
Copper-Promoted Cross-Coupling Reactions for the Synthesis of Aryl(difluoromethyl)phosphonates Using Trimethylsilyl(difluoromethyl)phosphonate


Abstract
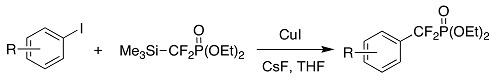
:
1. Introduction
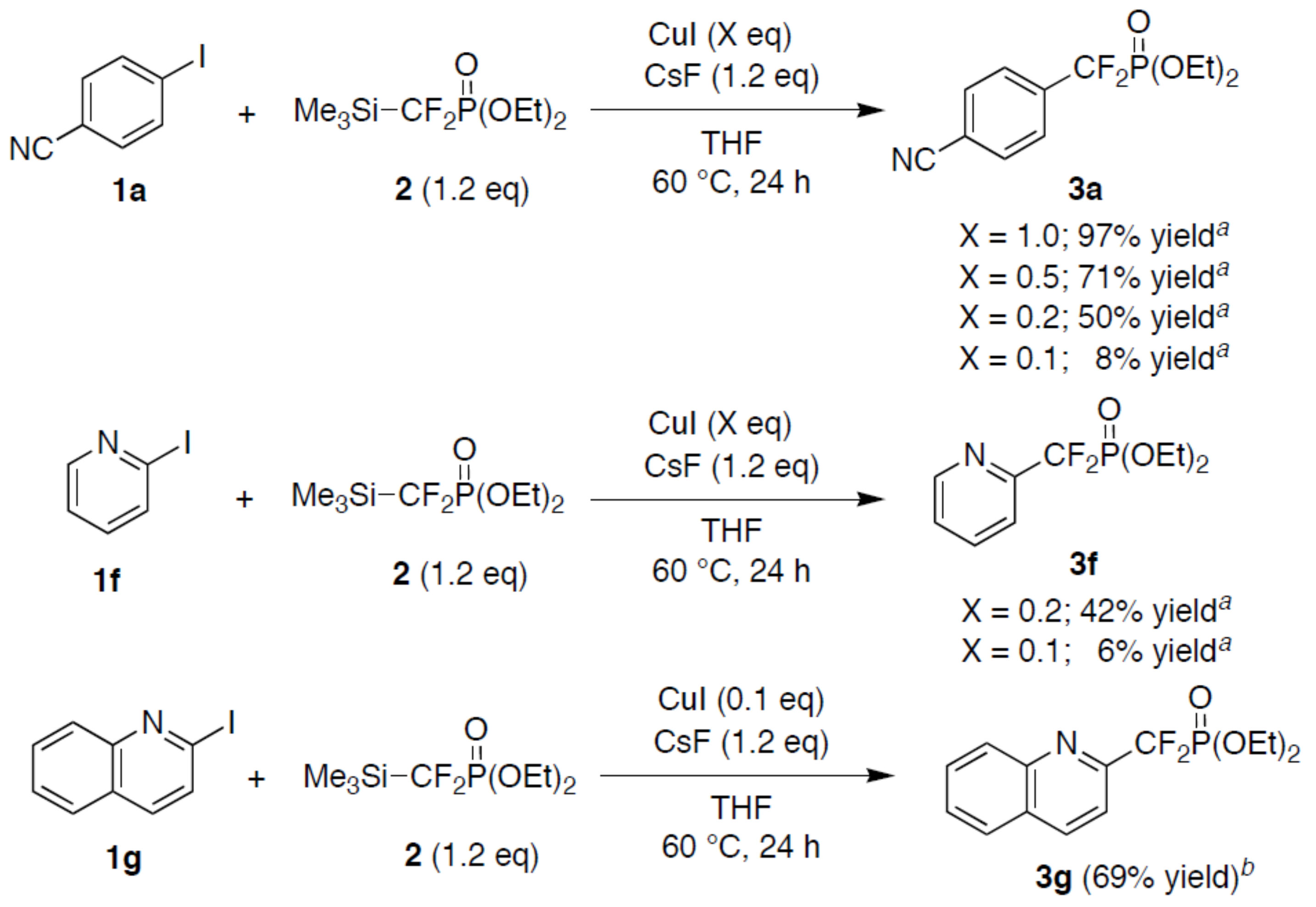
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Cross-Coupling of Aryl Iodides with Me3SiCF2PO(OEt)2
4. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Banks, R.E.; Smart, B.E.; Tatlow, J.C. Organofluorine Chemistry: Principles and Commercial Applications; Plenum Press: New York, NY, USA, 1994. [Google Scholar]
- Hiyama, T.; Kanie, K.; Kusumoto, T.; Morizawa, Y.; Shimizu, M. Organofluorine Compounds: Chemistry and Application; Springer-Verlag: Berlin, Germany, 2000. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Chambers, R.D. Fluorine in Organic Chemistry; Blackwell: Oxford, UK, 2004. [Google Scholar]
- Uneyama, K. Organofluorine Chemistry; Blackwell: Oxford, UK, 2006. [Google Scholar]
- Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Gouverneur, V.; Müller, K. Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications; World Scientific Publishing Company: London, UK, 2012. [Google Scholar]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, C.M.; England, D.A.; Kolkmann, F. Monofluoro- and difluoro-methylenebisphosphonic acids: Isopolar analogues of pyrophosphoric acid. J. Chem. Soc. Chem. Commun. 1981, 930–932. [Google Scholar] [CrossRef]
- Blackburn, G.M.; Kent, D.E.; Kolkmann, F. The synthesis and metal binding characteristics of novel, isopolar phosphonate analogues of nucleotides. J. Chem. Soc. Perkin Trans. 1 1984, 1119–1125. [Google Scholar] [CrossRef]
- Romanenko, V.D.; Kukhar, V.P. Fluorinated phosphonates: Synthesis and biomedical application. Chem. Rev. 2006, 106, 3868–93. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.R.; Lee, J.R.K. Phosphotyrosyl mimetics in the development of signal transduction inhibitors. Acc. Chem. Res. 2003, 36, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y. Chemical and mechanistic approaches to the study of protein tyrosine phosphatases. Acc. Chem. Res. 2003, 36, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Hikishima, S.; Hashimoto, M.; Magnowska, L.; Bzowska, A.; Yokomatsu, T. Synthesis and biological evaluation of 9-deazaguanine derivatives connected by a Linker to difluoromethylene phosphonic acid as multi-substrate analogue Inhibitors of PNP 2007. Bioorg. Med. Chem. Lett. 2010, 20, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Liao, W.S.-L.; McMurray, J.S. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org. Lett. 2009, 11, 3394–3397. [Google Scholar] [CrossRef]
- Mitra, S.; Barrios, A.M. Identifying selective protein tyrosine phosphatase substrates and inhibitors from a fluorogenic, combinatorial peptide library. ChemBioChem 2008, 9, 1216–1219. [Google Scholar] [CrossRef]
- De Meijere, A.; Diederich, F. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, Germany 2004. [Google Scholar]
- Braun, T.; Hughes, R.P. Organometallic Fluorine Chemistry (Topics in Organometallic Chemistry 52); Springer International Publishing: Basel, Switzerland, 2015. [Google Scholar]
- Jin, Z.; Hammond, G.B.; Xu, B. Transition-metal-mediated fluorination, difluoromethylation, and trifluoromethylation. Aldrichimica Acta 2012, 45, 67–83. [Google Scholar]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Burton, D.J. A facile and general preparation of α,α-difluoro benzylic phosphonates by the CuCl promoted coupling reaction of the (diethyl phosphonyl)difluoromethylcadmium reagent with aryl iodides. Tetrahedron Lett. 1996, 37, 2745–2748. [Google Scholar] [CrossRef]
- Yokomatsu, T.; Murano, T.; Suemune, K.; Shibuya, S. Facile Synthesis of aryl(difluoromethyl)phosphonates through CuBr-mediated cross coupling reactions of [(diethoxyphosphinyl)difluoromethyl]zinc bromide with aryl iodides. Tetrahedron 1997, 53, 815–822. [Google Scholar] [CrossRef]
- Murakami, S.; Ishii, H.; Tajima, T.; Fuchigami, T. Photochemical substitution of olefins and aromatic compounds with difluoromethyl radicals bearing ester and phosphonate groups. Tetrahedron 2006, 62, 3761–3769. [Google Scholar] [CrossRef]
- Qiu, W.; Burton, D.J. Cuprous chloride promoted coupling reaction of diethoxyphosphinyl-difluoromethylcadmium reagent with aryl iodides: A practical and convenient preparation of α,α-difluoro benzylic phosphonates. J. Fluorine Chem. 2013, 155, 45–51. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, F.; Zhang, X. Copper catalyzed cross-coupling of iodobenzoates with bromozinc-difluorophosphonate. Org. Lett. 2012, 14, 1938–1941. [Google Scholar] [CrossRef]
- Jiang, X.; Chu, L.; Qing, F.-L. Copper-mediated oxidative difluoromethylenation of aryl boronic acids with α-silyldifluoromethylphosphonates: A new method for aryldifluorophosphonates. New. J. Chem. 2013, 37, 1736–1741. [Google Scholar] [CrossRef]
- Feng, Z.; Xiao, Y.-L.; Zhang, X. Copper-catalyzed cross-coupling of bromozinc-difluoromethylphosphonate with iodo/bromo-aryl triazenes. Org. Chem. Front. 2014, 1, 113–116. [Google Scholar] [CrossRef]
- Murakami, S.; Kim, S.; Ishii, H.; Fuchigami, T. Aromatic substitution with photochemically generated difluoromethyl radicals bearing electron-withdrawing group. Synlett. 2014, 5, 815–818. [Google Scholar] [CrossRef]
- Wang, L.; Wei, X.-J.; Lei, W.-L.; Chen, H.; Wub, L.-Z.; Liu, Q. Direct C-H difluoromethylenephosphonation of arenes and heteroarenes with bromodifluoromethyl phosphonate via visible-light photocatalysis. Chem. Commun. 2014, 50, 15916–15919. [Google Scholar] [CrossRef]
- Feng, Z.; Min, Q.-Q.; Xiao, Y.-L.; Zhang, B.; Zhang, X. Palladium-catalyzed difluoroalkylation of aryl boronic acids: A new method for the synthesis of aryldifluoromethylated phosphonates and carboxylic acid derivatives. Angew. Chem. Int. Ed. 2014, 53, 1669–1673. [Google Scholar] [CrossRef] [PubMed]
- Bayle, A.; Cocaud, C.; Nicolas, C.; Martin, O.R.; Poisson, T.; Pannecoucke, X. Copper-mediated synthesis of aryldifluoromethylphosphonates: A Sandmeyer approach. Eur. J. Org. Chem. 2015, 3787–3792. [Google Scholar] [CrossRef]
- Ivanova, M.V.; Bayle, A.; Besset, T.; Poisson, T.; Pannecoucke, X. Copper-mediated formation of aryl, heteroaryl, vinyl and alkynyl difluoromethylphosphonates: A general approach to fluorinated phosphate mimics. Angew. Chem. Int. Ed. 2015, 54, 13406–13410. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wan, W.; Ma, G.; Chen, Y.; Hu, Q.; Kang, K.; Jiang, H.; Hao, J. Silver-mediated C-H difluoromethylation of arenes. Eur. J. Org. Chem. 2016, 4916–4921. [Google Scholar] [CrossRef]
- Ivanova, M.V.; Bayle, A.; Besset, T.; Pannecoucke, X.; Poisson, T. Copper-mediated introduction of the CF2PO(OEt)2 motif: Scope and limitations. Chem. Eur. J. 2017, 23, 17318–17338. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.V.; Besset, T.; Pannecoucke, X.; Poisson, T. Palladium-catalyzed synthesis of aryl and heteroaryl difluoromethylated phosphonates. Synthesis 2018, 50, 778–784. [Google Scholar] [CrossRef]
- Reily, M.D.; Robosky, L.C.; Manning, M.L.; Butler, A.; Baker, J.D.; Winters, R.T. DFTMP, an NMR reagent for assessing the near-neutral pH of biological samples. J. Am. Chem. Soc. 2006, 128, 12360–12361. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Fluoride (MF) | Yield 2 |
|---|---|---|---|
| 1 | toluene | KF | 0 |
| 2 | DMSO | KF | 43 |
| 3 | NMP | KF | 55 |
| 4 | DMF | KF | 76 |
| 5 | THF | KF | 74 |
| 6 | DMF | TBAF | 0 |
| 7 | DMF | CsF | 40 |
| 8 | THF | CsF | 97 (84) 3 |
 |
 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komoda, K.; Iwamoto, R.; Kasumi, M.; Amii, H. Copper-Promoted Cross-Coupling Reactions for the Synthesis of Aryl(difluoromethyl)phosphonates Using Trimethylsilyl(difluoromethyl)phosphonate. Molecules 2018, 23, 3292. https://doi.org/10.3390/molecules23123292
Komoda K, Iwamoto R, Kasumi M, Amii H. Copper-Promoted Cross-Coupling Reactions for the Synthesis of Aryl(difluoromethyl)phosphonates Using Trimethylsilyl(difluoromethyl)phosphonate. Molecules. 2018; 23(12):3292. https://doi.org/10.3390/molecules23123292
Chicago/Turabian StyleKomoda, Kazuki, Rei Iwamoto, Masakazu Kasumi, and Hideki Amii. 2018. "Copper-Promoted Cross-Coupling Reactions for the Synthesis of Aryl(difluoromethyl)phosphonates Using Trimethylsilyl(difluoromethyl)phosphonate" Molecules 23, no. 12: 3292. https://doi.org/10.3390/molecules23123292
APA StyleKomoda, K., Iwamoto, R., Kasumi, M., & Amii, H. (2018). Copper-Promoted Cross-Coupling Reactions for the Synthesis of Aryl(difluoromethyl)phosphonates Using Trimethylsilyl(difluoromethyl)phosphonate. Molecules, 23(12), 3292. https://doi.org/10.3390/molecules23123292