
Pharmacological Inhibition of LSD1 for Cancer Treatment




Abstract
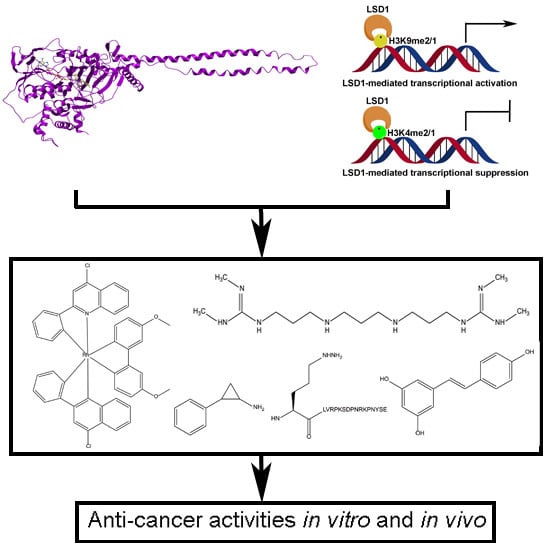
1. Introduction
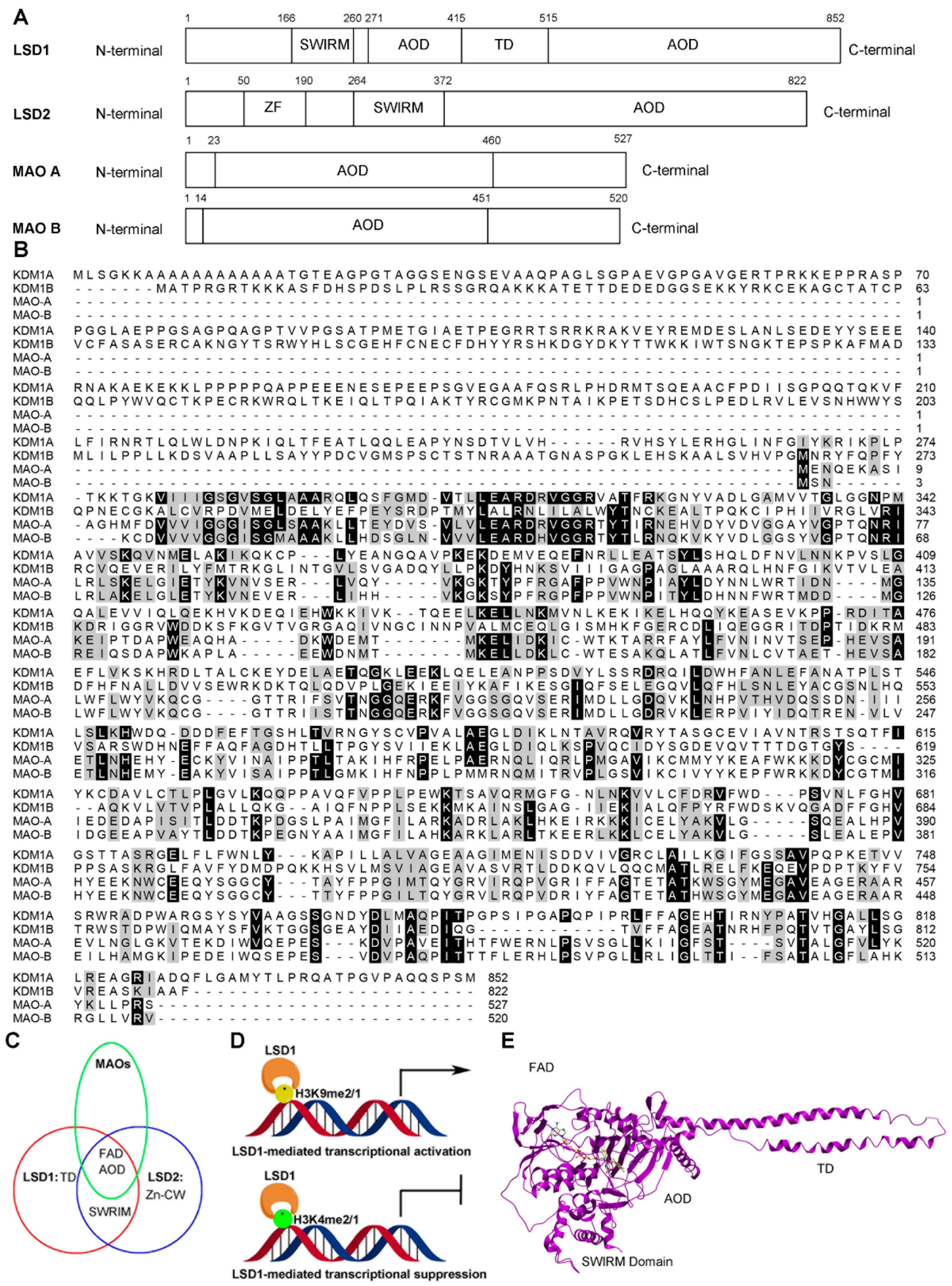
2. Structure and Function of LSD1
2.1. Structure of LSD1
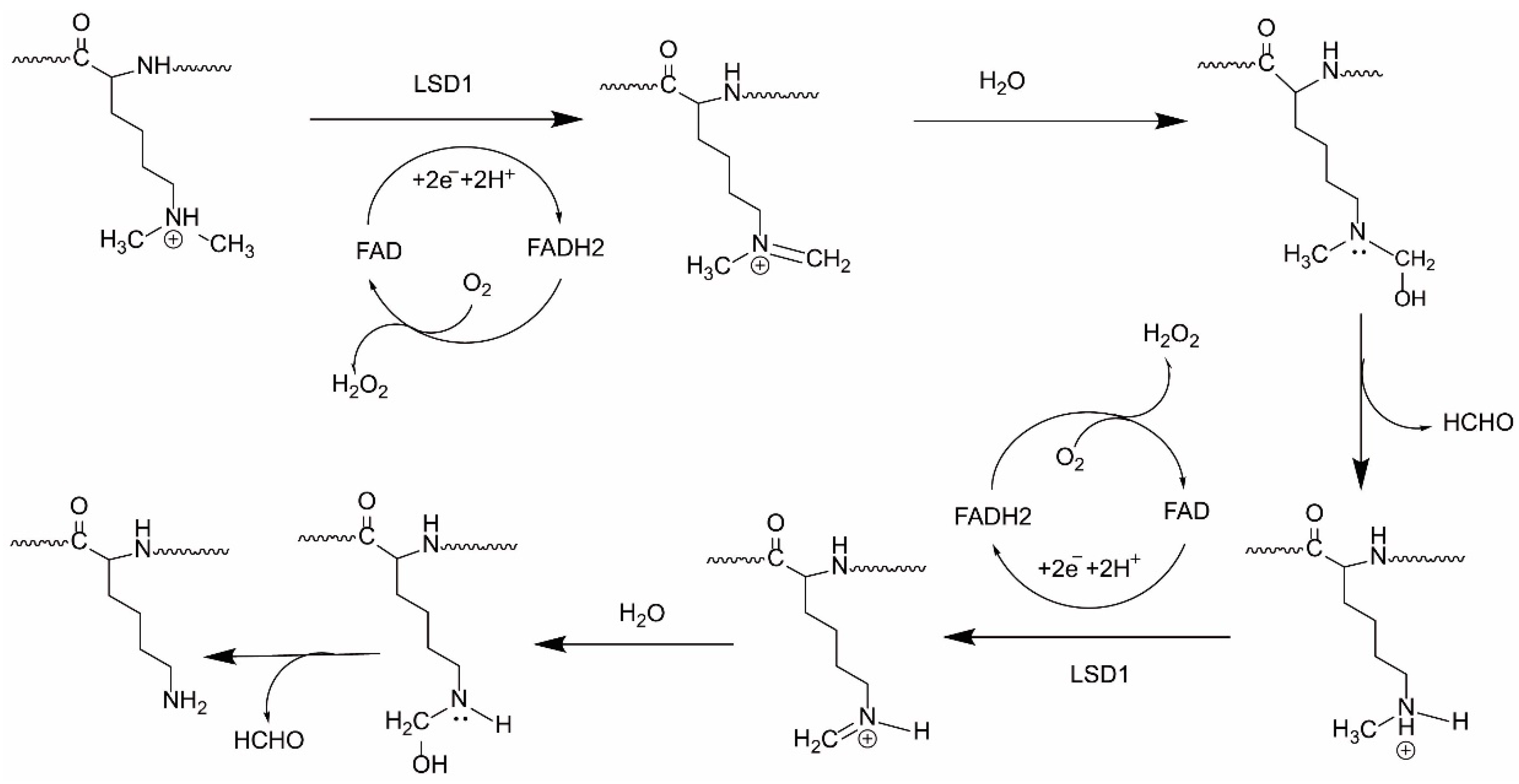
2.2. Functions of LSD1
2.2.1. LSD1 as a Transcription Co-Repressor
2.2.2. LSD1 as a Transcription Co-Activator
2.2.3. LSD1 as a Demethylase of Non-Histone Proteins
3. LSD1 and Cancer
3.1. LSD1 in Breast Cancer
3.2. LSD1 in Prostate Cancer
3.3. LSD1 in AML
4. Screening Methods for LSD1 Inhibitors
4.1. Target-Based Assay
4.2. Substrate-Based Assay
4.3. Byproduct-Based Assay
4.4. PPI-Based Assay
5. Pharmacological Inhibition of LSD1 for Cancer Therapy
5.1. MAO Inactivators and Their Derivatives
5.2. Natural Products and Their Derivatives
5.3. Peptide-Based Inhibitors
5.4. Polyamine-Based Inhibitors
5.5. Metal Complex Inhibitors
5.6. Others
6. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. [Google Scholar] [CrossRef]
- Xie, Q.; Bai, Y.; Wu, J.; Sun, Y.; Wang, Y.; Zhang, Y.; Mei, P.; Yuan, Z. Methylation-mediated regulation of E2F1 in DNA damage-induced cell death. J. Recept. Signal Transduct. Res. 2011, 31, 139–146. [Google Scholar] [CrossRef]
- Kim, Y.; Nam, H.J.; Lee, J.; Park, D.Y.; Kim, C.; Yu, Y.S.; Kim, D.; Park, S.W.; Bhin, J.; Hwang, D.; et al. Methylation-dependent regulation of HIF-1α stability restricts retinal and tumour angiogenesis. Nat. Commun. 2016, 7, 10347. [Google Scholar] [CrossRef]
- Kozub, M.M.; Carr, R.M.; Lomberk, G.L.; Fernandez-Zapico, M.E. LSD1, a double-edged sword, confers dynamic chromatin regulation but commonly promotes aberrant cell growth. F1000Res 2017, 6, 2016. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef]
- Kontaki, H.; Talianidis, I. Lysine methylation regulates E2F1-induced cell death. Mol. Cell. 2010, 39, 152–160. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, J.H.; Choi, H.J.; Won, H.Y.; Joo, H.S.; Shin, D.H.; Park, M.K.; Han, B.; Kim, K.P.; Lee, T.J.; et al. LSD1 demethylates HIF1α to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene. 2017, 36, 5512–5521. [Google Scholar] [CrossRef]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef]
- Laurent, B.; Ruitu, L.; Murn, J.; Hempel, K.; Ferrao, R.; Xiang, Y.; Liu, S.; Garcia, B.A.; Wu, H.; Wu, F.; et al. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol. Cell 2015, 57, 957–970. [Google Scholar] [CrossRef]
- Jotatsu, T.; Yagishita, S.; Tajima, K.; Takahashi, F.; Mogushi, K.; Hidayat, M.; Wirawan, A.; Ko, R.; Kanemaru, R.; Shimada, N.; et al. LSD1/KDM1 isoform LSD1+ 8a contributes to neural differentiation in small cell lung cancer. Biochem. Biophys. Rep. 2017, 9, 86–94. [Google Scholar] [CrossRef]
- Pedersen, M.T.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar] [CrossRef]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 2012, 21, 473–487. [Google Scholar] [CrossRef]
- Schenk, T.; Chen, W.C.; Göllner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef]
- Magliulo, D.; Bernardi, R.; Messina, S. Lysine-Specific Demethylase 1A as a Promising Target in Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 255. [Google Scholar] [CrossRef]
- Pilotto, S.; Speranzini, V.; Tortorici, M.; Durand, D.; Fish, A.; Valente, S.; Forneris, F.; Mai, A.; Sixma, T.K.; Vachette, P.; et al. Interplay among nucleosomal DNA, histone tails, and corepressor CoREST underlies LSD1-mediated H3 demethylation. Proc. Natl. Acad. Sci. USA 2015, 112, 2752–2757. [Google Scholar] [CrossRef]
- Marabelli, C.; Marrocco, B.; Mattevi, A. The growing structural and functional complexity of the LSD1/KDM1A histone demethylase. Curr. Opin. Struc. Biol. 2016, 41, 135–144. [Google Scholar] [CrossRef]
- Da, G.; Lenkart, J.; Zhao, K.; Shiekhattar, R.; Cairns, B.R.; Marmorstein, R. Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc. Natl. Acad. Sci. USA 2006, 103, 2057–2062. [Google Scholar] [CrossRef]
- Aravind, L.; Iyer, L.M. The SWIRM domain: A conserved module found in chromosomal proteins points to novel chromatin-modifying activities. Genome Biol. 2002, 3, research0039. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Stavropoulos, P.; Blobel, G.; Hoelz, A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat. Struct. Mol. Biol. 2006, 13, 626–632. [Google Scholar] [CrossRef]
- Forneris, F.; Battaglioli, E.; Mattevi, A.; Binda, C. New roles of flavoproteins in molecular cell biology: Histone demethylase LSD1 and chromatin. FEBS J. 2009, 276, 4304–4312. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Y.; Wang, F.; Wan, K.; Yamane, K.; Zhang, Y.; Lei, M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proc. Natl. Acad. Sci. USA 2006, 103, 13956–13961. [Google Scholar] [CrossRef]
- Yoneyama, M.; Tochio, N.; Umehara, T.; Koshiba, S.; Inoue, M.; Yabuki, T.; Aoki, M.; Seki, E.; Matsuda, T.; Watanabe, S.; et al. Structural and functional differences of SWIRM domain subtypes. J. Mol. Biol. 2007, 369, 222–238. [Google Scholar] [CrossRef]
- Zhou, C.; Kang, D.; Xu, Y.; Zhang, L.; Zha, X. Identification of Novel Selective Lysine-Specific Demethylase 1 (LSD 1) Inhibitors Using a Pharmacophore-Based Virtual Screening Combined with Docking. Chem. Biol. Drug Des. 2015, 85, 659–671. [Google Scholar] [CrossRef]
- Ota, Y.; Suzuki, T. Drug Design Concepts for LSD1-Selective Inhibitors. Chem. Rec. 2018, 1–11. [Google Scholar] [CrossRef]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef]
- Baron, R.; Vellore, N.A. LSD1/CoREST is an allosteric nanoscale clamp regulated by H3-histone-tail molecular recognition. Proc. Natl. Acad. Sci. USA 2012, 109, 12509–12514. [Google Scholar] [CrossRef]
- Sun, G.; Alzayady, K.; Stewart, R.; Ye, P.; Yang, S.; Li, W.; Shi, Y. Histone demethylase LSD1 regulates neural stem cell proliferation. Mol.Cell. Biol. 2010, 30, 1997–2005. [Google Scholar] [CrossRef]
- Hakimi, M.A.; Bochar, D.A.; Chenoweth, J.; Lane, W.S.; Mandel, G.; Shiekhattar, R. A core–BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc. Natl. Acad. Sci. USA 2002, 99, 7420–7425. [Google Scholar] [CrossRef]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 7420–7425. [Google Scholar] [CrossRef]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]
- Bennesch, M.A.; Segala, G.; Wider, D.; Picard, D. LSD1 engages a corepressor complex for the activation of the estrogen receptor α by estrogen and cAMP. Nucleic Acids Res. 2016, 44, 8655–8670. [Google Scholar] [CrossRef]
- Hino, S.; Kohrogi, K.; Nakao, M. Histone demethylase LSD1 controls the phenotypic plasticity of cancer cells. Cancer Sci. 2016, 107, 1187–1192. [Google Scholar] [CrossRef]
- Sorna, V.; Theisen, E.R.; Stephens, B.; Warner, S.L.; Bearss, D.J.; Vankayalapati, H.; Sharma, S. High-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J. Med. Chem. 2013, 56, 9496–9508. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef]
- Rivenbark, A.G.; Coleman, W.B. Field cancerization in mammary carcinogenesis—Implications for prevention and treatment of breast cancer. Exp. Mol. Pathol. 2012, 93, 391–398. [Google Scholar] [CrossRef]
- Serce, N.; Gnatzy, A.; Steiner, S.; Lorenzen, H.; Kirfel, J.; Buettner, R. Elevated expression of LSD1 (Lysine-specific demethylase 1) during tumour progression from pre-invasive to invasive ductal carcinoma of the breast. BMC Clin. Pathol. 2012, 12, 13. [Google Scholar] [CrossRef]
- Bradley, C.; van der Meer, R.; Roodi, N.; Yan, H.; Chandrasekharan, M.B.; Sun, Z.W.; Mernaugh, R.L.; Parl, F.F. Carcinogen-induced histone alteration in normal human mammary epithelial cells. Carcinogenesis 2007, 28, 2184–2192. [Google Scholar] [CrossRef]
- Jordan, V.C. Selective estrogen receptor modulation: Concept and consequences in cancer. Cancer Cell 2004, 5, 207–213. [Google Scholar] [CrossRef]
- Mann, M.; Cortez, V.; Vadlamudi, R.K. Epigenetics of estrogen receptor signaling: Role in hormonal cancer progression and therapy. Cancers 2011, 3, 1691–1707. [Google Scholar] [CrossRef]
- Hu, Q.; Kwon, Y.-S.; Nunez, E.; Cardamone, M.D.; Hutt, K.R.; Ohgi, K.A. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc. Natl. Acad. Sci. USA 2008, 105, 19199–19204. [Google Scholar] [CrossRef]
- Pollock, J.A.; Larrea, M.D.; Jasper, J.S.; McDonnell, D.P.; McCafferty, D.G. Lysine-specific histone demethylase 1 inhibitors control breast cancer proliferation in ERα-dependent and-independent manners. ACS Chem. Biol. 2012, 7, 1221–1231. [Google Scholar] [CrossRef]
- Bennani-Baiti, I.M. Integration of ERα-PELP1-HER2 signaling by LSD1 (KDM1A/AOF2) offers combinatorial therapeutic opportunities to circumventing hormone resistance in breast cancer. Breast Cancer Res. 2012, 14, 112. [Google Scholar] [CrossRef]
- Kim, J.; Park, U.H.; Moon, M.; Um, S.J.; Kim, E.J. Negative regulation of ERα by a novel protein CAC1 through association with histone demethylase LSD1. FEBS Lett. 2013, 587, 17–22. [Google Scholar] [CrossRef]
- Park, U.H.; Kang, M.R.; Kim, E.J.; Kwon, Y.S.; Hur, W.; Yoon, S.K.; Song, B.J.; Park, J.H.; Hwang, J.T.; Jeong, J.C.; et al. ASXL2 promotes proliferation of breast cancer cells by linking ERα to histone methylation. Oncogene 2016, 35, 3742–3752. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. Epigenetic regulation of LSD1 during mammary carcinogenesis. Molecular & cellular oncology. Mol. Cell. Oncol. 2014, 1, e963426. [Google Scholar]
- Vasilatos, S.N.; Katz, T.A.; Oesterreich, S.; Wan, Y.; Davidson, N.E.; Huang, Y. Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis 2013, 34, 1196–1207. [Google Scholar] [CrossRef]
- Huang, Y.; Vasilatos, S.N.; Boric, L.; Shaw, P.G.; Davidson, N.E. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res. Treat. 2012, 131, 777–789. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, W.; Qiu, R.; Liu, R.; Zeng, Y.; Gao, J.; Zheng, Y.; Hou, Y.; Wang, S.; Yu, W.; et al. LSD1 coordinates with the SIN3A/HDAC complex and maintains sensitivity to chemotherapy in breast cancer. J. Mol. Cell. Biol. 2018, 10, 285–301. [Google Scholar] [CrossRef]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell. Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef]
- Hartwell, K.A.; Muir, B.; Reinhardt, F.; Carpenter, A.E.; Sgroi, D.C.; Weinberg, R.A. The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 18969–18974. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef]
- Lin, Y.; Kang, T.; Zhou, B.P. Doxorubicin enhances Snail/LSD1-mediated PTEN suppression in a PARP1-dependent manner. Cell Cycle 2014, 13, 1708–1716. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Li, X.Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA 2012, 109, 16654–16659. [Google Scholar] [CrossRef]
- Zheng, Y.; Zeng, Y.; Qiu, R.; Liu, R.; Huang, W.; Hou, Y.; Wang, S.; Leng, S.; Feng, D.; Yang, Y.; et al. The Homeotic Protein SIX3 Suppresses Carcinogenesis and Metastasis through Recruiting the LSD1/NuRD (MTA3) Complex. Theranostics 2018, 8, 972–989. [Google Scholar] [CrossRef]
- Munkley, J.; Vodak, D.; Livermore, K.E.; James, K.; Wilson, B.T.; Knight, B.; Mccullagh, P.; Mcgrath, J.; Crundwell, M.; Harries, L.W.; et al. Glycosylation is an androgen-regulated process essential for prostate cancer cell viability. EBioMedicine. 2016, 8, 103–116. [Google Scholar] [CrossRef]
- Willmann, D.; Lim, S.; Wetzel, S.; Metzger, E.; Jandausch, A.; Wilk, W.; Jung, M.; Forne, I.; Imhof, A.; Janzer, A.; et al. Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. Intl. J. Cancer 2012, 131, 2704–2709. [Google Scholar] [CrossRef]
- Kashyap, V.; Ahmad, S.; Nilsson, E.M.; Helczynski, L.; Kenna, S.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. The lysine specific demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate cancer. Mol. Oncol. 2013, 7, 555–566. [Google Scholar] [CrossRef]
- Ketscher, A.; Jilg, C.A.; Willmann, D.; Hummel, B.; Imhof, A.; Rüsseler, V.; Hölz, S.; Metzger, E.; Müller, J.M.; Schüle, R. LSD1 controls metastasis of androgen-independent prostate cancer cells through PXN and LPAR6. Oncogenesis 2014, 3, e120. [Google Scholar] [CrossRef]
- Rozan, L.M.; El-Deiry, W.S. p53 downstream target genes and tumor suppression: a classical view in evolution. Cell Death Differ. 2007, 14, 3–9. [Google Scholar] [CrossRef]
- Hotte, S.J.; Saad, F. Current management of castrate-resistant prostate cancer. Curr. Oncol. 2010, 17, S72–S79. [Google Scholar] [CrossRef]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef]
- ahl, P.; Gullotti, L.; Heukamp, L.C.; Wolf, S.; Friedrichs, N.; Vorreuther, R.; Solleder, G.; Bastian, P.J.; Ellinger, J.; Metzger, E.; et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006, 66, 11341–11347. [Google Scholar]
- Wissmann, M.; Yin, N.; Müller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Günther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef]
- Regufe da Mota, S.; Bailey, S.; Strivens, R.A.; Hayden, A.L.; Douglas, L.R.; Duriez, P.J.; Borrello, M.T.; Benelkebir, H.; Ganesan, A.; Packham, G.; et al. LSD1 inhibition attenuates androgen receptor V7 splice variant activation in castration resistant prostate cancer models. Cancer Cell. Int. 2018, 18, 71. [Google Scholar] [CrossRef]
- Liang, Y.; Ahmed, M.; Guo, H.; Soares, F.; Hua, J.T.; Gao, S.; Lu, C.; Poon, C.; Han, W.; Langstein, J.; et al. LSD1-Mediated Epigenetic Reprogramming Drives CENPE Expression and Prostate Cancer Progression. Cancer Res. 2017, 77, 5479–5490. [Google Scholar] [CrossRef]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A.; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl. Acad. Sci. USA 2018, 115, E4179–E4188. [Google Scholar] [PubMed]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 inhibitors in acute myeloid leukemia: Current status and future directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Saijo, K.; Skola, D.; Jin, C.; Ma, Q.; Merkurjev, D.; Glass, C.K.; Rosenfeld, M.G. Histone demethylase LSD1 regulates hematopoietic stem cells homeostasis and protects from death by endotoxic shock. Proc. Natl. Acad. Sci. USA 2018, 115, E244–E252. [Google Scholar] [CrossRef] [PubMed]
- Sprüssel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Händschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; König, K.; Diehl, L.; et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Gore, SD.; Zeidan, A.M. Epigenetic therapy in acute myeloid leukemia: Current and future directions. Semin. Hematol. 2015, 52, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Lania, L.; Majello, B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochem. Biophys. Acta 2013, 1829, 981–986. [Google Scholar] [CrossRef]
- Takeuchi, M.; Fuse, Y.; Watanabe, M.; Andrea, C.S.; Takeuchi, M.; Nakajima, H.; Ohashi, K.; Kaneko, H.; Kobayashi-Osaki, M.; Yamamoto, M.; et al. LSD1/KDM1A promotes hematopoietic commitment of hemangioblasts through downregulation of Etv2. Proc. Natl. Acad. Sci. USA 2015, 112, 13922–13927. [Google Scholar] [CrossRef]
- Velinder, M.; Singer, J.; Bareyan, D.; Meznarich, J.; Tracy, C.M.; Fulcher, J.M. GFI1 functions in transcriptional control and cell fate determination require SNAG domain methylation to recruit LSD1. Biochem. J. 2016, 474, 2951. [Google Scholar] [CrossRef]
- Thambyrajah, R.; Mazan, M.; Patel, R.; Moignard, V.; Stefanska, M.; Marinopoulou, E.; Li, Y.; Lancrin, C.; Clapes, T.; Möröy, T.; et al. GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell. Biol. 2016, 18, 21–32. [Google Scholar] [CrossRef]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 2007, 27, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Altucci, L.; Gronemeyer, H. The promise of retinoids to fight against cancer. Nat. Rev. Cancer 2001, 1, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.T.; Spencer, G.J.; Harris, W.J.; Maiques-Díaz, A.; Ciceri, F.; Huang, X.; Somervaille, T.C.P. Pharmacological inhibitors of LSD1 promote differentiation of myeloid leukemia cells through a mechanism independent of histone demethylation. Blood 2014, 124, 167. [Google Scholar]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.P.; Bremberg, U.; Jordan, A.M.; Geitmann, M.; Maiques-Diaz, A.; McGonagle, A.E.; Small, H.F.; Somervaille, T.C.P.; Ogilvie, D. Development of 5-hydroxypyrazole derivatives as reversible inhibitors of lysine specific demethylase 1. Bioorg. Med. Chem. Lett. 2017, 27, 3190–3195. [Google Scholar] [CrossRef]
- Metzger, E.; Schüle, R. Interaction of Methylated lsd1 and Chd1, a Compound Inhibiting this Interaction for use in Therapy, and a Screening Method for such a Compound. U.S. Patent 20180036256A1, 8 February 2018. [Google Scholar]
- Syafrizayanti, B.C.; Hoheisel, J.D.; Kastelic, D. Methods for analyzing and quantifying protein–protein interaction. Expert Rev. Proteom. 2014, 11, 107–120. [Google Scholar] [CrossRef]
- Zheng, Y.C.; Chang, J.; Zhang, T.; Suo, F.Z.; Chen, X.B.; Liu, Y.; Zhao, B.; Yu, B.; Liu, H.M. An Overview on Screening Methods for Lysine Specific Demethylase 1 (LSD1) Inhibitors. Curr. Med. Chem. 2017, 24, 2496–2504. [Google Scholar] [CrossRef]
- Wigle, T.J.; Swinger, K.K.; Campbell, J.E.; Scholle, M.D.; Sherrill, J.; Admirand, E.A.; Boriack-Sjodin, P.A.; Kuntz, K.W.; Chesworth, R.; Moyer, M.P.; et al. A high-throughput mass spectrometry assay coupled with redox activity testing reduces artifacts and false positives in lysine demethylase screening. J. Biomol. Screen. 2015, 20, 810–820. [Google Scholar] [CrossRef]
- Plant, M.; Dineen, T.; Cheng, A.; Long, A.M.; Chen, H.; Morgenstern, K.A. Screening for lysine-specific demethylase-1 inhibitors using a label-free high-throughput mass spectrometry assay. Anal. Biochem. 2011, 419, 217–227. [Google Scholar] [CrossRef]
- Huang, Y.; Greene, E.; Murray Stewart, T.; Goodwin, A.C.; Baylin, S.B.; Woster, P.M.; Casero, R.A. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc. Natl. Acad. Sci. USA 2007, 104, 8023–8028. [Google Scholar] [CrossRef]
- Zhou, M.; Diwu, Z.; Panchuk-Voloshina, N.; Haugland, R.P. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: Applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal. Biochem. 1997, 253, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Trinder, P. Determination of glucose in blood using glucose oxidase with an alternative oxygen acceptor. Ann. Clin. Biochem. 1969, 6, 24–27. [Google Scholar] [CrossRef]
- Hauser, A.T.; Bissinger, E.M.; Metzger, E.; Repenning, A.; Bauer, U.M.; Mai, A.; Schüle, R.; Jung, M. Screening assays for epigenetic targets using native histones as substrates. J. Biomol. Screen. 2012, 17, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided. Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Mimasu, S.; Umezawa, N.; Sato, S.; Higuchi, T.; Umehara, T.; Yokoyama, S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry 2010, 49, 6494–6503. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Doyle, K.; Mosbruger, T.L.; Butterfield, A.; Weston, A.; Ast, A.; Kaadige, M.; Verma, A.; Sharma, S. Reversible LSD1 inhibition with HCI-2509 induces the p53 gene expression signature and disrupts the MYCN signature in high-risk neuroblastoma cells. Oncotarget 2018, 9, 9907–9924. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Shah, B.; Portier, B.P.; Devaraj, S.G.; Liu, K.; Iyer, S.P.; Bearss, D.; Bhalla, K.N. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2014, 28, 2155–2164. [Google Scholar] [CrossRef]
- Gupta, S.; Weston, A.; Bearrs, J.; Thode, T.; Neiss, A.; Soldi, R.; Sharma, S. Reversible lysine-specific demethylase 1 antagonist HCI-2509 inhibits growth and decreases c-MYC in castration- and docetaxel-resistant prostate cancer cells. Prostate Cancer Prostatic Dis. 2016, 19, 349–357. [Google Scholar] [CrossRef]
- Malo, N.; Hanley, J.A.; Cerquozzi, S.; Pelletier, J.; Nadon, R. Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 2006, 24, 167–175. [Google Scholar] [CrossRef]
- Bibette, J. Gaining confidence in high-throughput screening. Proc. Natl. Acad. Sci. USA 2012, 109, 649–650. [Google Scholar] [CrossRef]
- Yang, M.; Culhane, J.C.; Szewczuk, L.M.; Jalili, P.; Ball, H.L.; Machius, M.; Cole, P.A.; Yu, H. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 2007, 46, 8058–8065. [Google Scholar] [CrossRef]
- Culhane, J.C.; Szewczuk, L.M.; Liu, X.; Da, G.; Marmorstein, R.; Cole, P.A. A mechanism-based inactivator for histone demethylase LSD1. J. Am. Chem Soc. 2006, 128, 4536–4537. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Unzeta, M.; Tipton, K.F. A spectrophotometric method for determining the oxidative deamination of methylamine by the amine oxidases. Anal. Biochem. 2000, 286, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, S.; Pachaiyappan, B.; Steinbergs, N.; Nowotarski, S.; Hanson, A.S.; Casero, R.A.; Woster, P.M. Low molecular weight amidoximes that act as potent inhibitors of lysine-specific demethylase 1. J. Med. Chem. 2012, 55, 7378–7391. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Stewart, T.M.; Wu, Y.; Baylin, S.B.; Marton, L.J.; Perkins, B.; Jones, R.J.; Woster, P.M.; Casero, R.A. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin. Cancer Res. 2009, 15, 7217–7228. [Google Scholar] [CrossRef]
- Wu, Y.; Steinbergs, N.; Murray-Stewart, T.; Marton, L.J.; Casero, R.A. Oligoamine analogues in combination with 2-difluoromethylornithine synergistically induce re-expression of aberrantly silenced tumour-suppressor genes. Biochem. J. 2012, 442, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.; Fisch, T.; Long, A.M.; Tang, J.; Lee, J.H.; Hierl, M.; Chen, H.; Yakowec, P.; Schwandner, R.; Emkey, R. High-throughput TR-FRET assays for identifying inhibitors of LSD1 and JMJD2C histone lysine demethylases. J. Biomol. Screen. 2012, 17, 27–38. [Google Scholar] [CrossRef]
- Gauthier, N.; Caron, M.; Pedro, L.; Arcand, M.; Blouin, J.; Labonté, A.; Normand, C.; Paquet, V.; Rodenbrock, A.; Roy, M.; et al. Development of homogeneous nonradioactive methyltransferase and demethylase assays targeting histone H3 lysine 4. J. Biomol. Screen. 2012, 17, 49–58. [Google Scholar] [CrossRef]
- Takagi, T.; Shum, D.; Parisi, M.; Santos, R.E.; Radu, C.; Calder, P.; Rizvi, Z.; Frattini, M.G.; Djaballah, H. Comparison of luminescence ADP production assay and radiometric scintillation proximity assay for Cdc7 kinase. Comb. Chem. High Throughput Screen. 2011, 14, 669–687. [Google Scholar] [CrossRef]
- Yu, W.; Eram, M.S.; Hajian, T.; Szykowska, A.; Burgess-Brown, N.; Vedadi, M.; Brown, P.J. A scintillation proximity assay for histone demethylases. Anal. Biochem. 2014, 463, 54–60. [Google Scholar] [CrossRef]
- Khan, M.N.A.; Tsumoto, H.; Itoh, Y.; Ota, Y.; Suzuki, M.; Ogasawara, D.; Nakagawa, H.; Mizukami, T.; Miyata, N.; Suzuki, T. Design, synthesis, and biological activity of N-alkylated analogue of NCL1, a selective inhibitor of lysine-specific demethylase 1. MedChemComm 2015, 6, 407–412. [Google Scholar] [CrossRef]
- Zheng, Y.C.; Ma, J.; Wang, Z.; Li, J.; Jiang, B.; Zhou, W.; Shi, X.; Wang, X.; Zhao, W.; Liu, H.M. A systematic review of histone lysine-specific demethylase 1 and its inhibitors. Med. Res. Rev. 2015, 35, 1032–1071. [Google Scholar] [CrossRef] [PubMed]
- Ueda, R.; Suzuki, T.; Mino, K.; Tsumoto, H.; Nakagawa, H.; Hasegawa, M.; Sasaki, R.; Mizukami, T.; Miyata, N. Identification of cell-active lysine specific demethylase 1-selective inhibitors. J. Am. Chem. Soc. 2009, 131, 17536–17537. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Maes, T.; Carceller, E.; Salas, J.; Ortega, A.; Buesa, C. Advances in the development of histone lysine demethylase inhibitors. Curr. Opin. Pharmacol. 2015, 23, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 2018, 33, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.; Mann, M.; Tekmal, S.; Suzuki, T.; Miyata, N.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Sood, A.K.; Vadlamudi, R.K. Targeting the PELP1-KDM1 axis as a potential therapeutic strategy for breast cancer. Breast Cancer Res. 2012, 14, R108. [Google Scholar] [CrossRef]
- Etani, T.; Suzuki, T.; Naiki, T.; Naiki-Ito, A.; Ando, R.; Iida, K.; Kawai, N.; Tozawa, K.; Miyata, N.; Kohri, K. NCL1, a highly selective lysine-specific demethylase 1 inhibitor, suppresses prostate cancer without adverse effect. Oncotarget 2015, 6, 2865–2878. [Google Scholar] [CrossRef]
- Sareddy, G.; Viswanadhapalli, S.; Surapaneni, P.; Suzuki, T.; Brenner, A.; Vadlamudi, R. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene 2017, 36, 2423–2434. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Nair, B.C.; Krishnan, S.K.; Gonugunta, V.K.; Zhang, Q.G.; Suzuki, T.; Miyata, N.; Brenner, A.J.; Brann, D.W.; Vadlamudi, R.K. KDM1 is a novel therapeutic target for the treatment of gliomas. Oncotarget 2013, 4, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Prusevich, P.; Kalin, J.H.; Ming, S.A.; Basso, M.; Givens, J.; Li, X.; Hu, J.; Taylor, M.S.; Cieniewicz, A.M.; Hsiao, P.Y.; et al. A selective phenelzine analogue inhibitor of histone demethylase LSD1. ACS Chem. Biol. 2014, 9, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S. Target identification using drug affinity responsive target stability (DARTS). Curr. Protoc. Chem. Biol. 2011, 3, 163–180. [Google Scholar] [PubMed]
- Abdulla, A.; Zhao, X.; Yang, F. Natural polyphenols inhibit lysine-specific demethylase-1 in vitro. J. Biochem. Pharmacol. Res. 2013, 1, 56–63. [Google Scholar] [PubMed]
- Sakane, C.; Okitsu, T.; Wada, A.; Sagami, H.; Shidoji, Y. Inhibition of lysine-specific demethylase 1 by the acyclic diterpenoid geranylgeranoic acid and its derivatives. Biochem. Biophys. Res. Commun. 2014, 444, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, P.; Berlicki, L. Peptide-based inhibitors of protein–protein interactions. Bioorganic Med. Chem. Lett. 2016, 26, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Binda, C.; Adamo, A.; Battaglioli, E.; Mattevi, A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J. Biol. Chem. 2007, 282, 20070–20074. [Google Scholar] [CrossRef] [PubMed]
- Kumarasinghe, I.R.; Woster, P.M. Synthesis and evaluation of novel cyclic Peptide inhibitors of lysine-specific demethylase 1. ACS Med. Chem. Lett. 2014, 5, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Aihara, K.; Mellini, P.; Tojo, T.; Ota, Y.; Tsumoto, H.; Solomon, V.R.; Zhan, P.; Suzuki, M.; Ogasawara, D.; et al. Identification of SNAIL1 peptide-based irreversible lysine-specific demethylase 1-selective inactivators. J. Med. Chem. 2016, 59, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Wu, Y.; Steinbergs, N.; Crowley, M.L.; Hanson, A.S.; Casero, R.A.; Woster, P.M. (Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. J. Med. Chem. 2010, 53, 5197–5212. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Li, S.; Song, W.; Li, X.; Li, Q.; Zhang, Z.; Han, Y.; Zhang, X.; Miao, S.; Du, R.; et al. Lysine-specific demethylase 1 (LSD1/KDM1A) contributes to colorectal tumorigenesis via activation of the Wnt/β-catenin pathway by down-regulating Dickkopf-1 (DKK1). PLoS ONE 2013, 8, e70077. [Google Scholar] [CrossRef]
- Yang, C.; Wang, W.; Liang, J.X.; Li, G.; Vellaisamy, K.; Wong, C.Y.; Ma, D.L.; Leung, C.H. A Rhodium(III)-Based Inhibitor of Lysine-Specific Histone Demethylase 1 as an Epigenetic Modulator in Prostate Cancer Cells. J. Med. Chem. 2017, 60, 2597–2603. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, F.; Ren, Q.; Sun, H.; Xu, Z.; Lan, R.; Liu, Y.; Ward, D.; Quan, J.; Ye, T.; et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011, 71, 7238–7249. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.P.; Williamson, K.E.; Balasubramanian, S.; Odate, S.; Arora, S.; Hatton, C.; Edwards, T.M.; O’Brien, T.; Magnuson, S.; Stokoe, D.; et al. Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res. 2016, 76, 1975–1988. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.P.; McGonagle, A.E.; Wiseman, D.H.; Williams, E.L.; Jordan, A.M. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: Clinical significance and progress to date. Med. Res. Rev. 2015, 35, 586–618. [Google Scholar] [CrossRef] [PubMed]
- Hitchin, J.R.; Blagg, J.; Burke, R.; Burns, S.; Cockerill, M.J.; Fairweather, E.E. Development and evaluation of selective, reversible LSD1 inhibitors derived from fragments. MedChemComm. 2013, 4, 1513–1522. [Google Scholar] [CrossRef]
- Theisen, E.R.; Gajiwala, S.; Bearss, J.; Sorna, V.; Sharma, S.; Janat-Amsbury, M. Reversible inhibition of lysine specific demethylase 1 is a novel anti-tumor strategy for poorly differentiated endometrial carcinoma. BMC Cancer 2014, 14, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Sankar, S.; Theisen, E.R.; Bearss, J.; Mulvihill, T.; Hoffman, L.M.; Sorna, V.; Beckerle, M.C.; Sharma, S.; Lessnick, S.L. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin. Cancer Res. 2014, 20, 4584–4597. [Google Scholar] [CrossRef]
- Yang, G.J.; Zhong, H.J.; Ko, C.N.; Wong, S.Y.; Vellaisamy, K.; Ye, M.; Ma, D.L.; Leung, C.H. Identification of a rhodium(iii) complex as a Wee1 inhibitor against TP53-mutated triple-negative breast cancer cells. Chem. Commun. 2018, 54, 2463–2466. [Google Scholar] [CrossRef]
- Yang, G.J.; Wang, W.; Mok, S.W.F.; Wu, C.; Law, B.Y.K.; Miao, X.M.; Wu, K.J.; Zhong, H.J.; Wong, C.Y.; Wong, V.K.W.; et al. Selective inhibition of lysine-specific demethylase 5A (KDM5A) using a rhodium (III) complex for triple-negative breast cancer therapy. Angew. Chem. Int. Ed. Engl.. 2018, 57, 13091–13095. [Google Scholar] [CrossRef]
- Leung, C.-H.; Lin, S.; Zhong, H.-J.; Ma, D.-L. Metal complexes as potential modulators of inflammatory and autoimmune responses. Chem. Sci. 2015, 6, 871–884. [Google Scholar] [CrossRef]
- Leung, C.-H.; He, H.-Z.; Liu, L.-J.; Wang, M.; Chan, D.S.-H.; Ma, D.-L. Metal complexes as inhibitors of transcription factor activity. Coord. Chem. Rev. 2013, 257, 3139–31351. [Google Scholar] [CrossRef]
- Liu, L.J.; Lu, L.; Zhong, H.J.; He, B.; Kwong, D.W.; Ma, D.L.; Leung, C.H. An iridium (III) complex inhibits JMJD2 activities and acts as a potential epigenetic modulator. J. Med. Chem. 2015, 58, 6697–6703. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.C.; Ma, Y.C.; Qin, W.P.; Ding, L.N.; Zheng, Y.C.; Zhu, Y.L.; Zhai, X.Y.; Yang, J.; Ma, C.Y.; Guan, Y.Y. Design and synthesis of tranylcypromine derivatives as novel LSD1/HDACs dual inhibitors for cancer treatment. Eur. J. Med. Chem. 2017, 140, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Wang, J.; Liu, P.; Li, Y.; Lu, W.; Hu, Y.; Liu, J.; He, Z.; Huang, P. Novel combination of histone methylation modulators with therapeutic synergy against acute myeloid leukemia in vitro and in vivo. Cancer Lett. 2018, 413, 35–45. [Google Scholar] [CrossRef]
- Dent, S.Y.; Chandra, J. Hematopoietic Stem Cells: The lasting influence of LSD1 in the blood. Elife. 2013, 2, e00963. [Google Scholar] [CrossRef]
- Feng, Z.; Yao, Y.; Zhou, C.; Chen, F.; Wu, F.; Wei, L.; Liu, W.; Dong, S.; Redell, M.; Mo, Q.; et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J. Hematol. Oncol. 2016, 9, 24. [Google Scholar] [CrossRef]
- Lee, M.G.; Wynder, C.; Schmidt, D.M.; McCafferty, D.G.; Shiekhattar, R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 2006, 13, 563–567. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
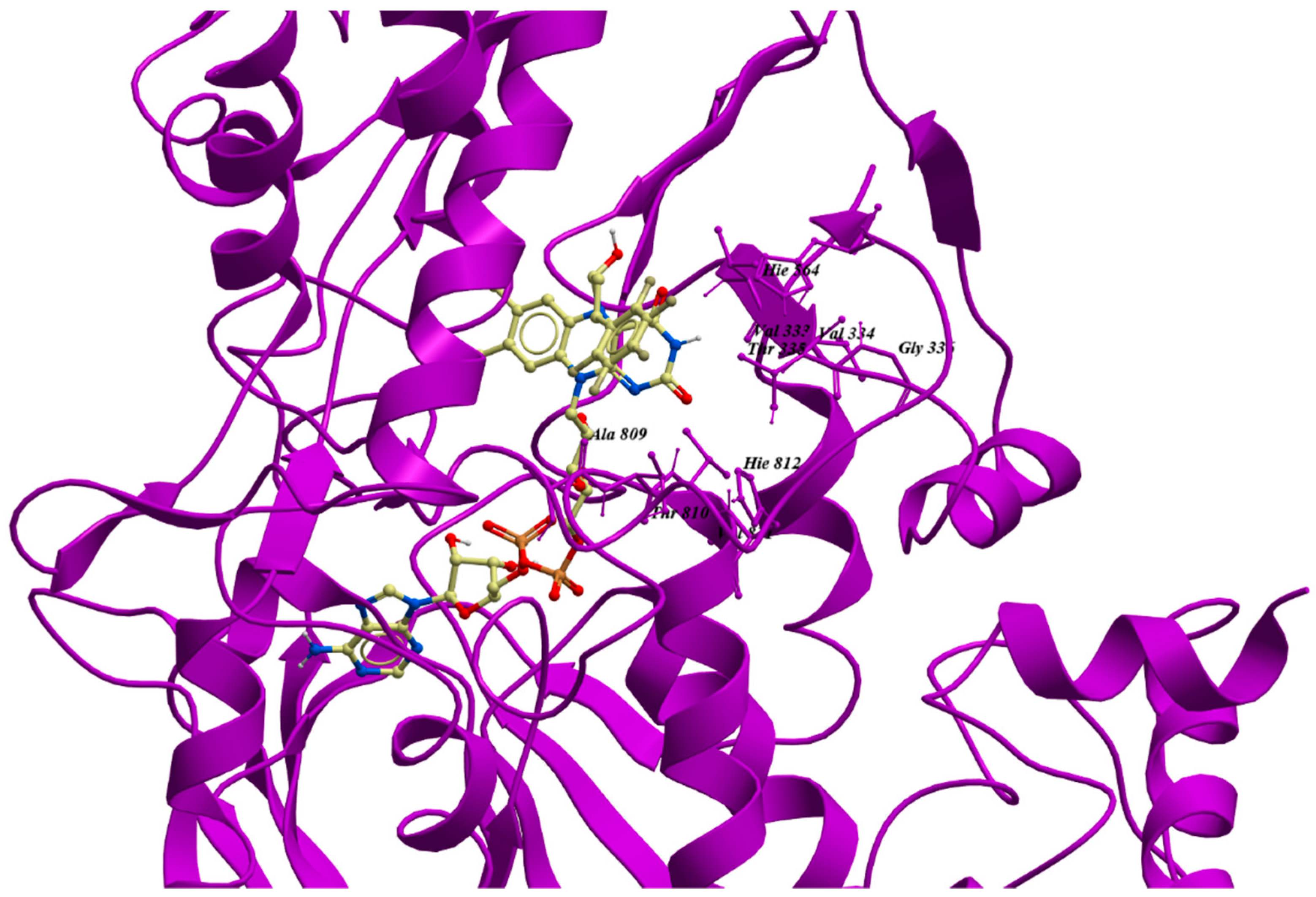{kind=link}
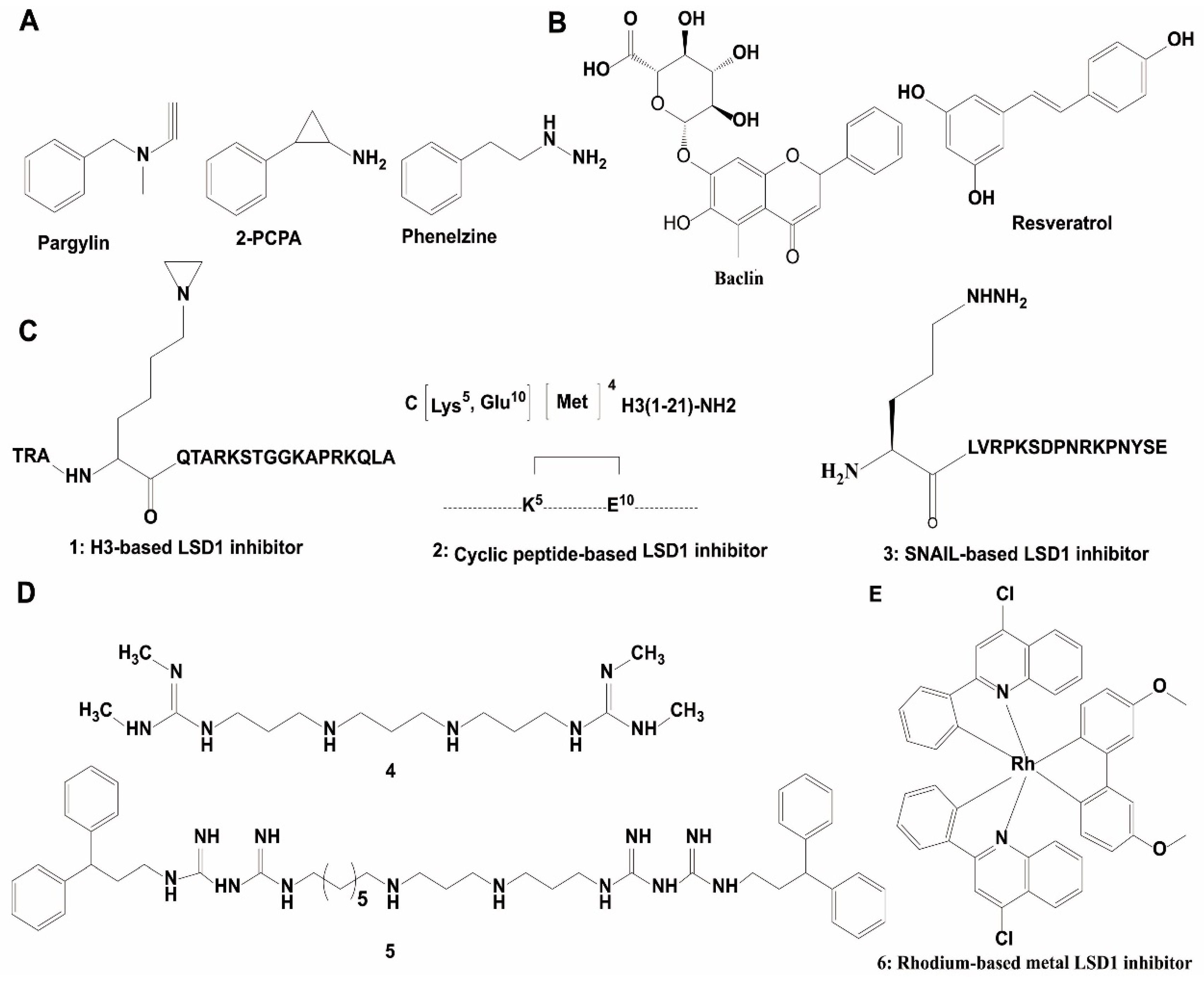{kind=link}
| Type of Substrates | Protein | Subunits of Complex | Functions |
|---|---|---|---|
| Histone | H3K4me2/me1 | RCOR2 | Regulates ESC property and reprogramming somatic cells to pluripotency |
| NuRD (MTA3) | Nucleosome remodeling and H3K4 demethylation | ||
| TLX | Regulation of neuronal stem cell proliferation | ||
| CtBP/CoREST | Repression of growth hormone expression in hypophysis | ||
| HDAC/CoREST | Regulation of pluripotency of embryonic stem/carcinoma cells | ||
| HDAC/SIN3A | Maintenance of sensitivity to chemotherapy in breast cancer | ||
| Core-BRAF35 | Mediates repression of neuronal specific genes in nonneuronal tissues | ||
| HP1/SU(VAR)3–9 | Heterochromatic gene silencing | ||
| HOTAIR/PRC2 | HOX gene silencing | ||
| H3K9me2/me1 | ERα | Activation of estrogen receptor alpha-dependent genes | |
| spLSD1/2 | Control of replication, imprinting and heterochromatin propagation | ||
| AR | LSD1 functions as an activator of androgen receptor-responsive genes | ||
| Non-histone | p53 | - | Suppresses tumor and activates transcription |
| DNMT1 | - | Maintains DNA methylation | |
| E2F1 | - | Regulates DNA damage-induced stabilization and apoptotic function. | |
| HIF-1α | - | Regulates transcription of VEGF | |
| STAT3 | - | Regulates gene expression through the formation of dimers |
| Category | DetectedSpecies | Methods | Benefits | Drawbacks |
|---|---|---|---|---|
| Target-based assay | LSD1 | Virtual screening | Low cost; High-throughput | High false positive rate; only applicable for primary screening |
| SPR | Label-free; low false positive rate | Low-throughput; high technical and equipment requirements; high cost | ||
| ITC | ||||
| BLI | ||||
| Substrate-based assay | Truncated H3 | MS-based assay | Label-free; direct detection of product | Requirement of expensive instrumentation, dedicated operation professionals, and complicated sample preparation steps |
| Byproduct-based assay | H2O2 | Luminol couple assay | Low cost; high-throughput; label-free; high sensitivity | Only applicable for compounds that do not interact with H2O2 |
| Amplex red- coupled assay | Only applicable for compounds that do not interact with H2O2 and without autofluorescence or quenching ability | |||
| 4-aminoantipyrine-coupled assay | Low cost; high-throughput; label-free | Low sensitivity; Only applicable for compounds that do not interact with H2O2 | ||
| HCHO | FDH-coupled assay | Only be applicable for compounds that do not interact with HCHO | ||
| PPI-based assay | LSD-peptide PPI | HTRF-based assay | High-throughput, easy to operate; high sensitivity | Labeling requirement; interference by compounds with autofluorescence or fluorescence quenching ability |
| HTRF-based assay | ||||
| Scintillation proximity assay | high-throughput; high sensitivity | Heat required; special instrument required; additional enzyme introduced |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.-J.; Lei, P.-M.; Wong, S.-Y.; Ma, D.-L.; Leung, C.-H. Pharmacological Inhibition of LSD1 for Cancer Treatment. Molecules 2018, 23, 3194. https://doi.org/10.3390/molecules23123194
Yang G-J, Lei P-M, Wong S-Y, Ma D-L, Leung C-H. Pharmacological Inhibition of LSD1 for Cancer Treatment. Molecules. 2018; 23(12):3194. https://doi.org/10.3390/molecules23123194
Chicago/Turabian StyleYang, Guan-Jun, Pui-Man Lei, Suk-Yu Wong, Dik-Lung Ma, and Chung-Hang Leung. 2018. "Pharmacological Inhibition of LSD1 for Cancer Treatment" Molecules 23, no. 12: 3194. https://doi.org/10.3390/molecules23123194
APA StyleYang, G.-J., Lei, P.-M., Wong, S.-Y., Ma, D.-L., & Leung, C.-H. (2018). Pharmacological Inhibition of LSD1 for Cancer Treatment. Molecules, 23(12), 3194. https://doi.org/10.3390/molecules23123194