Use of an UHPLC-MS/MS Method for Determination of Kuraridin and Characterization of Its Metabolites in Rat Plasma after Oral Administration


Abstract
:1. Introduction
2. Results and Discussion
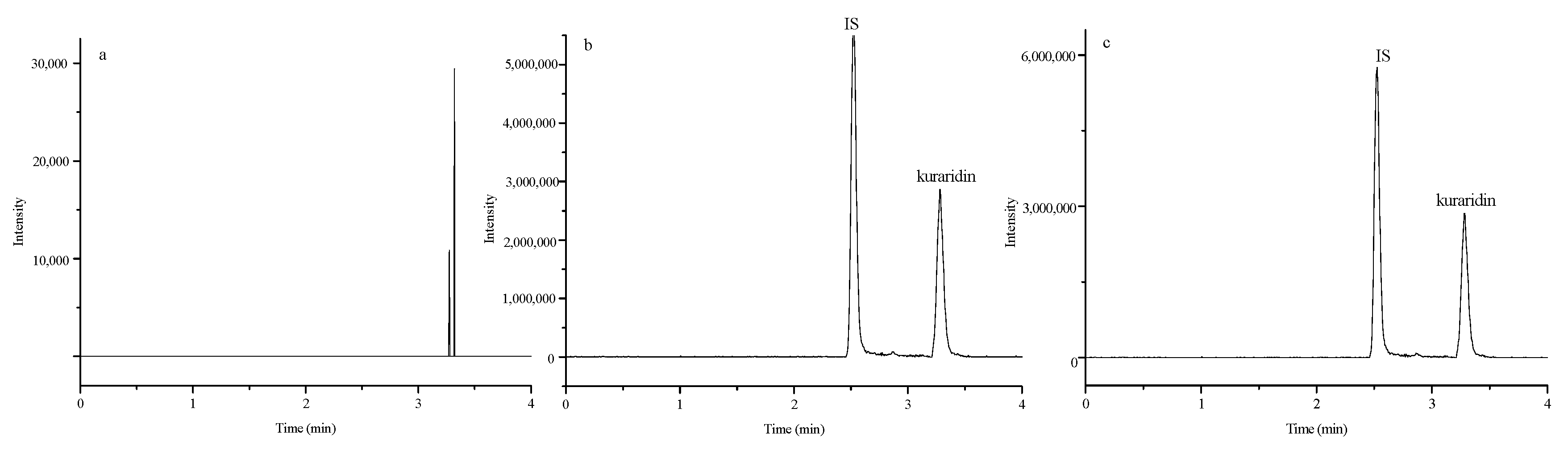
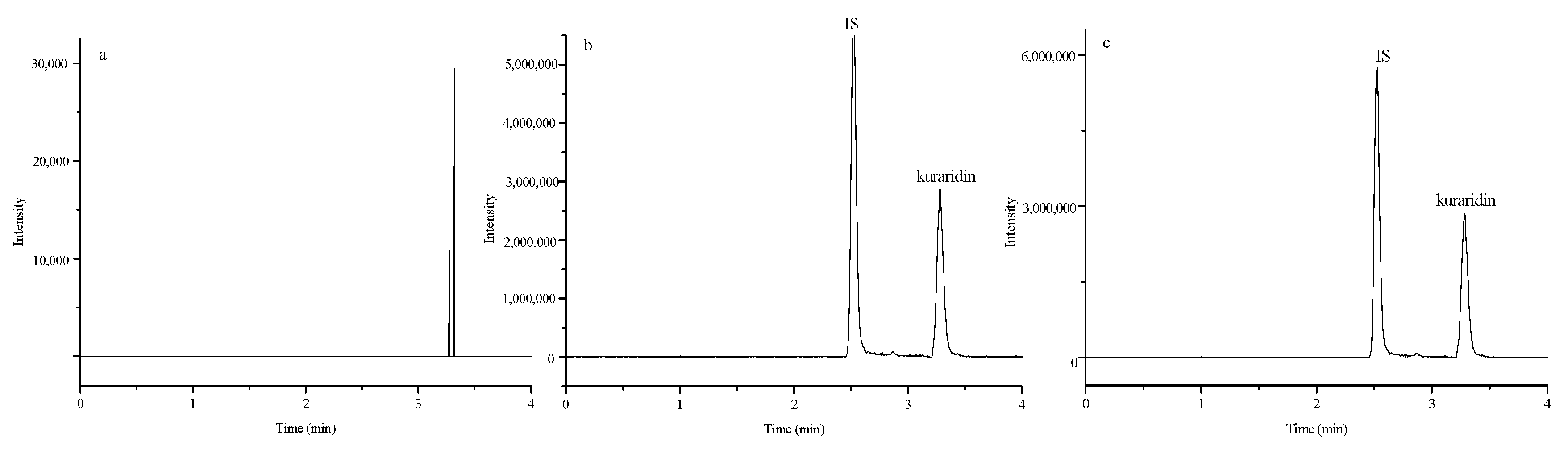
2.1. Method Development
2.2. Pharmacokinetic Method Validation
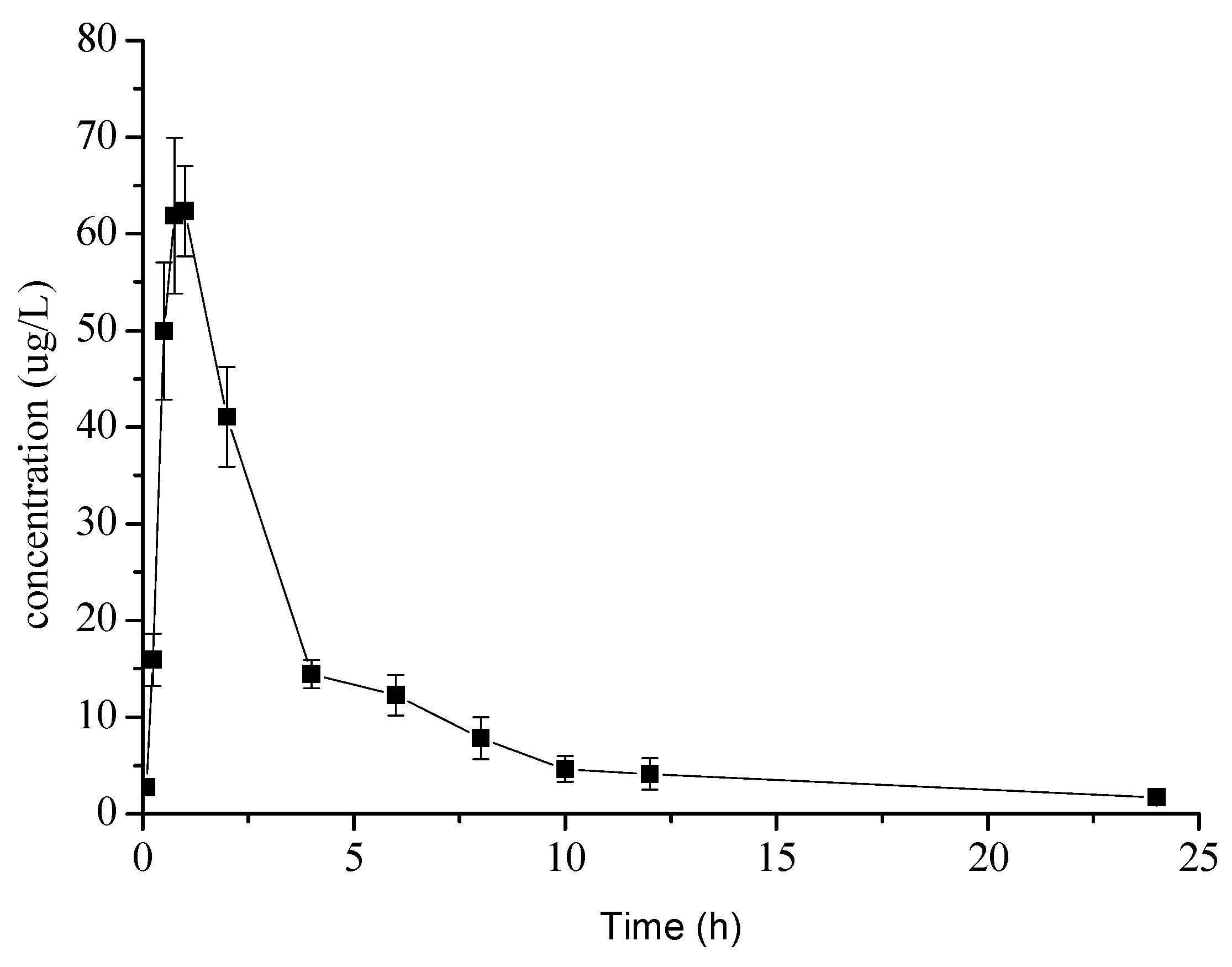
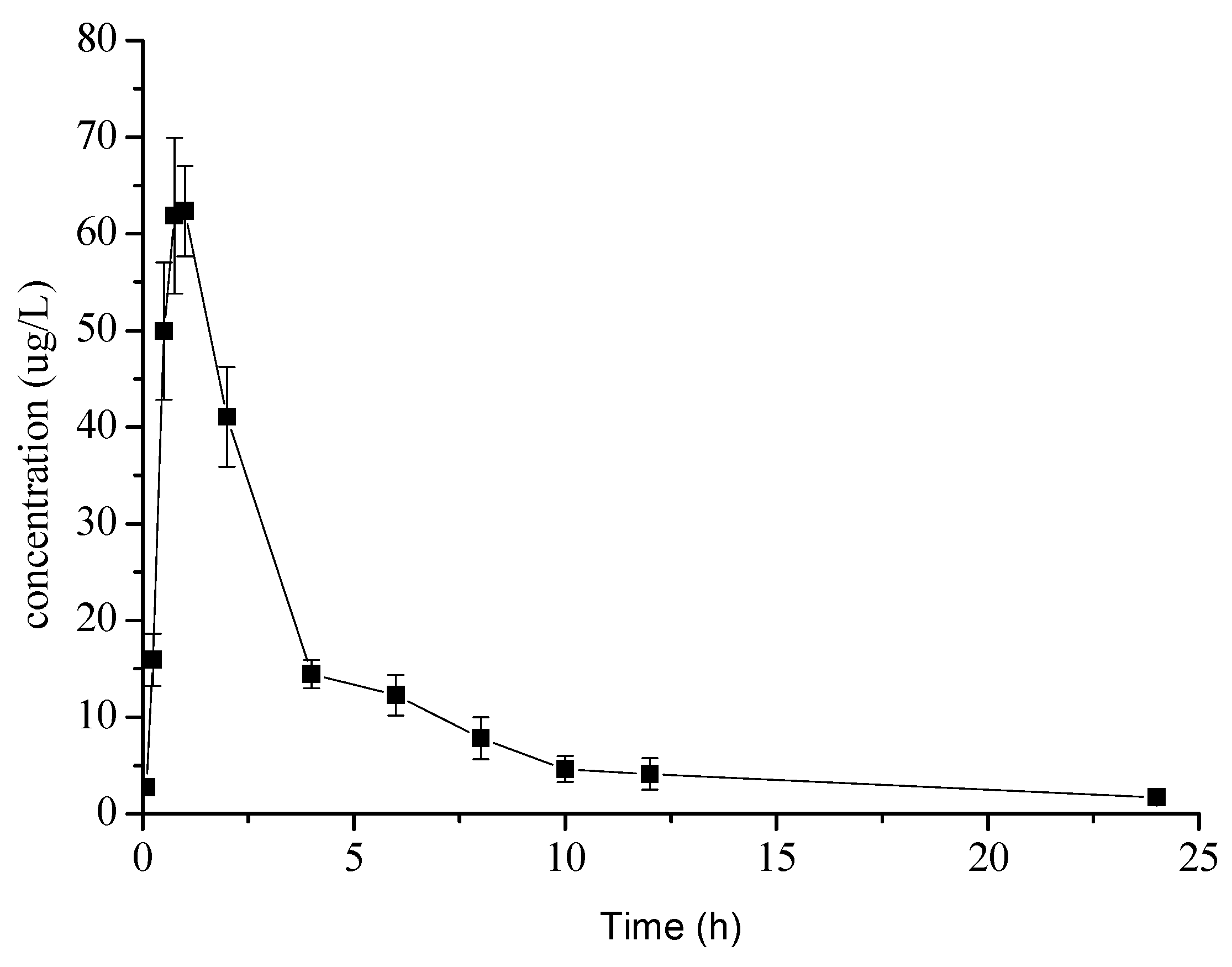
2.3. Pharmacokinetic Study
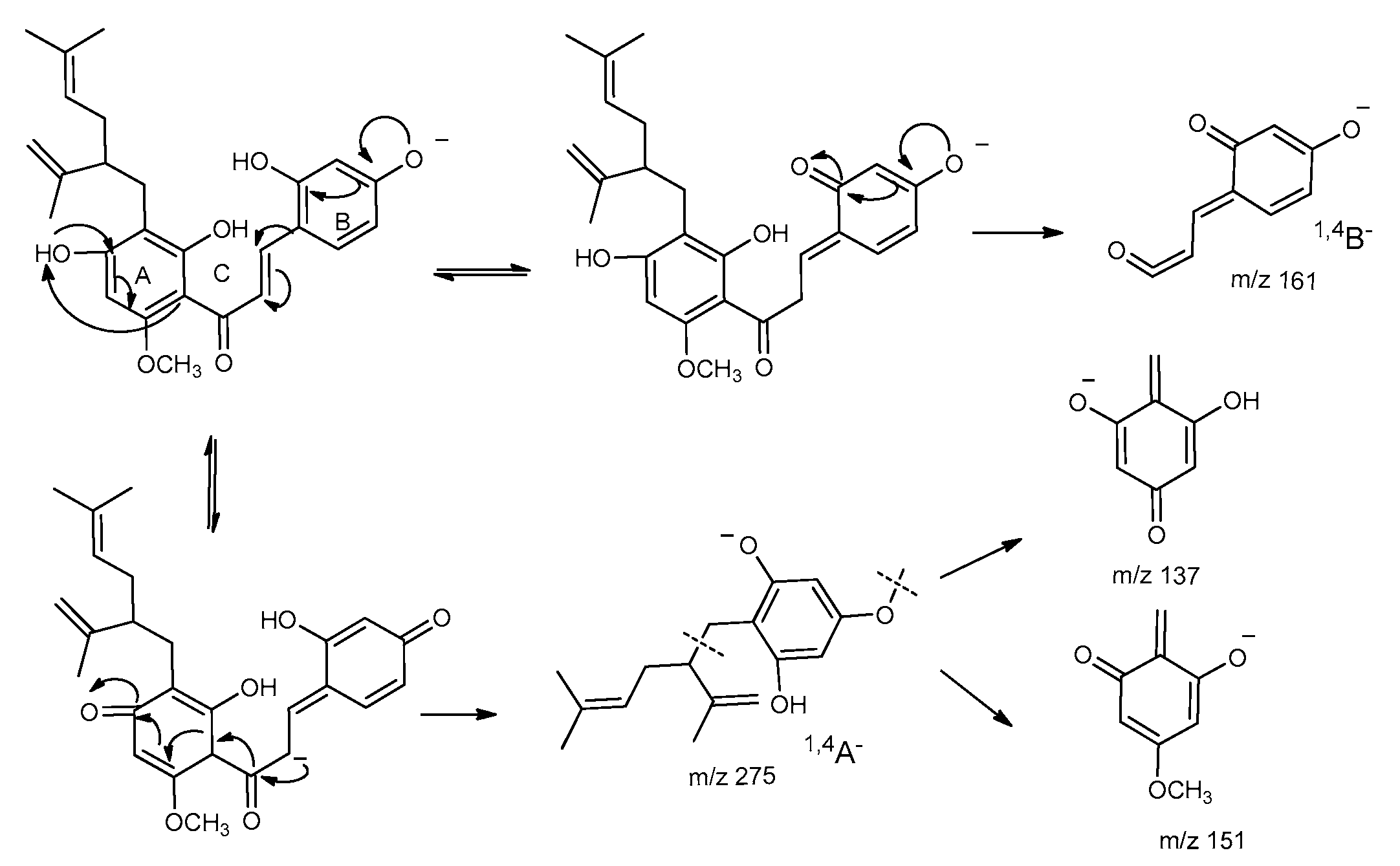
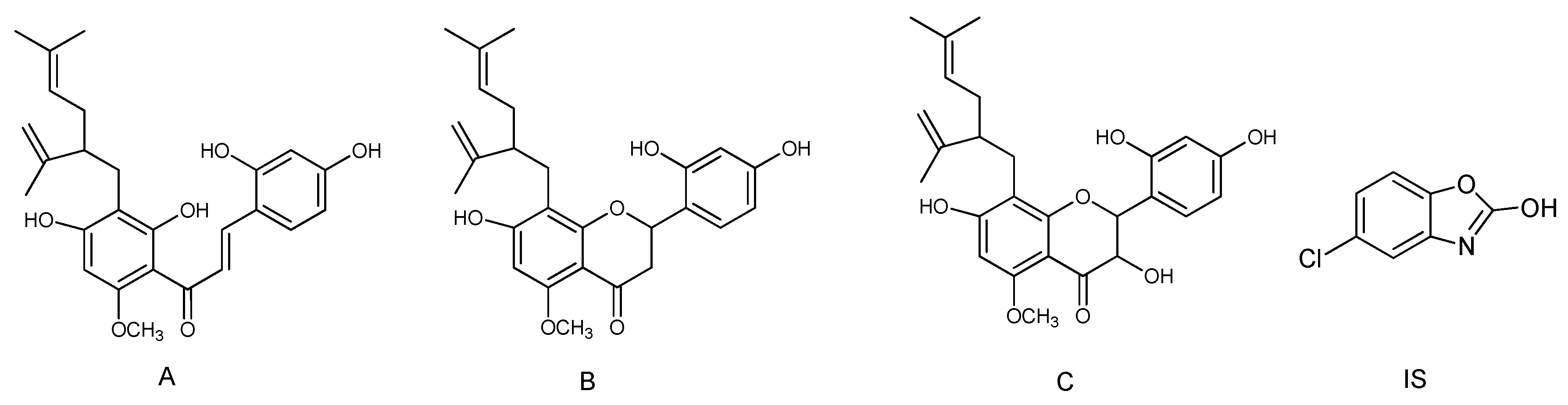
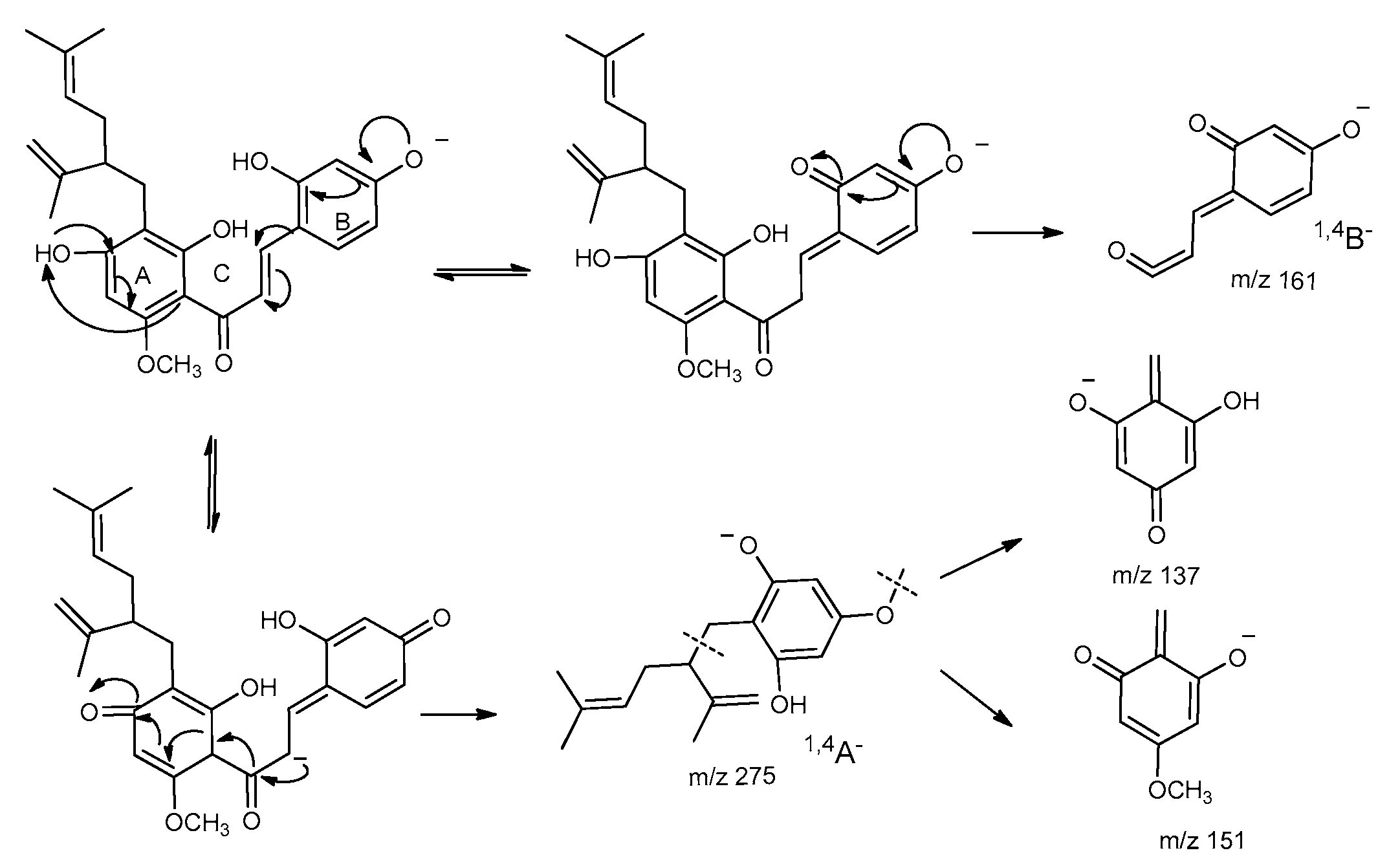
2.4. Fragmentation Pathway of Kuraridin in Negative Ion Mode
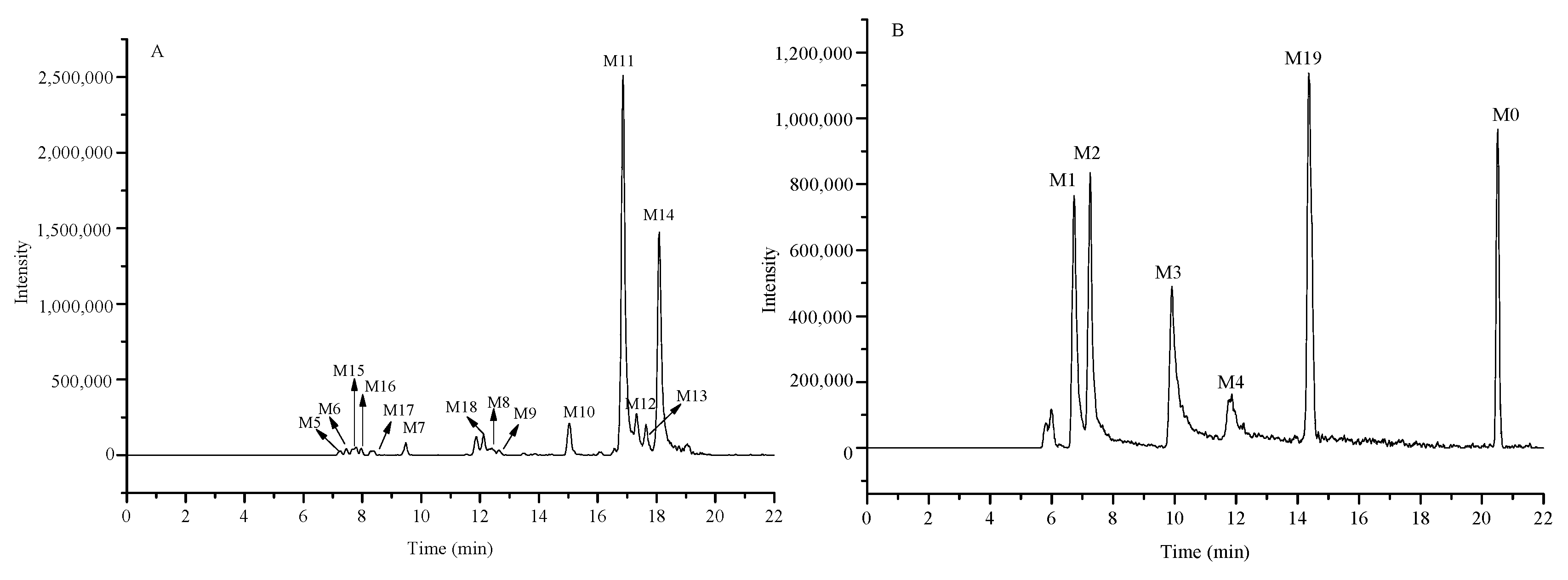
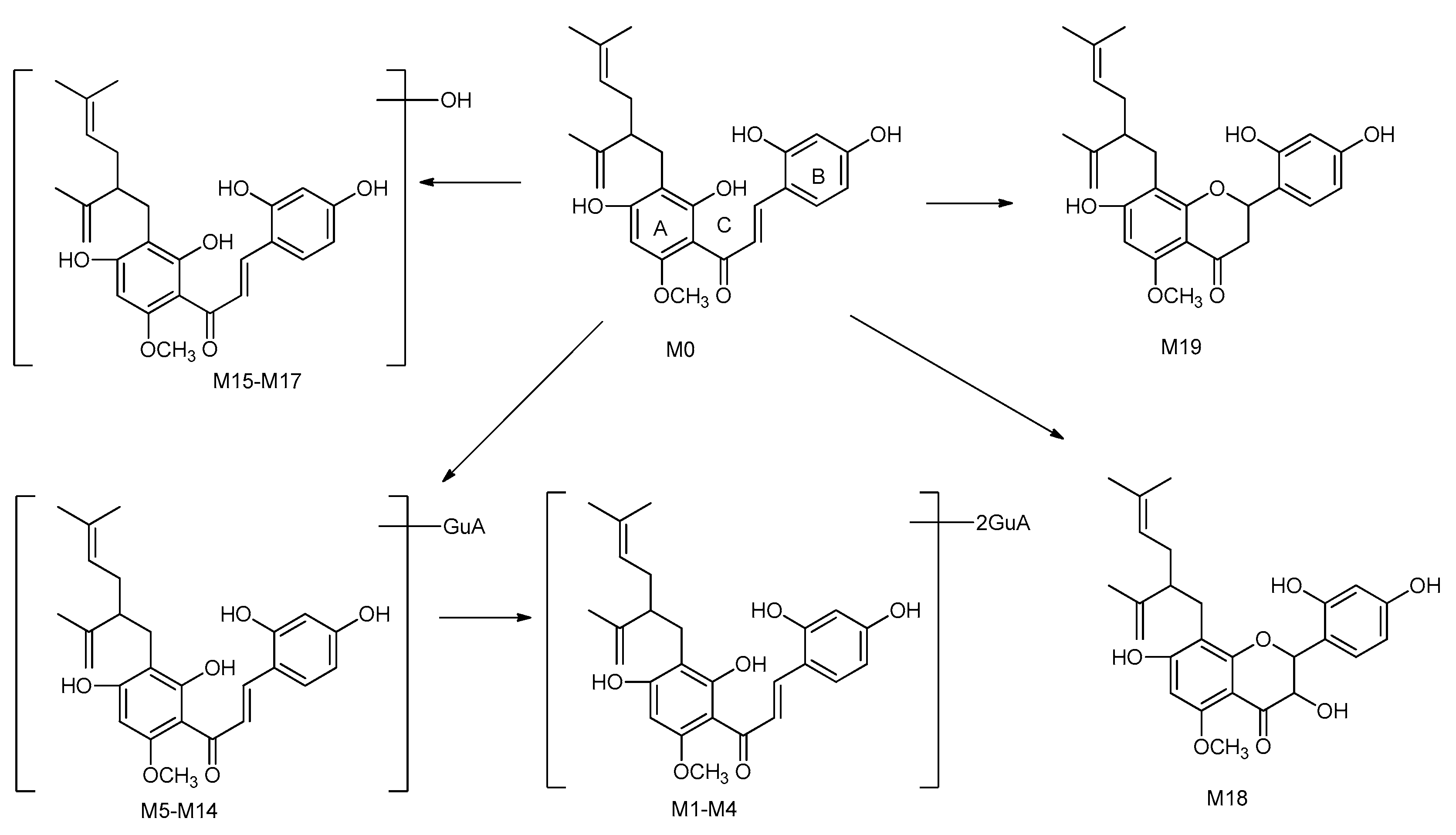
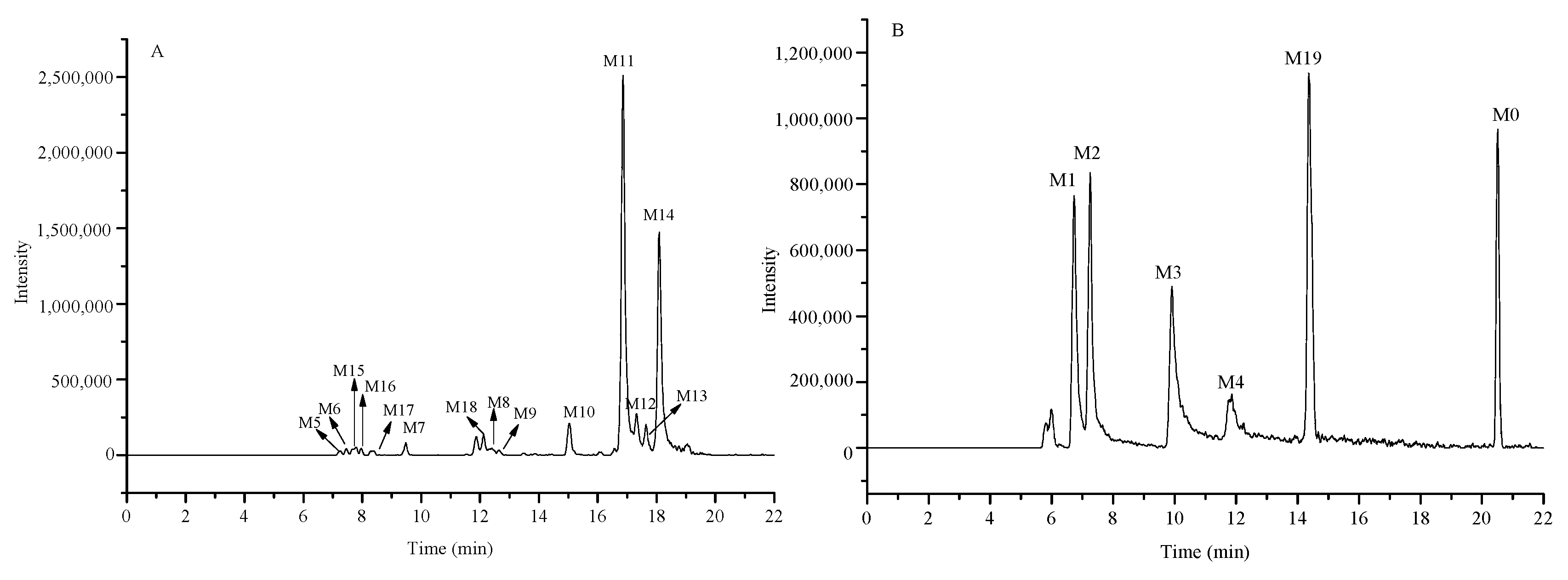
2.5. Identification of the Prototype (Kuraridin, M0) and Metabolites (M1–M19) in Rat Plasma
2.5.1. Identification of the Prototype (M0) in Rat Plasma
2.5.2. Identification of Metabolites M1–M4
2.5.3. Metabolites M5–M14
2.5.4. Metabolites M15–M18
2.5.5. Metabolite M19
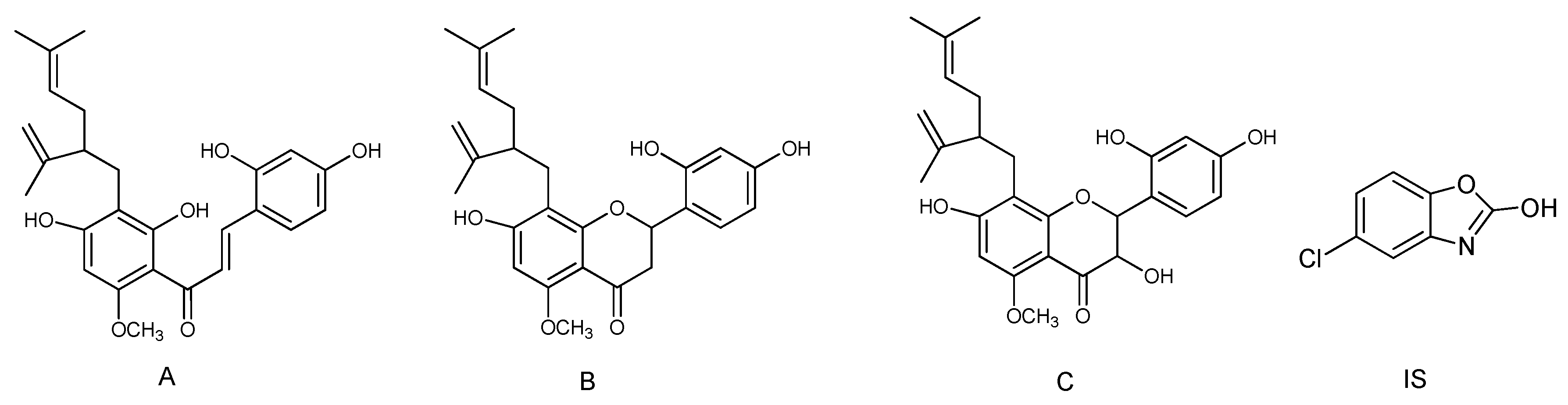
2.6. Proposed Metabolic Pathways of Kuraridin
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. LC-MS/MS Conditions
3.3. Pharmacokinetic Study
3.3.1. Preparation of Calibration Standards and Quality Control (QC) Samples
3.3.2. Sample Preparation
3.3.3. Method Validation
Specificity
Linearity and Lower Limits of Quantification (LLOQ)
Precision and Accuracy
Extraction Recovery and Matrix Effect
Stability
3.3.4. Application in Pharmacokinetic Study
3.4. Metabolites Study
3.4.1. Animals and Drug Administration
3.4.2. Sample Extraction
3.4.3. Peak Selections and Data Processing
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- He, X.; Fang, J.; Huang, L.; Wang, J.; Huang, X. Sophora flavescens Ait., Traditional usage, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 2015, 172, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shi, R.B.; Chen, S. HPLC determination of xanthohumol and kuraridin in Sophora flavescens Ait. and the effective fraction of Kushen decoction. Chin. J. Pharm. Anal. 2006, 5, 564–567. [Google Scholar]
- Sun, M.; Han, J.; Duan, J.; Cui, Y.; Wang, T.; Zhang, W.; Liu, W.; Hong, J.; Yao, M.; Xiong, S.; et al. Novel antitumor activities of Kushen flavonoids in vitro and in vivo. Phytother. Res. 2007, 21, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.Y.; Choi, S.U.; Kim, S.H. In vitro antitumor activity of flavoniod from Sophora flavascens. Phytother. Res. 1997, 11, 51–53. [Google Scholar] [CrossRef]
- Lee, H.S.; Ko, H.R.; Ryu, S.Y.; Oh, W.K.; Kim, B.Y.; Ahn, S.C.; Mheen, T.I.; Ahn, J.S. Inhibition of phospholipase Cγ1 by the prenylated flavonoids from Sophora flavescens. Planta Medica 1997, 63, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Ryu, Y.B.; Kang, N.S.; Lee, B.W.; Heo, J.S.; Jeong, I.Y.; Park, K.H. Glycosidase inhibitory flavonoids from Sophora flavescens. Biol. Pharm. Bull. 2006, 29, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Takeo, J.; Aoyama, C.; Kawahara, H. Na+-glucose cotransporter (SGLT) inhibitory flavonoids from the roots of Sophora flavescens. Bioorg. Med. Chem. 2007, 15, 3445–3449. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Li, W.; Higai, K.; Quang, T.H.; Kim, Y.H.; Koike, K. Protein Tyrosine Phosphatase 1B Inhibitory Activity of Lavandulyl Flavonoids from Roots of Sophora flavescens. Planta Medica 2014, 80, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.C.; Yu, H.; Wong, C.W.; Lui, S.L.; Jolivalt, C.; Ganem-Elbaz, C.; Paris, J.M.; Morleo, B.; Litaudon, M.; Lau, C.B.; et al. Quick identification of kuraridin, a noncytotoxic anti-MRSA (methicillin-resistant Staphylococcus aureus) agent from Sophora flavescens using high-speed counter-current chromatography. J. Chromatogr. B 2012, 880, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Jeong, J.H.; Lee, S.W.; Ryu, Y.B.; Jeong, H.J.; Jung, K.; Lim, J.S.; Cho, K.O.; Lee, W.S.; Rho, M.C.; et al. In vitro anti-reovirus activity of kuraridin isolated from Sophora flavescens against viral replication and hemagglutination. J. Pharmacol. Sci. 2015, 128, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, P.; Cheng, Y. Structural characterization of isoprenylated flavonoids from Kushen by electrospray ionization multistage tandem mass spectrometry. J. Mass Spectrom. 2008, 43, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Liu, Y.; Zhao, L.L.; Chen, X.X.; Wang, F.; Cai, W.; Chen, L. A new flavonoid from Sophora flavescens Ait. Nat. Prod. Res. 2017, 31, 2228–2232. [Google Scholar] [CrossRef] [PubMed]
- Havsteen, B.H. The biochemistry and medical significance of the flavonoids. Pharmacol. Ther. 2002, 96, 67–202. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Linear Range (ng/mL) | Regression Equation | r2 | LLOQ |
|---|---|---|---|---|
| Kuraridin | 1–500 | Y = 7.063 × 10−3 X− 2.585 × 10−3 | 0.9931 | 1 |
| Component | Concentration | Precision RSD (%) | Accuracy RE (%) | ||
|---|---|---|---|---|---|
| (ng/mL) | Intra-Day | Inter-Day | Intra-Day | Inter-Day | |
| Kuraridin | 1 | 8.95 | 2.18 | 4.12 | 2.09 |
| 2 | 5.21 | 6.08 | 3.07 | 6.48 | |
| 20 | 4.03 | 1.38 | −0.57 | 1.22 | |
| 400 | 4.35 | 3.50 | −4.97 | −6.27 | |
| Components | Concentration (ng/mL) | Recovery (Mean ± S.D) | Matrix Effect (Mean ± S.D) |
|---|---|---|---|
| 2 | 90.1 ± 4.7 | 87.1 ± 4.5 | |
| Kuraridin | 20 | 101.7 ± 5.7 | 87.4 ± 3.1 |
| 400 | 100.4 ± 2.9 | 84.8 ± 4.6 | |
| IS | 300 | 91.2 ± 2.9 | 90.1 ± 3.2 |
| Storage Conditions | High Concentrations | Medium Concentrations | Low Concentrations | |
|---|---|---|---|---|
| Autosampler 4 °C for 12 h | RE (%) | 1.27 | 7.66 | −8.86 |
| RSD (%) | 12.80 | 7.05 | 8.23 | |
| Short-term (at room temperature for 4 h) | RE (%) | 3.29 | 10.01 | 14.31 |
| RSD (%) | 12.23 | 5.51 | 10.68 | |
| Three freeze-thraw cycles | RE (%) | −9.97 | 8.1 | 10.82 |
| RSD (%) | 10.20 | 4.31 | 7.07 | |
| Storage at −75 °C for 21 days | RE (%) | 4.68 | 0.89 | 8.97 |
| RSD (%) | 8.28 | 4.18 | 8.17 | |
| Parameters | Kuraridin |
|---|---|
| Cmax (μg/L) | 65.43 ± 6.54 |
| Tmax (h) | 0.87 ± 0.14 |
| T1/2z (h) | 5.54 ± 3.17 |
| AUC0–t (μgh/L) | 284.05 ± 24.19 |
| AUC0–∞ (μgh/L) | 289.79 ± 24.08 |
| MRT0–t (h) | 7.08 ± 0.86 |
| MRT0–∞ (h) | 8.79 ± 1.22 |
| Peak | tR | Experimental | Theoretical | Error | Formula [M − H]− | MS/MS Fragment | Identification/Reactions |
|---|---|---|---|---|---|---|---|
| Mass (m/z) | Mass (m/z) | (ppm) | |||||
| M1 | 6.74 | 789.2626 | 789.2612 | 1.82 | C38H45O18 | MS2[789]: 161.0235(100),613.2296(36),275.1656(33),437.1964(4) | Diglucuronidation |
| M2 | 7.26 | 789.2622 | 789.2612 | 1.36 | C38H45O18 | MS2[789]: 161.0235(100),613.2289(31),275.1659(31),437.1954(4) | Diglucuronidation |
| M3 | 9.91 | 789.2623 | 789.2612 | 1.51 | C38H45O18 | MS2[789]: 161.0235(100),275.1655(31),613.2299(28),437.1989(5) | Diglucuronidation |
| M4 | 11.87 | 789.2619 | 789.2612 | 0.97 | C38H45O18 | MS2[789]: 161.0235(100),789.2916(36),275.1658 (28),613.2286(16) | Diglucuronidation |
| M5 | 7.44 | 613.2299 | 613.2291 | 1.32 | C32H37O12 | MS2[613]: 161.0237(100),275.1653(41),437.1976(5) | Glucuronidation |
| M6 | 7.66 | 613.2302 | 613.2291 | 1.91 | C32H37O12 | MS2[613]: 161.0236(100),275.1659(28),137.0235(23),437.1969(5) | Glucuronidation |
| M7 | 9.48 | 613.2302 | 613.2291 | 1.91 | C32H37O12 | MS2[613]: 161.0237(100),275.1659(26),151.0391(17) | Glucuronidation |
| M8 | 12.40 | 613.2299 | 613.2291 | 1.32 | C32H37O12 | MS2[613]: 161.0236(100),275.1653(13),151.0394(13) | Glucuronidation |
| M9 | 12.63 | 613.2296 | 613.2291 | 0.82 | C32H37O12 | MS2[613]: 161.0237(100),275.1658(21),137.0236(6) | Glucuronidation |
| M10 | 15.02 | 613.2297 | 613.2291 | 1.23 | C32H37O12 | MS2[613]: 161.0236(100),275.1657(39),437.1979(15) | Glucuronidation |
| M11 | 16.85 | 613.2300 | 613.2291 | 1.61 | C32H37O12 | MS2[613]: 161.0236(100),275.1658(23),437.1980(7) | Glucuronidation |
| M12 | 17.31 | 613.2301 | 613.2291 | 1.71 | C32H37O12 | MS2[613]: 161.0237(100),275.1658(18),437.1980(14) | Glucuronidation |
| M13 | 17.64 | 613.2302 | 613.2291 | 1.91 | C32H37O12 | MS2[613]:161.0237(100),275.1662(19),437.1980(11) | Glucuronidation |
| M14 | 18.08 | 613.2301 | 613.2291 | 1.71 | C32H37O12 | MS2[613]: 161.0236(100),275.1658(26),151.0393(13),437.1979(5) | Glucuronidation |
| M15 | 7.78 | 453.1926 | 453.1919 | 1.58 | C26H29O7 | MS2[453]: 161.0237(100),151.0392(36),291.1607(16) | Hydroxylation |
| M16 | 7.94 | 453.1925 | 453.1919 | 1.38 | C26H29O7 | MS2[453]: 161.0236(100),151.0392(20),291.1609(15),137.0235(13) | Hydroxylation |
| M17 | 8.40 | 453.1928 | 453.1919 | 1.93 | C26H29O7 | MS2[453]: 161.0236(100),137.0236(36),291.1610(25),151.0393(13) | Hydroxylation |
| M18 | 12.11 | 453.1924 | 453.1919 | 1.24 | C26H29O7 | MS2[453]: 149.0237(100),177.0187(96),275.1658(63),137.0234(49),151.0394(38) | Kushenol N |
| M19 | 14.37 | 437.1975 | 437.19696 | 1.19 | C26H29O6 | MS2[437]: 161.0236(100),275.1656(33),137.0235(24),151.0392(14) | Kurarinone |
| M0 | 20.51 | 437.1982 | 437.19696 | 2.72 | C26H29O6 | MS2[437]: 161.0237(100),275.1657(39),137.0235(31),151.0393(20) | Kuraridin |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Chen, L.; Cai, W.; Zhao, L.-l.; Mo, Z.-x. Use of an UHPLC-MS/MS Method for Determination of Kuraridin and Characterization of Its Metabolites in Rat Plasma after Oral Administration. Molecules 2018, 23, 132. https://doi.org/10.3390/molecules23020132
Liu Y, Chen L, Cai W, Zhao L-l, Mo Z-x. Use of an UHPLC-MS/MS Method for Determination of Kuraridin and Characterization of Its Metabolites in Rat Plasma after Oral Administration. Molecules. 2018; 23(2):132. https://doi.org/10.3390/molecules23020132
Chicago/Turabian StyleLiu, Yi, Lei Chen, Wei Cai, Lin-lin Zhao, and Zhi-xian Mo. 2018. "Use of an UHPLC-MS/MS Method for Determination of Kuraridin and Characterization of Its Metabolites in Rat Plasma after Oral Administration" Molecules 23, no. 2: 132. https://doi.org/10.3390/molecules23020132
APA StyleLiu, Y., Chen, L., Cai, W., Zhao, L.-l., & Mo, Z.-x. (2018). Use of an UHPLC-MS/MS Method for Determination of Kuraridin and Characterization of Its Metabolites in Rat Plasma after Oral Administration. Molecules, 23(2), 132. https://doi.org/10.3390/molecules23020132