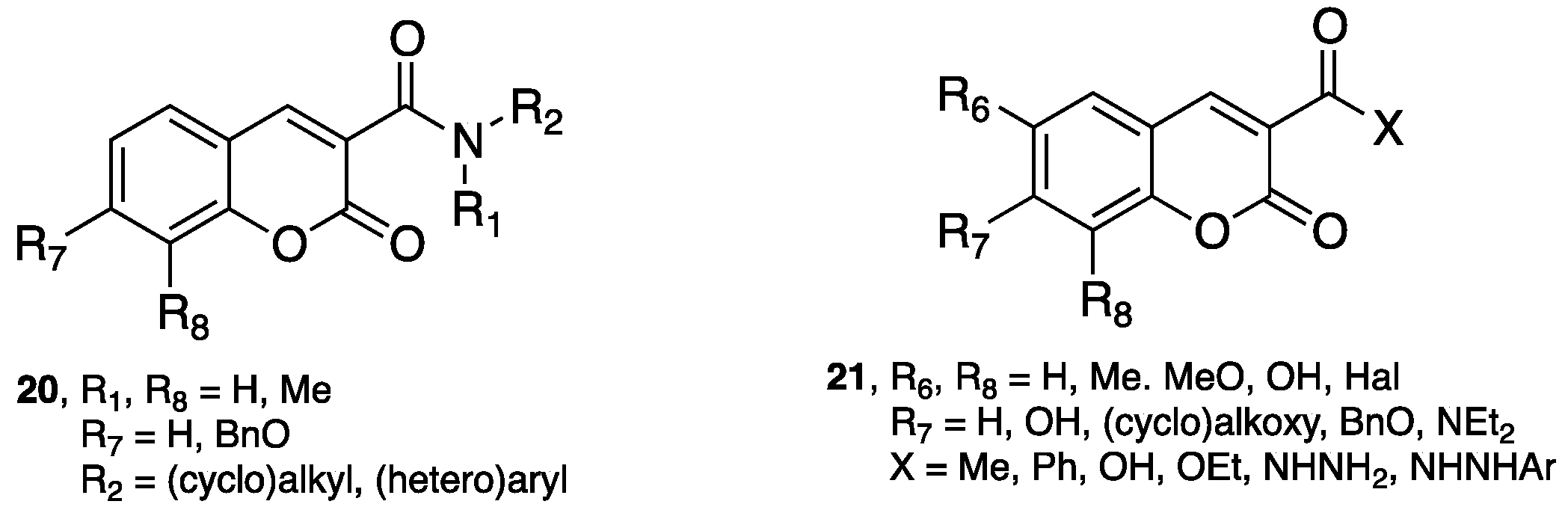
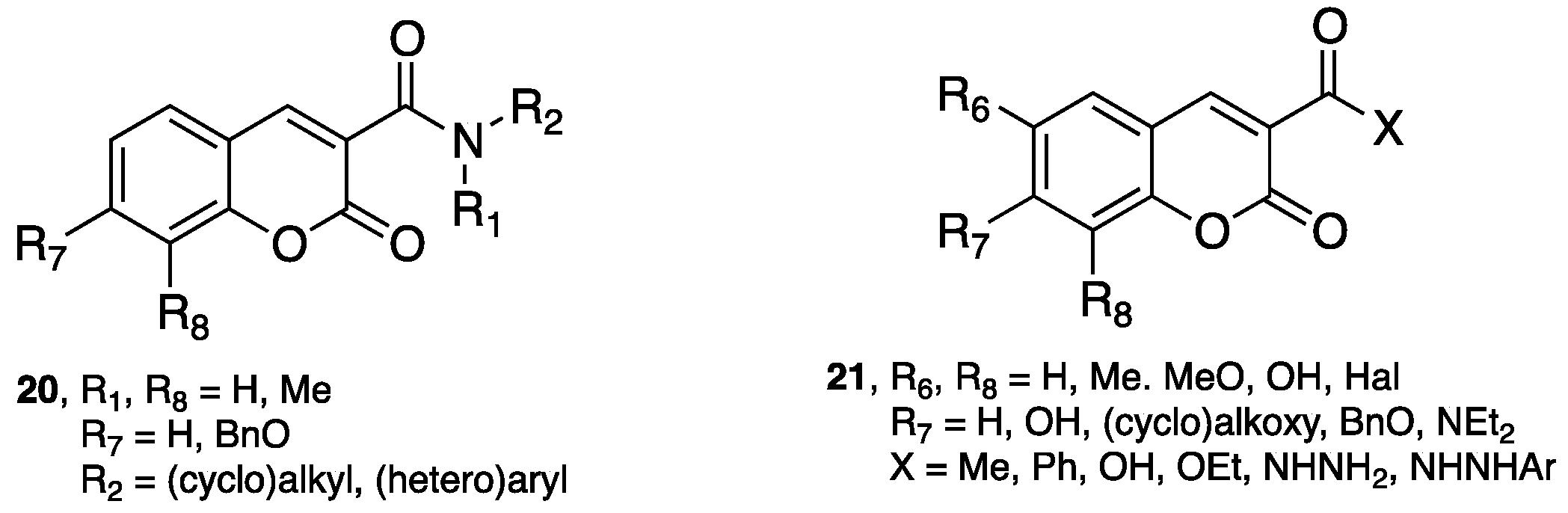
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Bizzarri, B.; Granese, A.; Carradori, S.; Yáñez, M.; Orallo, F.; Ortuso, F.; et al. Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J. Med. Chem. 2009, 52, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Secci, D.; Carradori, S.; Bolasco, A.; Chimenti, P.; Yáñez, M.; Ortuso, F.; Alcaro, S. Synthesis and selective human monoamine oxidase inhibition of 3-carbonyl, 3-acyl, and 3-carboxyhydrazido coumarin derivatives. Eur. J. Med. Chem. 2011, 46, 4846–4852. [Google Scholar] [CrossRef] [PubMed]
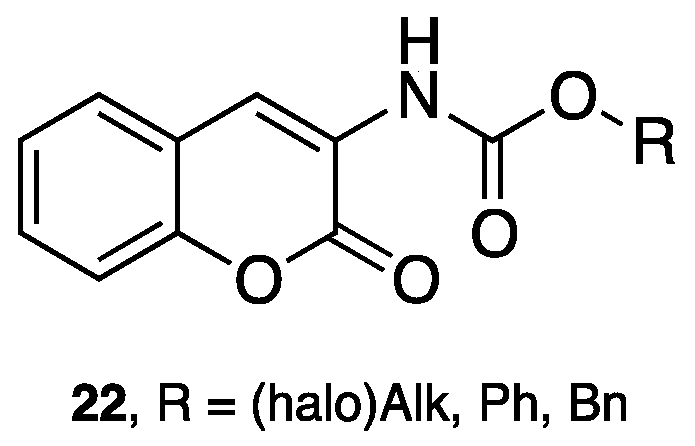
- Matos, M.J.; Vilar, S.; Gonzalez-Franco, R.M.; Uriarte, E.; Santana, L.; Friedman, C.; Tatonetti, N.P.; Viña, D.; Fontenla, J.A. Novel (coumarin-3-yl)carbamates as selective MAO-B inhibitors: Synthesis, in vitro and in vivo assays, theoretical evaluation of ADME properties and docking study. Eur. J. Med. Chem. 2013, 63, 151–161. [Google Scholar] [CrossRef] [PubMed]
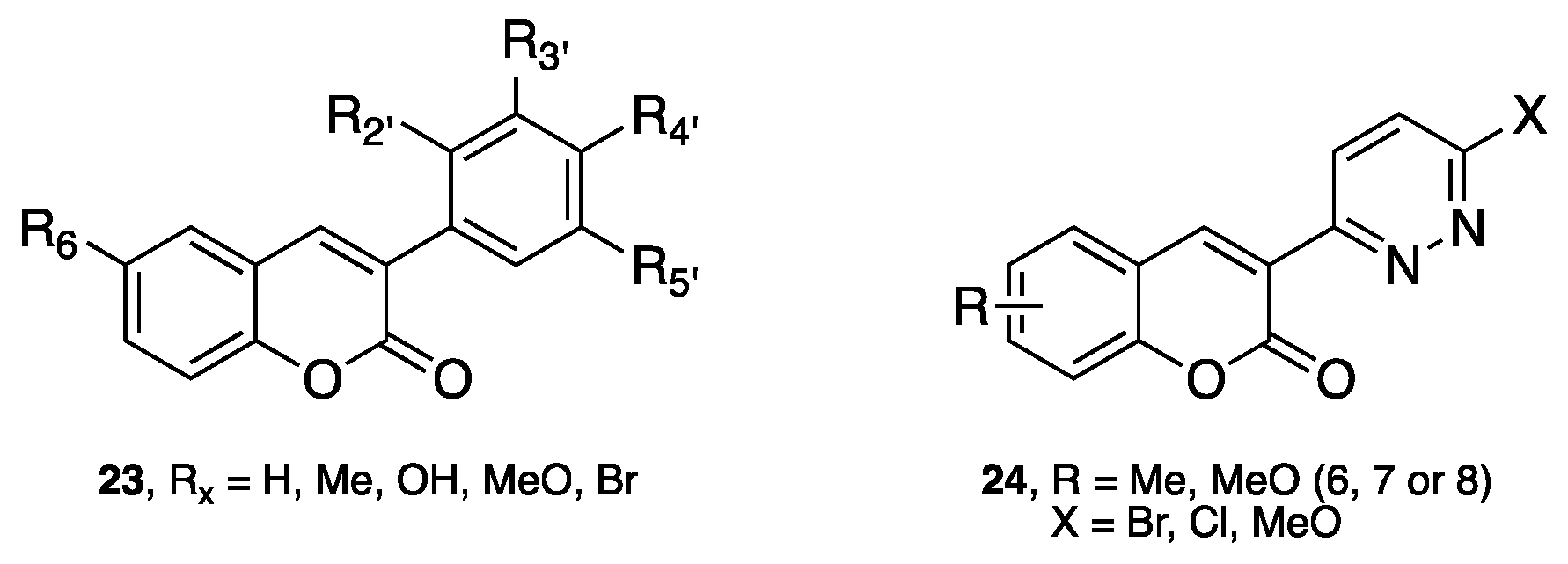
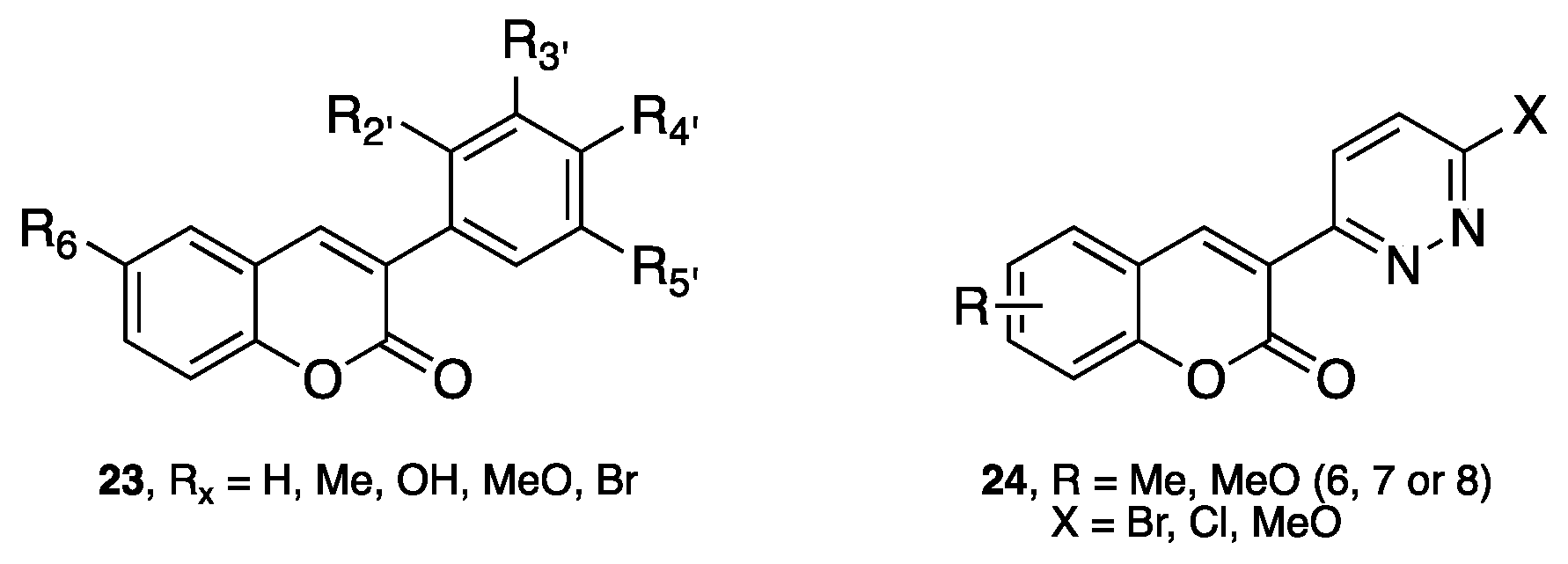
- Matos, M.J.; Terán, C.; Pérez-Castillo, Y.; Uriarte, E.; Santana, L.; Viña, D. Synthesis and study of a series of 3-arylcoumarins as potent and selective monoamine oxidase B inhibitors. J. Med. Chem. 2011, 54, 7127–7137. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Vilar, S.; García-Morales, V.; Tatonetti, N.P.; Uriarte, E.; Santana, L.; Viña, D. Insight into the functional and structural properties of 3-arylcoumarin as an interesting scaffold in monoamine oxidase B inhibition. ChemMedChem 2014, 9, 1488–1500. [Google Scholar] [CrossRef] [PubMed]
- Delogu, G.L.; Serra, S.; Quezada, E.; Uriarte, E.; Vilar, S.; Tatonetti, N.P.; Viña, D. Monoamine oxidase (MAO) inhibitory activity: 3-phenylcoumarins versus 4-hydroxy-3-phenylcoumarins. ChemMedChem 2014, 9, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Costas-Lago, M.C.; Besada, P.; Rodríguez-Enríquez, F.; Viña, D.; Vilar, S.; Uriarte, E.; Borges, F.; Terán, C. Synthesis and structure-activity relationship study of novel 3-heteroarylcoumarins based on pyridazine scaffold as selective MAO-B inhibitors. Eur. J. Med. Chem. 2017, 139, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Barletta, M.; Soto-Otero, R.; Nicolotti, O.; Mendez-Alvarez, E.; Catto, M.; Introcaso, A.; Stefanachi, A.; Cellamare, S.; Altomare, C.; Carotti, A. Discovery, biological evaluation, and structure-activity and -selectivity relationships of 6′-substituted (E)-2-(benzofuran-3(2H)-ylidene)-N-methylacetamides, a novel class of potent and selective monoamine oxidase inhibitors. J. Med. Chem. 2013, 56, 2651–2664. [Google Scholar] [CrossRef] [PubMed]
- Carotti, A.; Melloni, P.; Thaler, F.; Caccia, C.; Maestroni, S.; Salvati, P. Substituted Aminoalkyl and Amidoalkyl Benzopyran Derivatives. Patent WO102958 A1, 5 October 2006. [Google Scholar]
- Carotti, A.; Caccia, C.; Thaler, P.; Salvati, P.; Melloni, P.; Maestroni, S. Derivados de Amino-Alquil e Amido-Alquil Benzopiran Substitudos. Patent PI 0609265-9 A2, 9 March 2010. [Google Scholar]
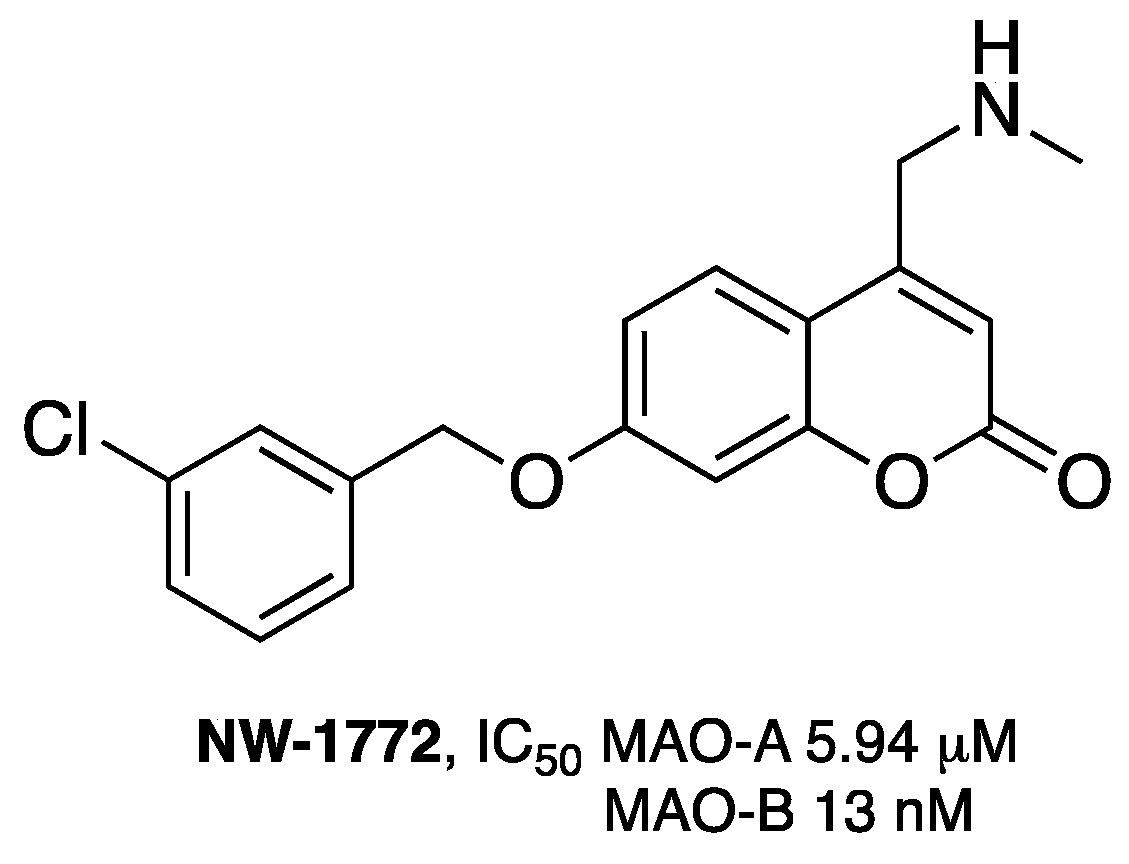
- Pisani, L.; Muncipinto, G.; Miscioscia, T.F.; Nicolotti, O.; Leonetti, F.; Catto, M.; Caccia, C.; Salvati, P.; Soto-Otero, R.; Mendez-Alvarez, E.; et al. Discovery of a novel class of potent coumarin monoamine oxidase B inhibitors: Development and biopharmacological profiling of 7-[(3-chlorobenzyl)oxy]-4-[(methylamino)methyl]-2H-chromen-2-one methanesulfonate (NW-1772) as a highly potent, selective, reversible, and orally active monoamine oxidase B inhibitor. J. Med. Chem. 2009, 52, 6685–6706. [Google Scholar] [PubMed]
- Borgohain, R.; Szasz, J.; Stanzione, P.; Meshram, C.; Bhatt, M.H.; Chirilineau, D.; Stocchi, F.; Lucini, V.; Giuliani, R.; Forrest, E.; et al. Two-year, randomized, controlled study of safinamide as add-on to levodopa in mid to late Parkinson's disease. Mov. Disord. 2014, 29, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, F.; Capaldi, C.; Pisani, L.; Muncipinto, G.; Nicolotti, O.; Caccia, C.; Carotti, A. Solid Phase Synthesis of Safinamide and Analogues as Potent and Selective MAO-B Inhibitors. J. Med. Chem. 2007, 50, 4909–4916. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Catto, M.; Nicolotti, O.; Grossi, G.; Di Braccio, M.; Soto-Otero, R.; Mendez-Alvarez, E.; Stefanachi, A.; Gadaleta, D.; Carotti, A. Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2013, 70, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Mladenović, M.; Patsilinakos, A.; Pirolli, A.; Sabatino, M.; Ragno, R. Understanding the molecular determinant of reversible human monoamine oxidase B inhibitors containing 2H-chromen-2-one Core: Structure-based and ligand-based derived three-dimensional quantitative structure-activity relationships predictive models. J. Chem. Inf. Model. 2017, 57, 787–814. [Google Scholar] [CrossRef] [PubMed]
- Santana, L.; Uriarte, E.; González-Díaz, H.; Zagotto, G.; Soto-Otero, R.; Méndez-Alvarez, E. A QSAR model for in silico screening of MAO-A inhibitors. Prediction, synthesis, and biological assay of novel coumarins. J. Med. Chem. 2006, 49, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Santana, L.; González-Díaz, H.; Quezada, E.; Uriarte, E.; Yáñez, M.; Viña, D.; Orallo, F. Quantitative structure-activity relationship and complex network approach to monoamine oxidase A and B inhibitors. J. Med. Chem. 2008, 51, 6740–6751. [Google Scholar] [CrossRef] [PubMed]
- Mangiatordi, G.F.; Alberga, D.; Pisani, L.; Gadaleta, D.; Trisciuzzi, D.; Farina, R.; Carotti, A.; Lattanzi, G.; Catto, M.; Nicolotti, O. A rational approach to elucidate human monoamine oxidase molecular selectivity. Eur. J. Pharm. Sci. 2017, 101, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V., Jr.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Sanson, B.; Colletier, J.P.; Xu, Y.; Lang, P.T.; Jiang, H.; Silman, I.; Sussman, J.L.; Weik, M. Backdoor opening mechanism in acetylcholinesterase based on X-ray crystallography and molecular dynamics simulations. Protein Sci. 2011, 20, 1114–1118. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, F.; Cappa, A.; Maccallini, C.; Carotti, A. Synthesis of potential dual binding site acetylcholinesterase inhibitors through an efficient solid phase approach based on Mitsunobu reaction. Arkivoc 2004, 272, 285. [Google Scholar]
- Leonetti, F.; Catto, M.; Nicolotti, O.; Pisani, L.; Cappa, A.; Stefanachi, A.; Carotti, A. Homo- and hetero-bivalent edrophonium-like ammonium salts as highly potent, dual binding site AChE inhibitors. Bioorg. Med. Chem. 2008, 16, 7450–7456. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Catto, M.; Giangreco, I.; Leonetti, F.; Nicolotti, O.; Stefanachi, A.; Cellamare, S.; Carotti, A. Design, Synthesis, and Biological Evaluation of Coumarin Derivatives Tethered to an Edrophonium-like Fragment as Highly Potent and Selective Dual Binding Site Acetylcholinesterase Inhibitors. ChemMedChem 2010, 5, 1616–1630. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Khoobi, M.; Foroumadi, A.; Nadri, H.; Moradi, A.; Sakhteman, A.; Ghandi, M.; Shafiee, A. Novel coumarin derivatives bearing N-benzyl pyridinium moiety: Potent and dual binding site acetylcholinesterase inhibitors. Bioorg. Med. Chem. 2012, 20, 7214–7222. [Google Scholar] [CrossRef] [PubMed]
- Catto, M.; Pisani, L.; Leonetti, F.; Nicolotti, O.; Pesce, P.; Stefanachi, A.; Cellamare, S.; Carotti, A. Design, synthesis and biological evaluation of coumarin alkylamines as potent and selective dual binding site inhibitors of acetylcholinesterase. Bioorg. Med. Chem. 2013, 20, 146–152. [Google Scholar] [CrossRef] [PubMed]
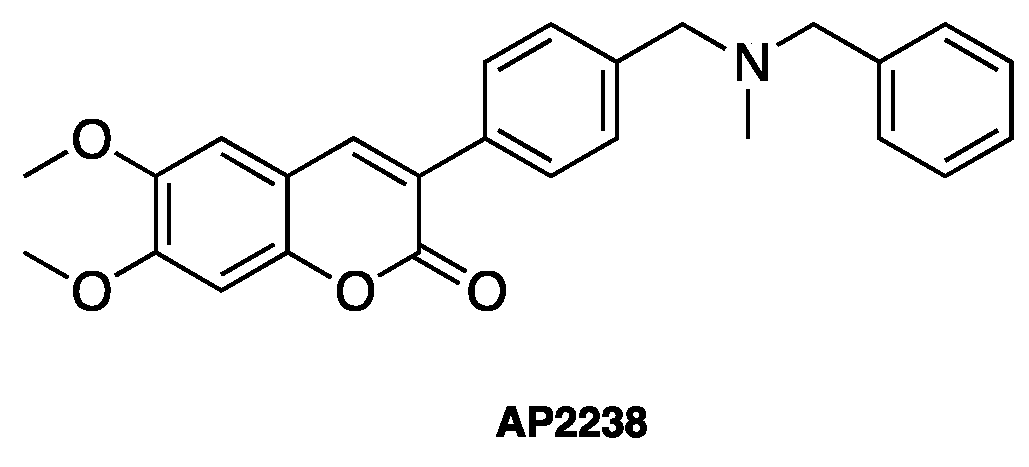
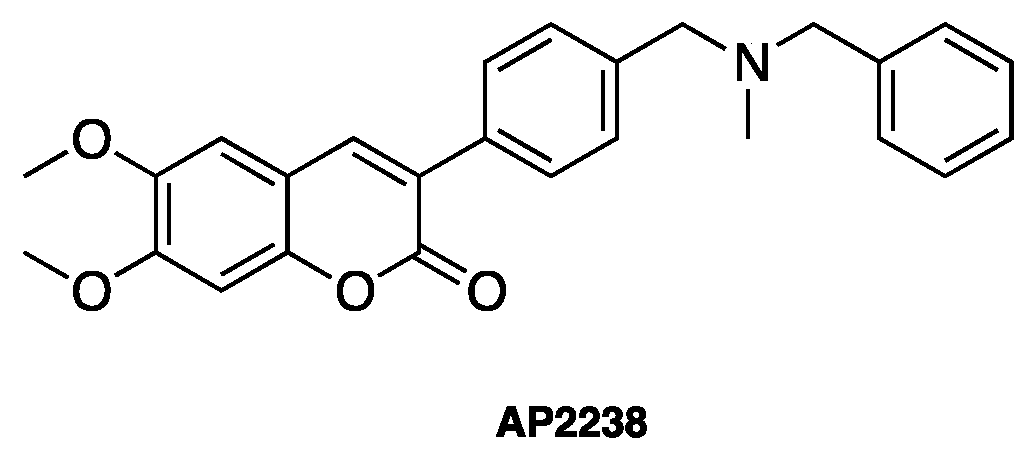
- Piazzi, L.; Rampa, A.; Bisi, A.; Gobbi, S.; Belluti, F.; Cavalli, A.; Bartolini, M.; Andrisano, V.; Valenti, P.; Recanatini, M. 3-(4-[[Benzyl(methyl)amino]methyl]phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation: A dual function lead for Alzheimer’s disease therapy. J. Med. Chem. 2003, 46, 2279–2282. [Google Scholar] [CrossRef] [PubMed]
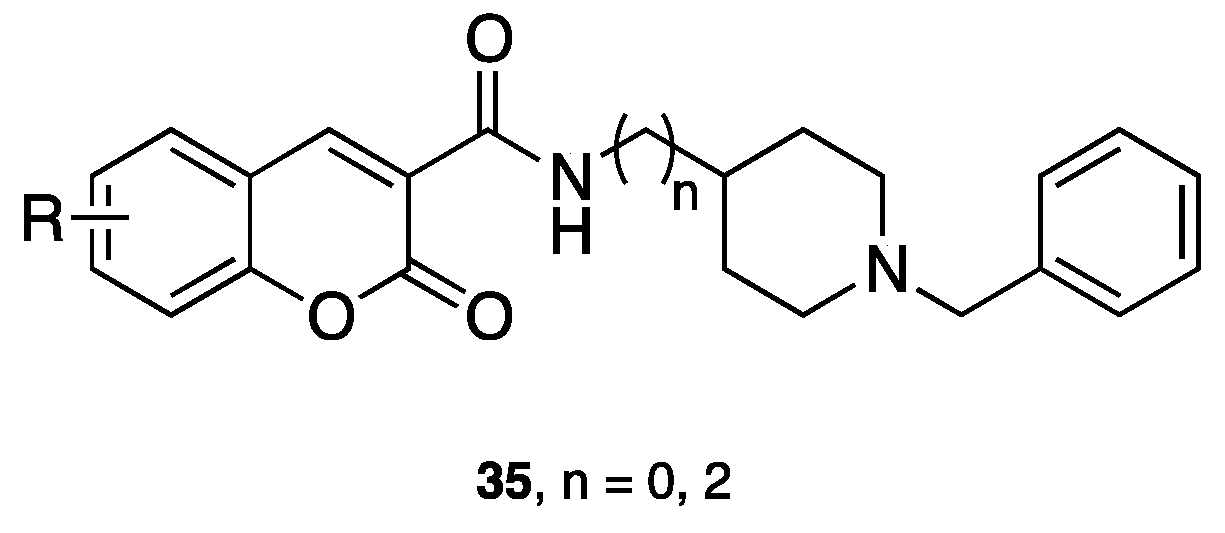
- Asadipour, A.; Alipour, M.; Jafari, M.; Khoobi, M.; Emami, S.; Nadri, H.; Sakhteman, A.; Moradi, A.; Sheibani, V.; Homayouni Moghadam, F.; et al. Novel coumarin-3-carboxamides bearing N-benzylpiperidine moiety as potent acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2013, 70, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Hamulakova, S.; Janovec, L.; Hrabinova, M.; Spilovska, K.; Korabecny, J.; Kristian, P.; Kuca, K.; Imrich, J. Synthesis and biological evaluation of novel tacrine derivatives and tacrine-coumarin hybrids as cholinesterase inhibitors. J. Med. Chem. 2014, 57, 7073–7084. [Google Scholar] [CrossRef] [PubMed]
- Morphy, J.R.; Harris, C.J. Designing Multi-Target Drugs; RSC Publishing: London, UK, 2012. [Google Scholar]
- Geldenhuys, W.J.; Van der Schyf, C.J. Designing drugs with multi-target activity: The next step in the treatment of neurodegenerative disorders. Expert Opin. Drug Discov. 2013, 8, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Nicolotti, O.; Giangreco, I.; Introcaso, A.; Leonetti, F.; Stefanachi, A.; Carotti, A. Strategies of multi-objective optimization in drug discovery and development. Expert Opin. Drug Discov. 2011, 6, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Catto, M.; Leonetti, F.; Nicolotti, O.; Stefanachi, A.; Campagna, F.; Carotti, A. Targeting monoamine oxidases with multipotent ligands: An emerging strategy in the search of new drugs against neurodegenerative diseases. Curr. Med. Chem. 2011, 18, 4568–4587. [Google Scholar] [CrossRef] [PubMed]
- Bruehlmann, C.; Ooms, F.; Carrupt, P.-A.; Testa, B.; Catto, M.; Leonetti, F.; Altomare, C.; Carotti, A. Coumarin derivatives as dual inhibitors of acetylcholinesterase and monoamine oxidase. J. Med. Chem. 2001, 44, 3195–3198. [Google Scholar] [CrossRef]
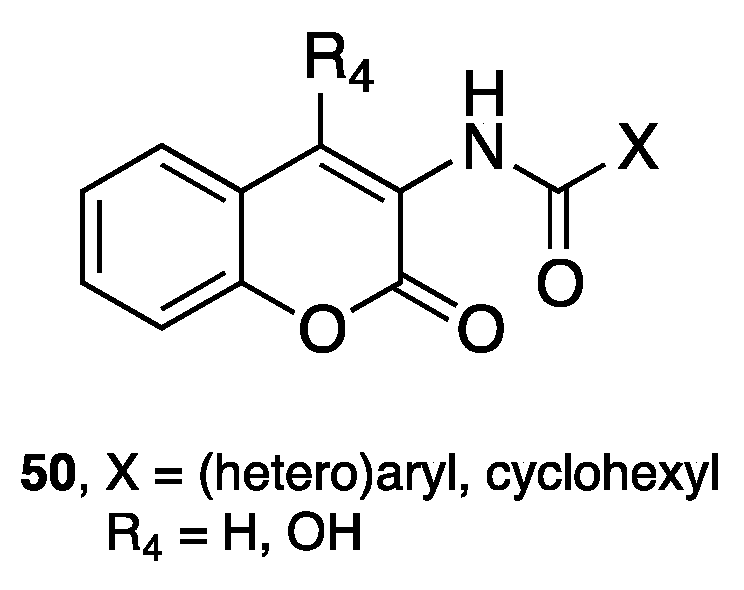
- Farina, R.; Pisani, L.; Catto, M.; Nicolotti, O.; Gadaleta, D.; Soto-Otero, R.; Méndez-Álvarez, E.; Passos, C.; Muncipinto, G.; Altomare, C.D.; et al. Structure-based design and optimization of multitarget-directed 2H-chromen-2-one derivatives as potent inhibitors of monoamine oxidase B and cholinesterases. J. Med. Chem. 2015, 58, 5561–5578. [Google Scholar] [CrossRef] [PubMed]
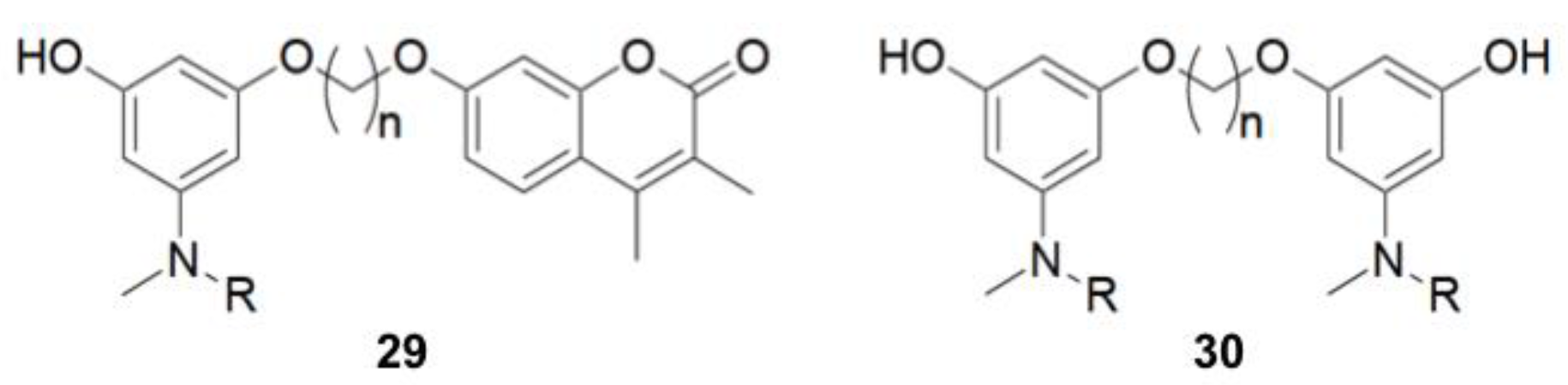
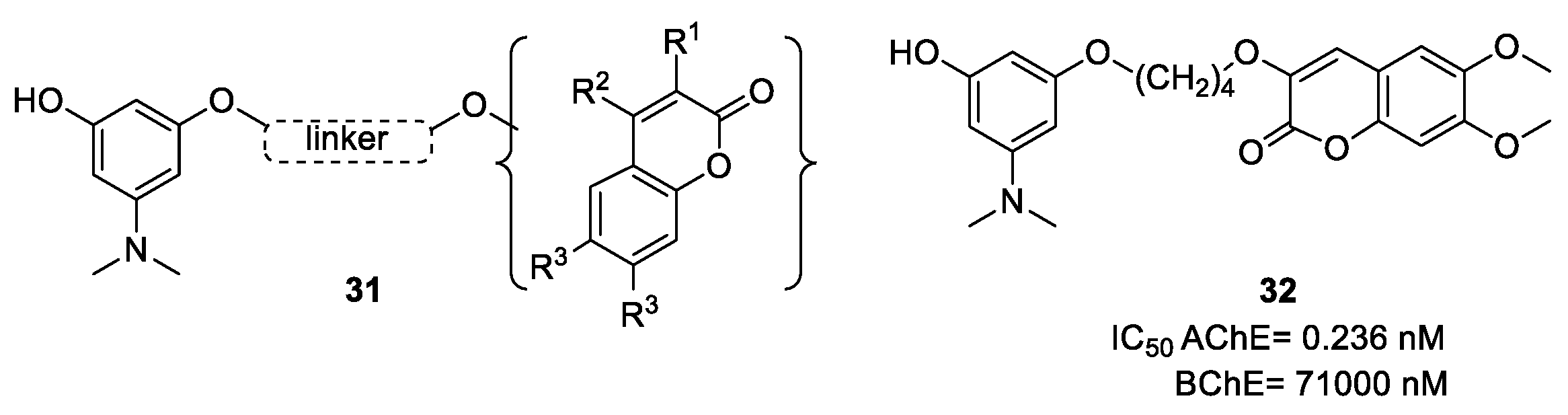
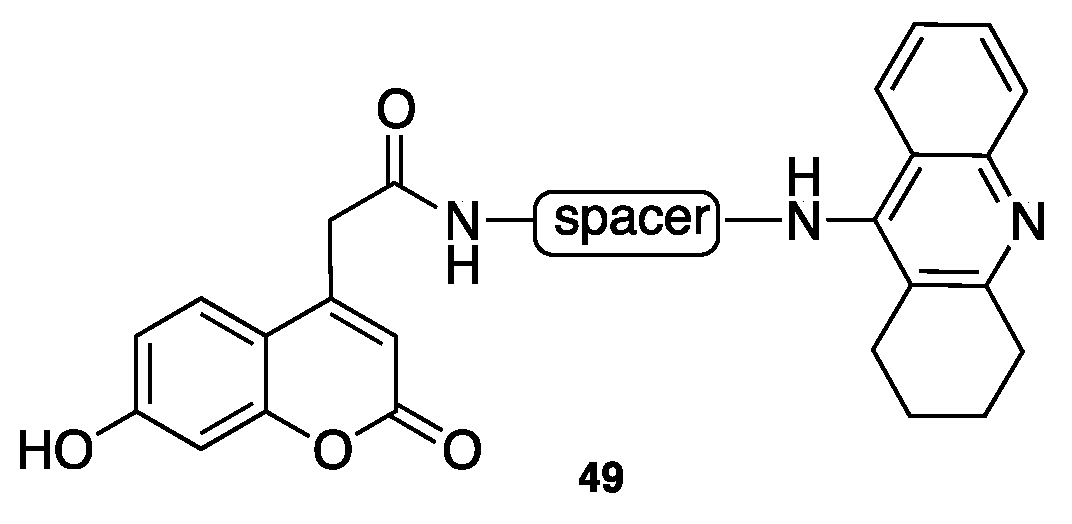
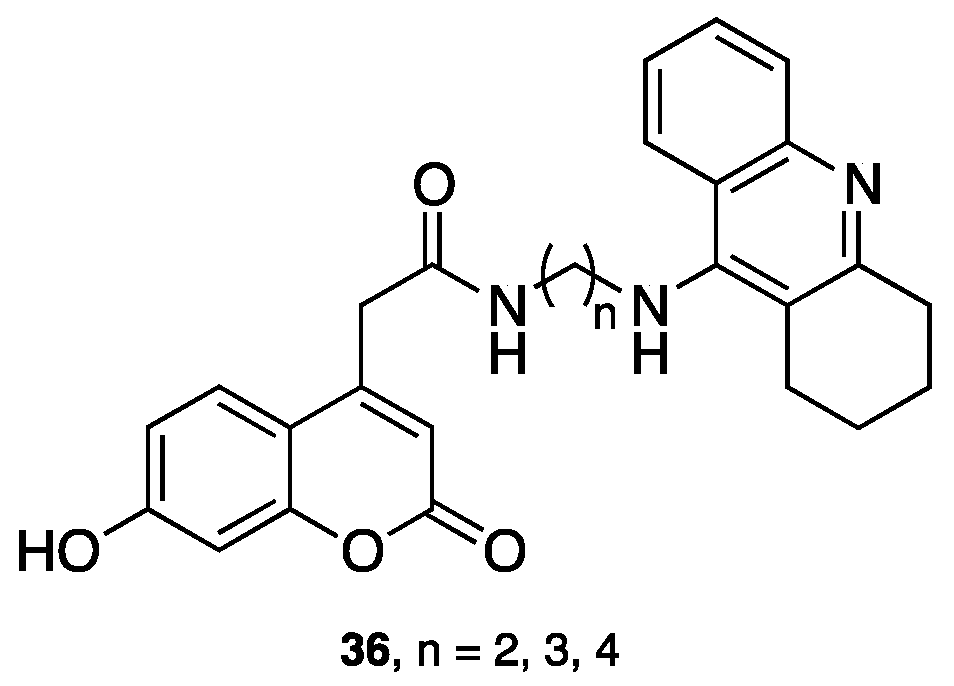
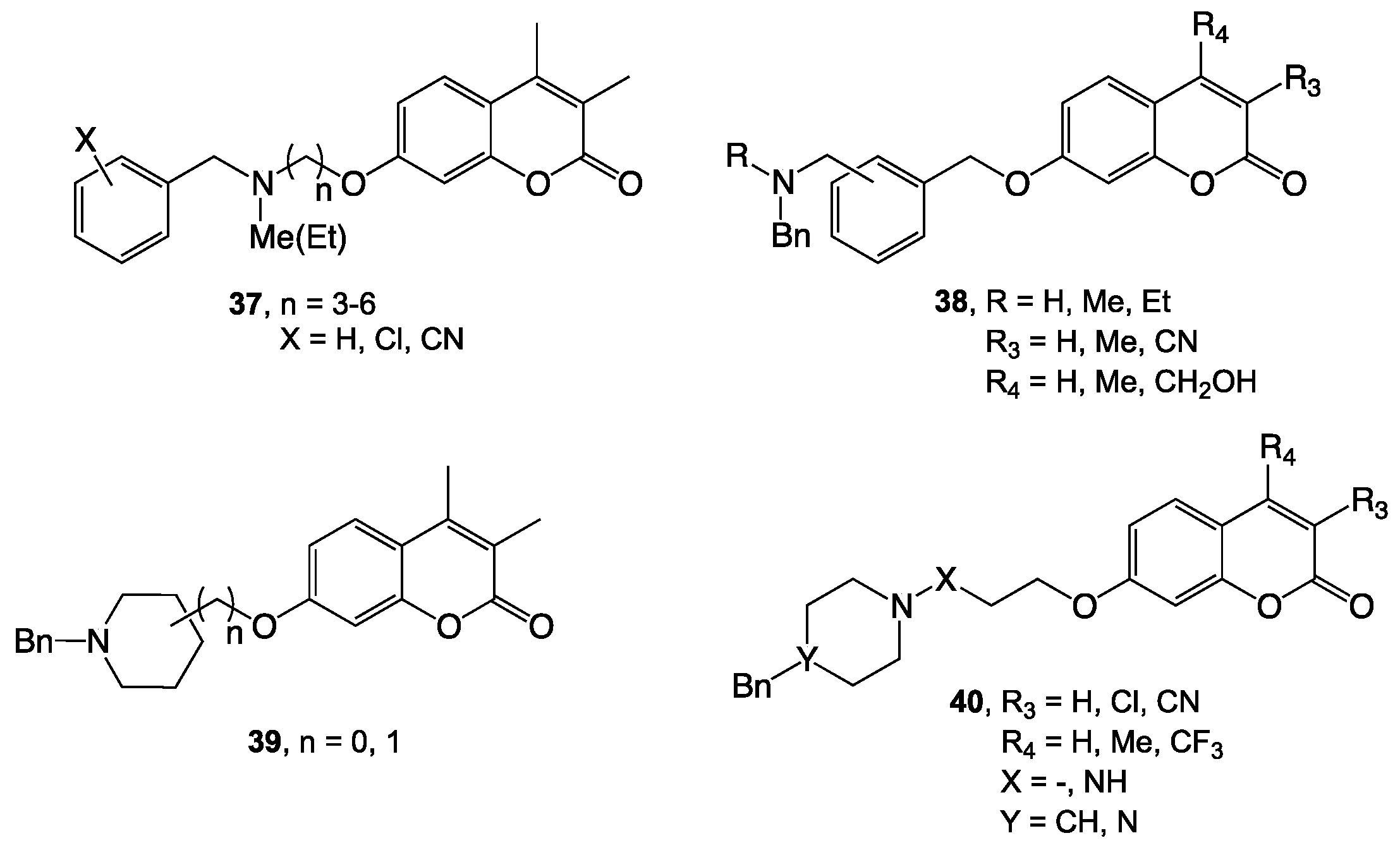
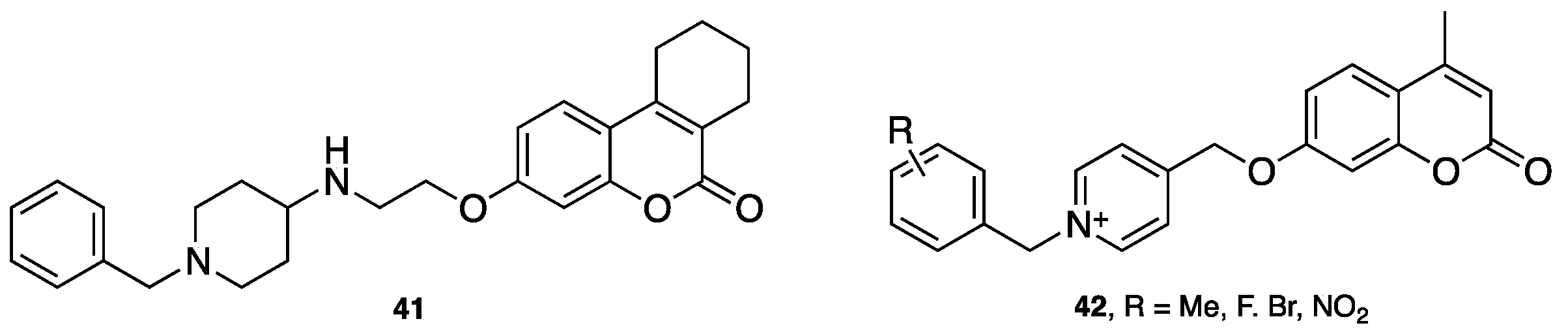
- Pisani, L.; Farina, R.; Catto, M.; Iacobazzi, R.; Nicolotti, O.; Cellamare, S.; Mangiatordi, G.; Denora, N.; Soto-Otero, R.; Siragusa, L.; et al. Exploring basic tail modifications of coumarin-based dual acetylcholinesterase-monoamine oxidase B inhibitors: Identification of water-soluble, brain-permeant neuroprotective multitarget agent. J. Med. Chem. 2016, 59, 6791–6806. [Google Scholar] [CrossRef] [PubMed]
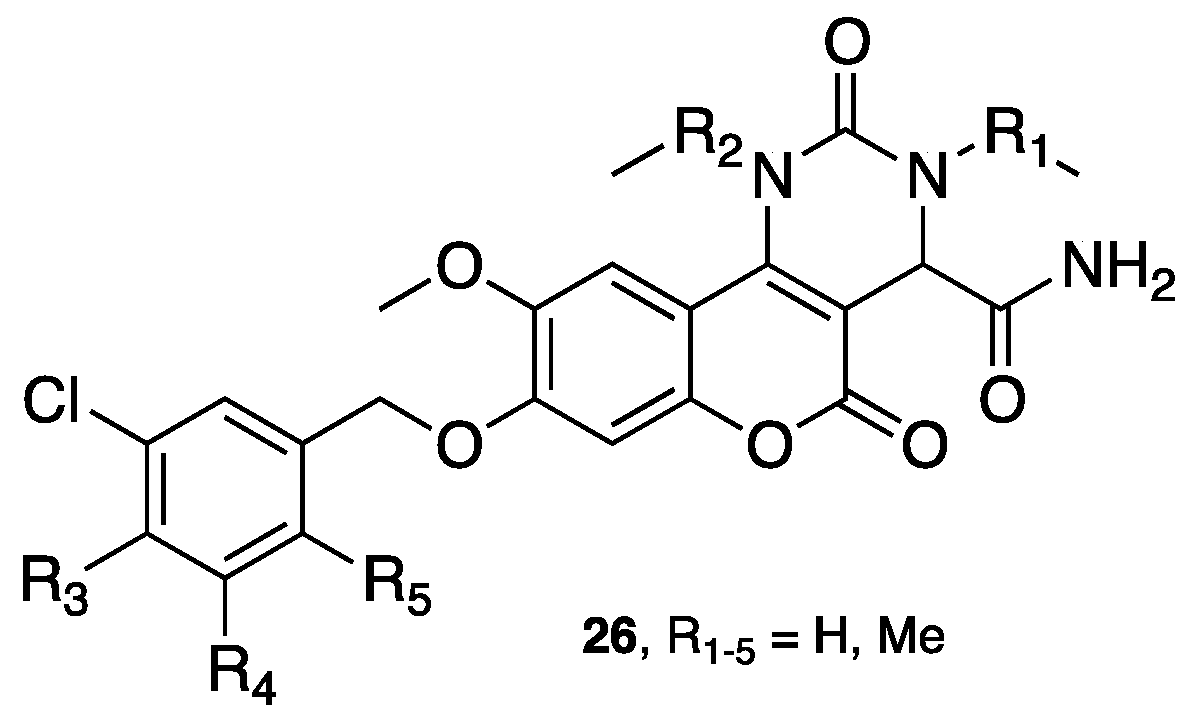
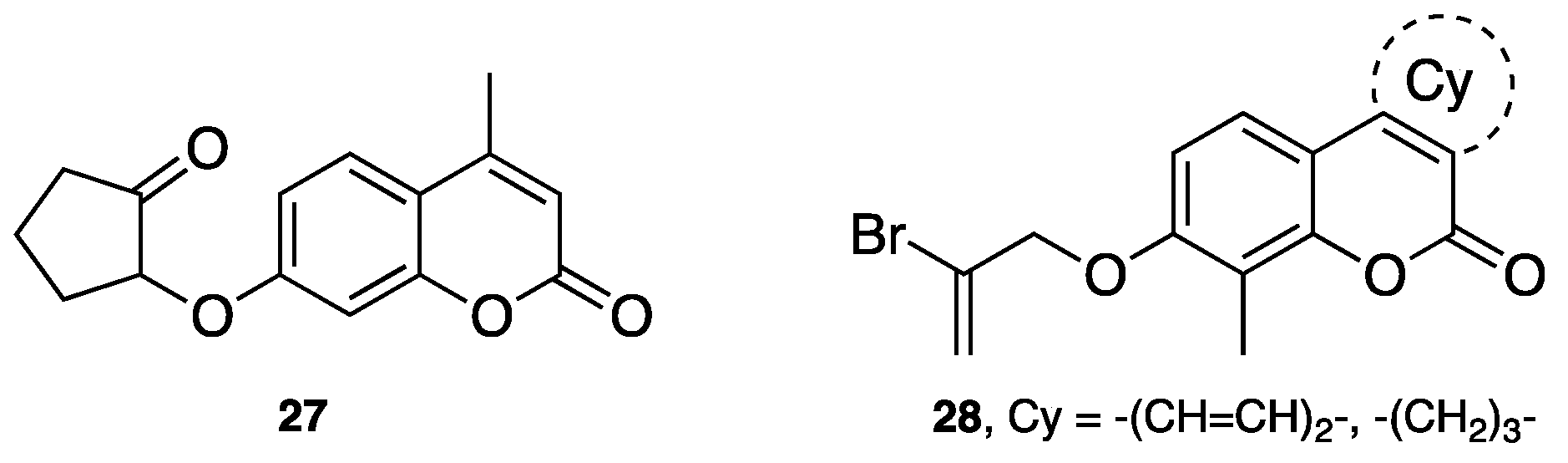
- Joubert, J.; Foka, G.B.; Repsold, B.P.; Oliver, D.W.; Kapp, E.; Malan, S.F. Synthesis and evaluation of 7-substituted coumarin derivatives as multimodal monoamine oxidase-B and cholinesterase inhibitors for the treatment of Alzheimer's disease. Eur. J. Med. Chem. 2017, 125, 853–864. [Google Scholar] [CrossRef] [PubMed]
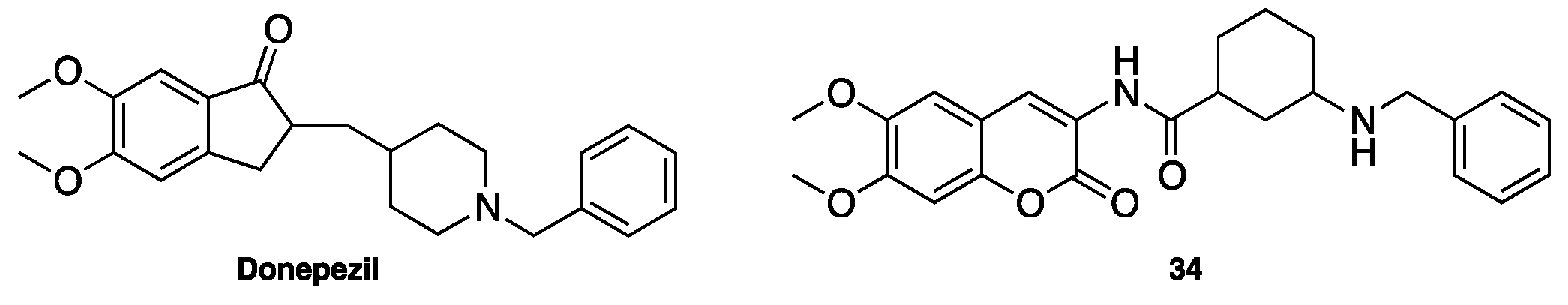
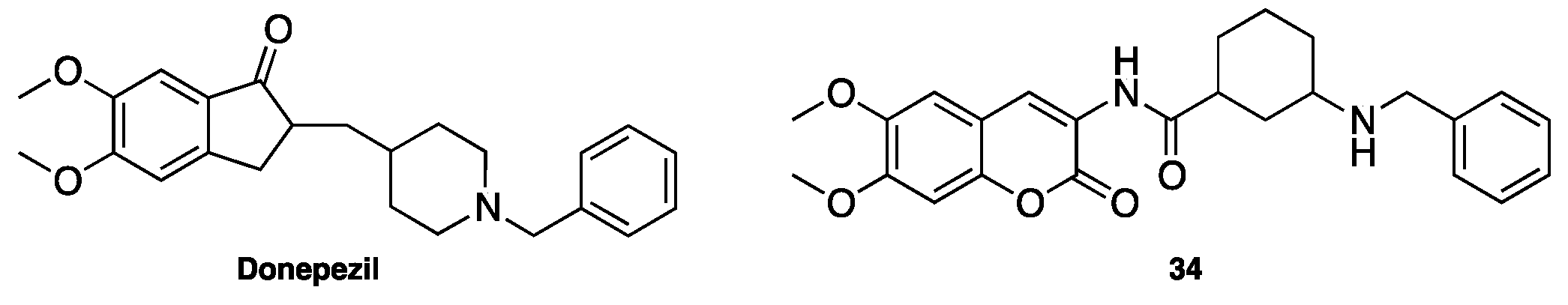
- Xie, S.S.; Lan, J.S.; Wang, X.; Wang, Z.M.; Jiang, N.; Li, F.; Wu, J.J.; Wang, J.; Kong, L.Y. Design, synthesis and biological evaluation of novel donepezil-coumarin hybrids as multi-target agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2016, 24, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
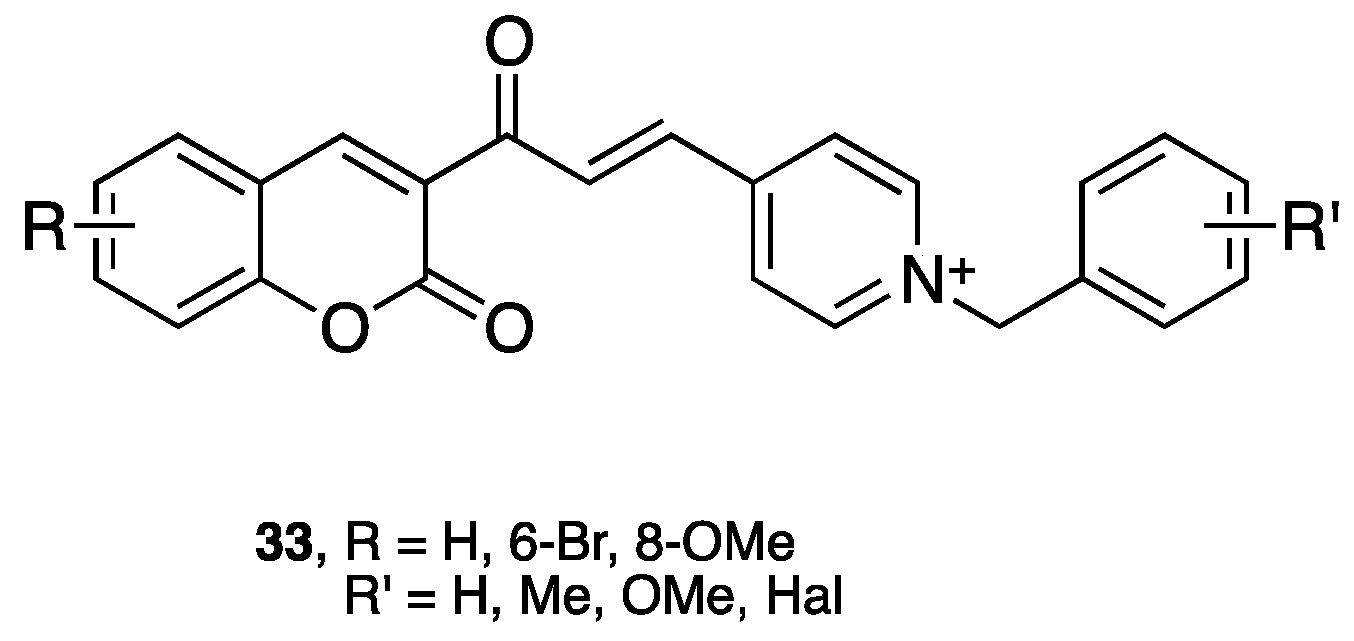
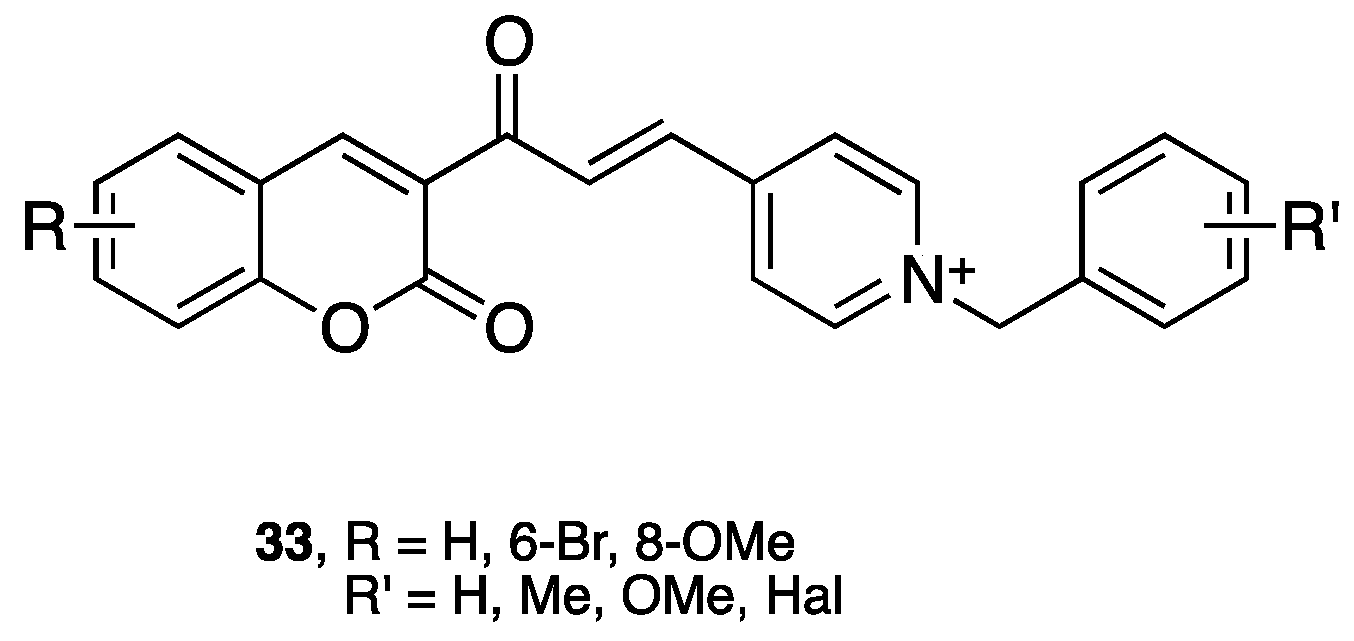
- Lan, J.S.; Ding, Y.; Liu, Y.; Kang, P.; Hou, J.W.; Zhang, X.Y.; Xie, S.S.; Zhang, T. Design, synthesis and biological evaluation of novel coumarin-N-benzyl pyridinium hybrids as multi-target agents for the treatment of Alzheimer's disease. Eur. J. Med. Chem. 2017, 139, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.S.; Wang, X.; Jiang, N.; Yu, W.; Wang, K.D.; Lan, J.S.; Li, Z.R.; Kong, L.Y. Multi-target tacrine-coumarin hybrids: Cholinesterase and monoamine oxidase B inhibition properties against Alzheimer’s disease. Eur. J. Med. Chem. 2015, 95, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Farina, R.; Soto Otero, R.; Denora, N.; Mangiatordi, G.F.; Nicolotti, O.; Mendez Alvarez, E.; Altomare, C.D.; Catto, M.; Carotti, A. Searching for multitargeting neurotherapeutics against Alzheimer’s: Discovery of potent AChE−MAO B inhibitors through the decoration of 2H-chromen-2-one structural motif. Molecules 2016, 21, 362. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Pei, J.; Lai, L. Computational multitarget drug design. J. Chem. Inf. Model. 2017, 57, 40–412. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.; Bansal, Y.; Silakari, O.; Bansal, G. Coumarin hybrids as novel therapeutic agents. Bioorg. Med. Chem. 2014, 22, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.S.; Wang, X.B.; Li, J.Y.; Yang, L.; Kong, L.Y. Design, synthesis and evaluation of novel tacrine-coumarin hybrids as multifunctional cholinesterase inhibitors against Alzheimer's disease. Eur. J. Med. Chem. 2013, 64, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Peng, D.Y.; Yang, S.G.; Zhu, X.L.; Yang, W.C.; Yang, G.F. Syntheses of coumarin-tacrine hybrids as dual-site acetylcholinesterase inhibitors and their activity against butylcholinesterase, Aβ aggregation, and β-secretase. Bioorg. Med. Chem. 2014, 22, 4784–4791. [Google Scholar] [CrossRef] [PubMed]
- Piazzi, L.; Cavalli, A.; Colizzi, F.; Belluti, F.; Bartolini, M.; Mancini, F.; Recanatini, M.; Andrisano, V.; Rampa, A. Multi-target-directed coumarin derivatives: HAChE and BACE1 inhibitors as potential anti-Alzheimer compounds. Bioorg. Med. Chem. Lett. 2008, 18, 423–426. [Google Scholar] [CrossRef] [PubMed]
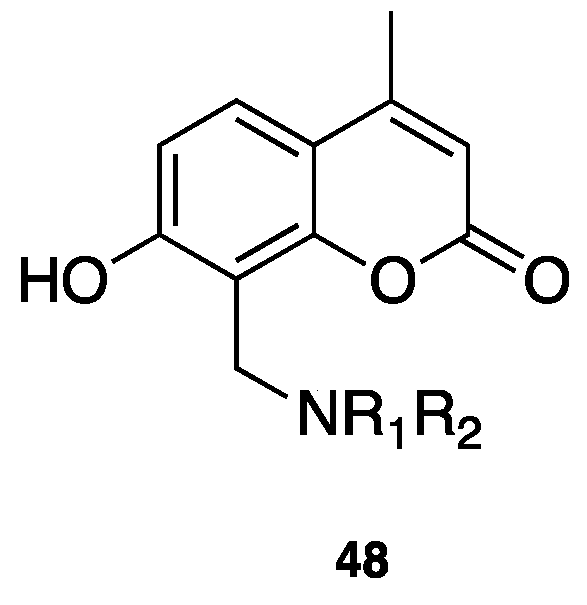
- Domínguez, J.L.; Fernández-Nieto, F.; Brea, J.M.; Catto, M.; Paleo, M.R.; Porto, S.; Sardina, F.J.; Castro, M.; Pisani, L.; Carotti, A.; et al. 8-Aminomethyl-7-hydroxy-4-methylcoumarins as multitarget leads for Alzheimer’s disease. ChemistrySelect 2016, 1, 2742–2749. [Google Scholar] [CrossRef]
- Weinreb, O.; Mandel, S.; Bar-Am, O.; Yogev-Falach, M.; Avramovich-Tirosh, Y.; Amit, T.; Youdim, M.B.H. Multifunctional neuroprotective derivatives of rasagiline as anti-Alzheimer’s disease drugs. Neurotherapeutics 2009, 6, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Hamulakova, S.; Poprac, P.; Jomova, K.; Brezova, V.; Lauro, P.; Drostinova, L.; Jun, D.; Sepsova, V.; Hrabinova, M.; Soukup, O.; et al. Targeting copper(II)-induced oxidative stress and the acetylcholinesterase system in Alzheimer’s disease using multifunctional tacrine-coumarin hybrid molecules. J. Inorg. Biochem. 2016, 161, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Rodríguez-Enríquez, F.; Borges, F.; Santana, L.; Uriarte, E.; Estrada, M.; Rodríguez-Franco, M.I.; Laguna, R.; Viña, D. 3-Amidocoumarins as potential multifunctional agents against neurodegenerative diseases. ChemMedChem 2015, 10, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Chen, W.J.; Zhou, Y.; Zhao, H.; Zhong, R.G. Rational design of coumarin derivatives as CK2 inhibitors by improving the interaction with the hinge region. Mol. Inform. 2016, 35, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Malini, B.; Purohit, A.; Ganeshapillai, D.; Woo, L.; Potter, B.V.; Reed, M. Inhibition of steroid sulphatase activity by tricyclic coumarin sulphamates. J. Steroid Biochem. Mol. Biol. 2000, 75, 253–258. [Google Scholar] [CrossRef]
- Woo, L.L.; Purohit, A.; Malini, B.; Reed, M.J.; Potter, B.V. Potent active site-directed inhibition of steroid sulphatase by tricyclic coumarin-based sulphamates. Chem. Biol. 2000, 7, 773–791. [Google Scholar] [CrossRef]
- Woo, L.L.; Purohit, A.; Reed, M.J.; Potter, B.V. Active site directed inhibition of estrone sulfatase by nonsteroidal coumarin suifamates. J. Med. Chem. 1996, 39, 1349–1351. [Google Scholar] [CrossRef] [PubMed]
- Danielian, P.; White, R.; Lees, J.; Parker, M. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992, 11, 1025–1033. [Google Scholar] [PubMed]
- Lewis, J.S.; Jordan, V.C. Selective estrogen receptor modulators (SERMs): Mechanisms of anticarcinogenesis and drug resistance. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 59, 1247–1263. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.K.; Ansari, M.I.; Yadav, N.; Gupta, P.K.; Gupta, A.K.; Saxena, R.; Fatima, L.; Manohar, M.; Kushwaha, P.; Khedgikar, V. Design and synthesis of ERa/ERB selective coumarin and chromene derivatives as potential antibreast cancer and anti-osteoporotic agents. RSC Adv. 2014, 4, 8828–8845. [Google Scholar] [CrossRef]
- Johnston, S.; Cheung, K. Fulvestrant-a novel endocrine therapy for breast cancer. Curr. Med. Chem. 2010, 17, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Yang, X.; Ren, Z.; McDonnell, D.P.; Norris, J.D.; Willson, T.M.; Greene, G.L. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell 2005, 18, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Degorce, S.L.; Bailey, A.; Callis, R.; De Savi, C.; Ducray, R.; Lamont, G.; MacFaul, P.; Maudet, M.; Martin, S.; Morgentin, R. Investigation of (E)-3-[4-(2-oxo-3-aryl-chromen-4-yl) oxyphenyl] acrylic acids as oral selective estrogen receptor down-reguiators. J. Med. Chem. 2015, 58, 3522–3533. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Kubo, H.; Tomigahara, Y.; Nishioka, K.; Takahashi, J.; Momose, M.; Inoue, S.; Kojima, A. Coumarins as novel 17-hydroxysteroid dehydrogenase type 3 inhibitors for potential treatment of prostate cancer. Bioorg. Med. Chem. Lett. 2010, 20, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Valente, S.; Bana, E.; Viry, E.; Bagrel, D.; Kirsch, G. Synthesis and biological evaluation of novel coumarin-based inhibitors of Cdc25 phosphatases. Bioorg. Med. Chem. Lett. 2010, 20, 5827–5830. [Google Scholar] [CrossRef] [PubMed]
- Saidu, N.E.B.; Valente, S.; Bana, E.; Kirsch, G.; Bagrel, D.; Montenarh, M. Coumarin polysulfides inhibit cell growth and induce apoptosis in HC7116 colon cancer cells. Bioorg. Med. Chem. 2012, 20, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Ganina, O.G.; Daras, E.; Bourgarel-Rey, V.; Peyrot, V.; Andresyuk, A.N.; Finer, J.-P.; Fedorov, A.Y.; Beletskaya, I.P.; Combes, S. Synthesis and biological evaluation of polymethoxylated 4-heteroarylcoumarins as tubulin assembly inhibitor. Bioorg. Med. Chem. 2008, 16, 8806–8812. [Google Scholar] [CrossRef] [PubMed]
- Bailiy, C.; Bal, C.; Barbier, P.; Combes, S.; Finet, J.-P.; Hildebrand, M.-P.; Peyrot, V.; Wattez, N. Synthesis and biological evaluation of 4-arylcoumarin. J. Med. Chem. 2003, 46, 5437–5444. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, S.; Yao, Y.; Zhou, L.; Zhao, J.; Gu, Y.; Wang, K.; Li, X. Design, synthesis, and anti-tumor activities of novel triphenylethvlene-coumarin hybrids, and their interactions with Ct-DNA. Bioorg. Med. Chem. Lett. 2013, 23, 4785–4789. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.D.; Nicolotti, O.; Stefanachi, A.; Leonetti, F.; Carotti, A. Computational methods for the design of potent aromatase inhibitors. Expert Opin. Drug Discov. 2013, 8, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cho, M.; Karlsberg, K.; Zhou, D.; Yuan, Y.-C. Biochemical and biological characterization of a novel anti-aromatase coumarin derivative. J. Biol. Chem. 2004, 279, 48071–48078. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, F.; Favia, A.; Rao, A.; Aliano, R.; Paluszcak, A.; Hartmann, R.W.; Carotti, A. Design, synthesis, and 3D QSAR of novel potent and selective aromatase inhibitors. J. Med. Chem. 2004, 47, 6792–6803. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.D.; Cavalli, A.; Masetti, M.; Carotti, A.; Recanatini, M. Three-dimensional model of the human aromatase enzyme and density functional parameterization of the iron-containing protoporphyrin IX for a molecular dynamics study of heme-cysteinato cytochromes. Proteins 2006, 62, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
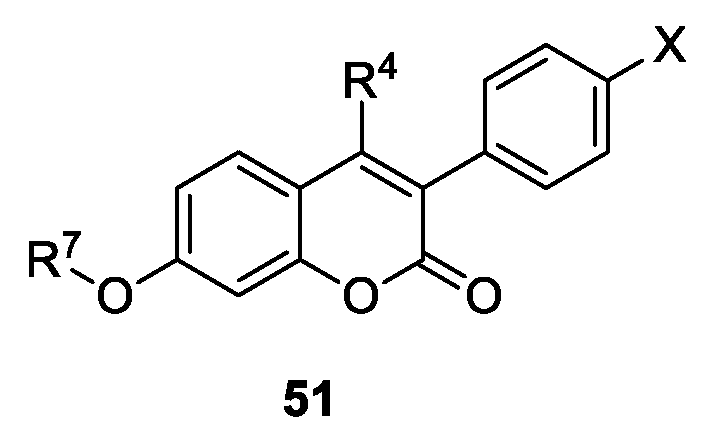
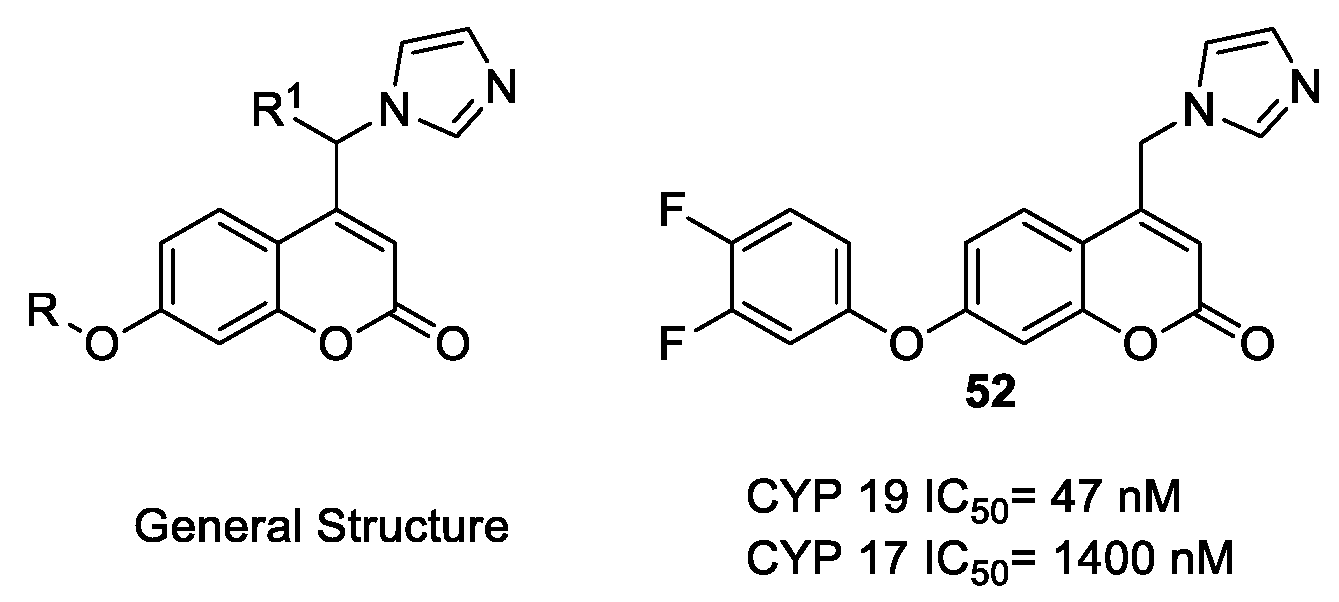
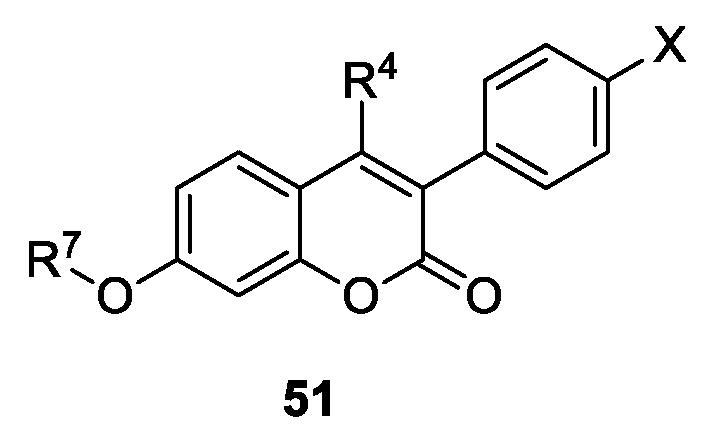
- Stefanachi, A.; Favia, A.D.; Nicolotti, O.; Leonetti, F.; Pisani, L.; Catto, M.; Zimmer, C.; Hartmann, R.W.; Carotti, A. Design, Synthesis, and Biological Evaluation of Imidazolyl Derivatives of 4,7-Disubstituted Coumarins as Selective Aromatase Inhibitors. J. Med. Chem. 2011, 54, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Hille, E.; Zimmer, C.; Vock, C.A.; Hartmann, R.W. First Selective CYP11B1 Inhibitors for the Treatment of Cortisol-Dependent Diseases. ACS Med. Chem. Lett. 2011, 2, 2–6. [Google Scholar] [CrossRef] [PubMed]
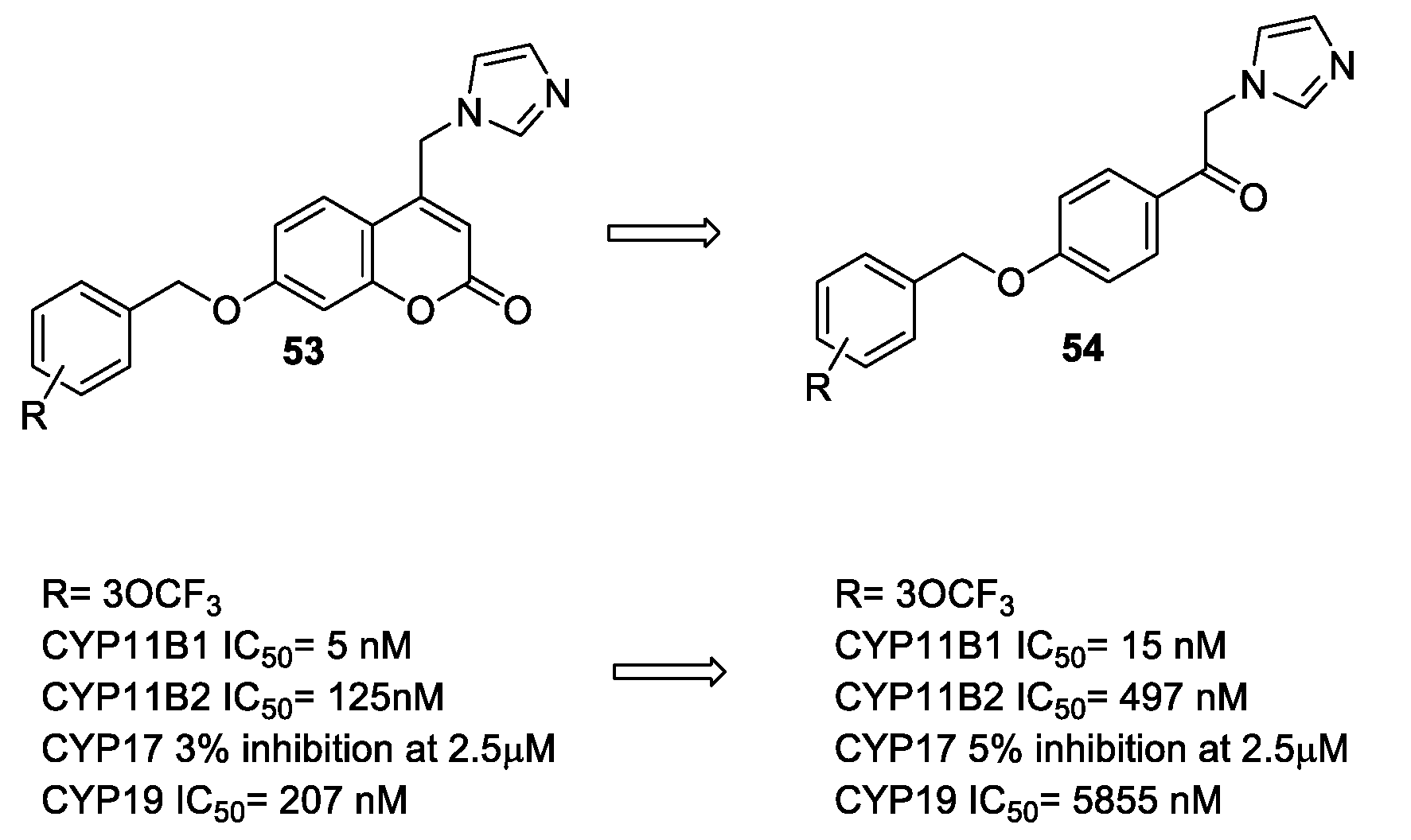
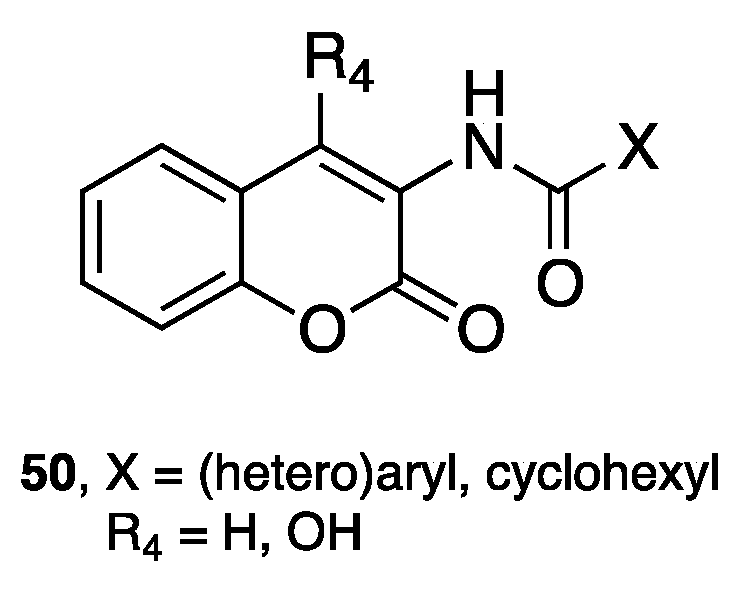
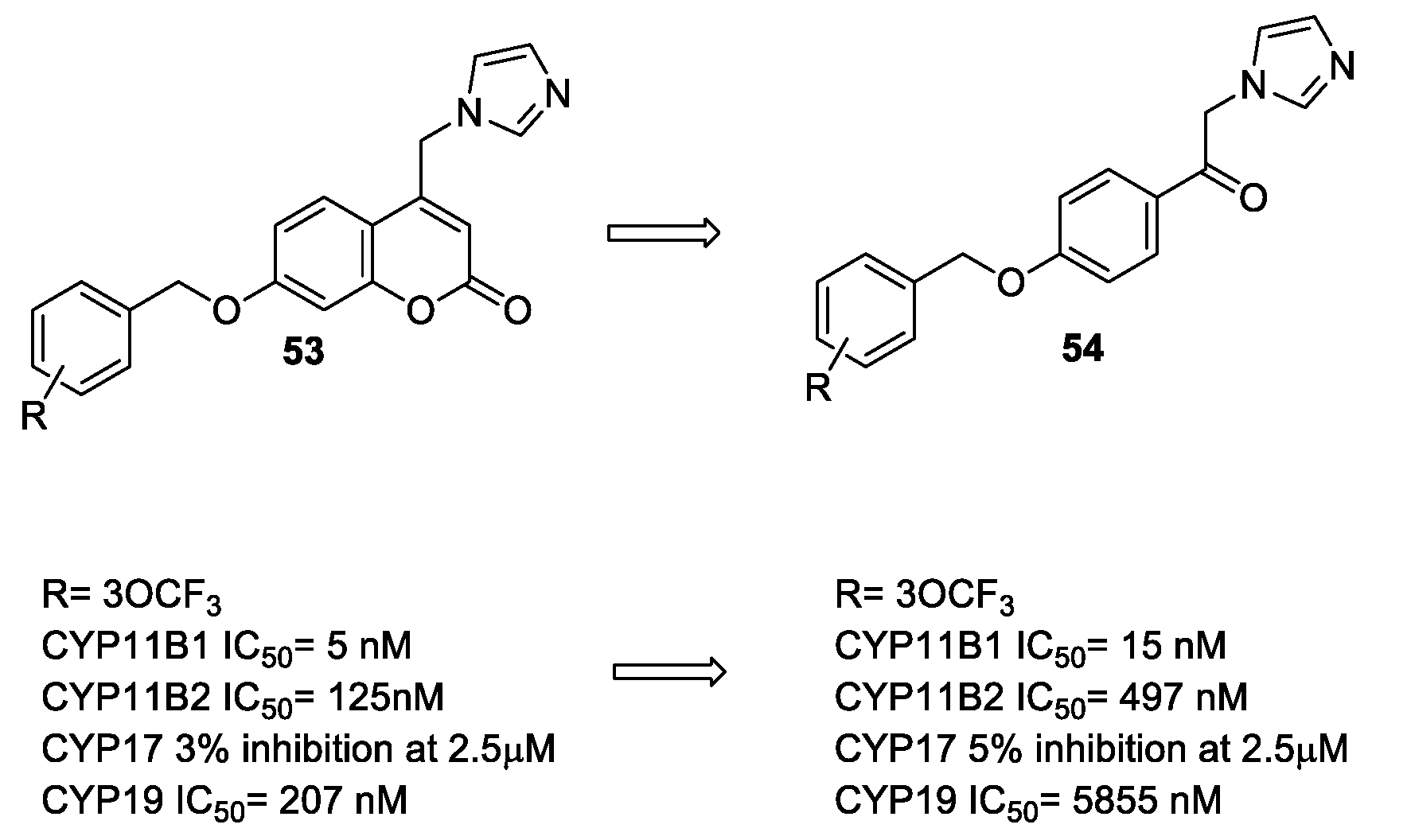
- Stefanachi, A.; Hanke, N.; Pisani, L.; Leonetti, F.; Nicolotti, O.; Cellamare, S.; Hartmann, R.W.; Carotti, A. Discovery of New 7-Substituted-4-imidazolylmethyl Coumarins and 4’-Susbstituted-2-Imidazolyl Acetophenones Open Analogues as Potent and Selective Inhibitors of Steroid-11β-hydroxylase. Eur. J. Med. Chem. 2015, 89, 106–114. [Google Scholar] [CrossRef] [PubMed]
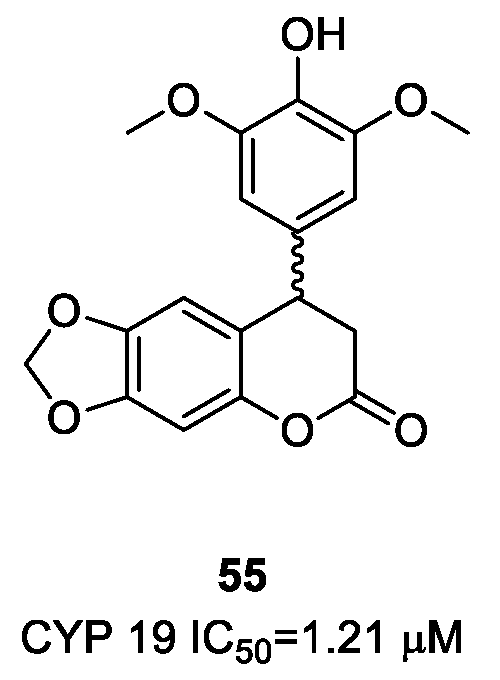
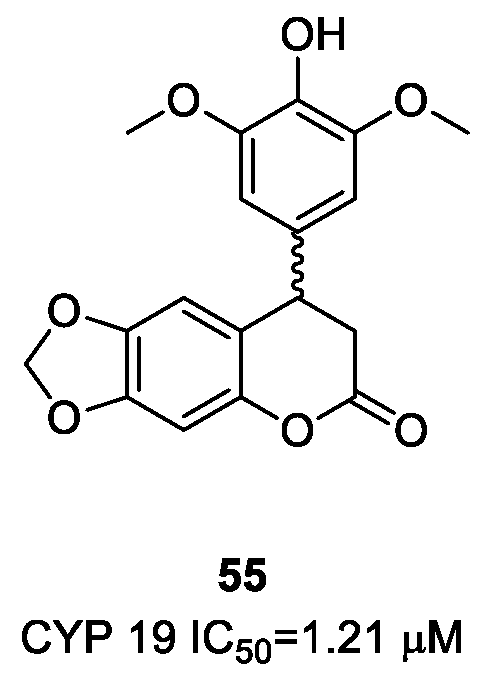
- Luqman, S.; Meena, A.; Singh, P.; Kondratyuk, T.P.; Marler, L.E.; Pezzuto, J.M.; Negi, A.S. Neoflavonoids and tetrahydroquinolones as possible cancer che-mopreventive agents. Chem. Biol. Drug Des. 2012, 80, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Nishizono, N.; Yoshimura, T.; Wada, K.; Kobayashi, D. Evaluation of synthesized coumarin derivatives on aromatase inhibitory activity. Bioorg. Med. Chem. Lett. 2017, 27, 2645–2649. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Price, T.; Aitken, J.; Mahendroo, M.; Simpson, E.R. A link between breast cancer and local estrogen biosynthesis suggested by quantification of breast adipose tissue aromatase cytochrome P450 transcripts using competitive polymerase chain reaction after reverse transcription. J. Clin. Endocrinol. Metab. 1993, 77, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
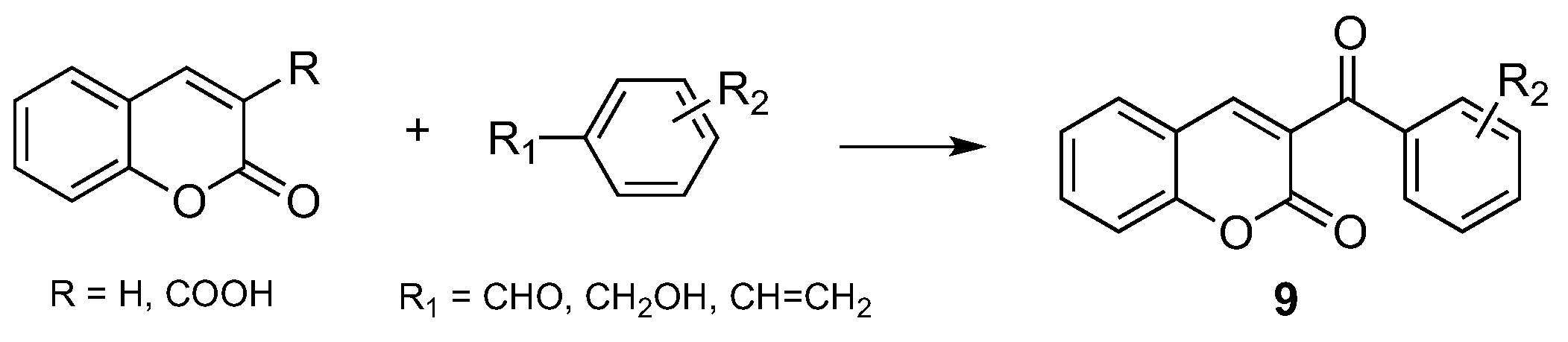

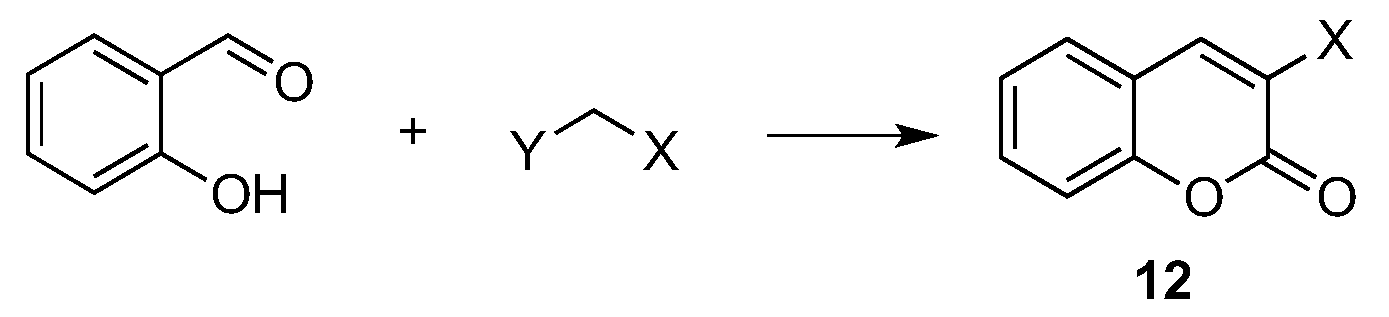














© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds. Molecules 2018, 23, 250. https://doi.org/10.3390/molecules23020250
Stefanachi A, Leonetti F, Pisani L, Catto M, Carotti A. Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds. Molecules. 2018; 23(2):250. https://doi.org/10.3390/molecules23020250
Chicago/Turabian StyleStefanachi, Angela, Francesco Leonetti, Leonardo Pisani, Marco Catto, and Angelo Carotti. 2018. "Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds" Molecules 23, no. 2: 250. https://doi.org/10.3390/molecules23020250
APA StyleStefanachi, A., Leonetti, F., Pisani, L., Catto, M., & Carotti, A. (2018). Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds. Molecules, 23(2), 250. https://doi.org/10.3390/molecules23020250