Discovery of Non-Peptidic Compounds against Chagas Disease Applying Pharmacophore Guided Molecular Modelling Approaches

Abstract
1. Introduction
2. Results
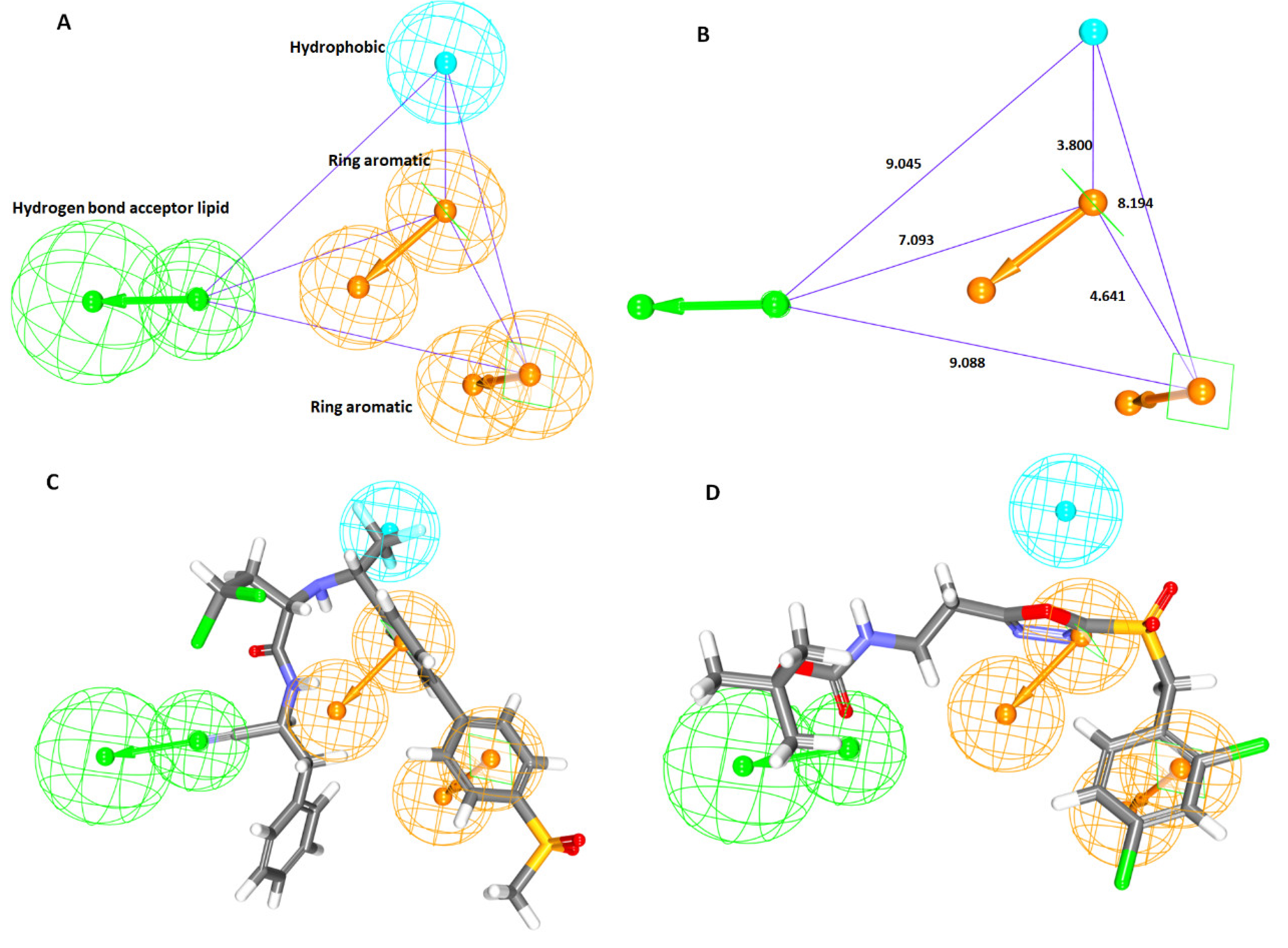
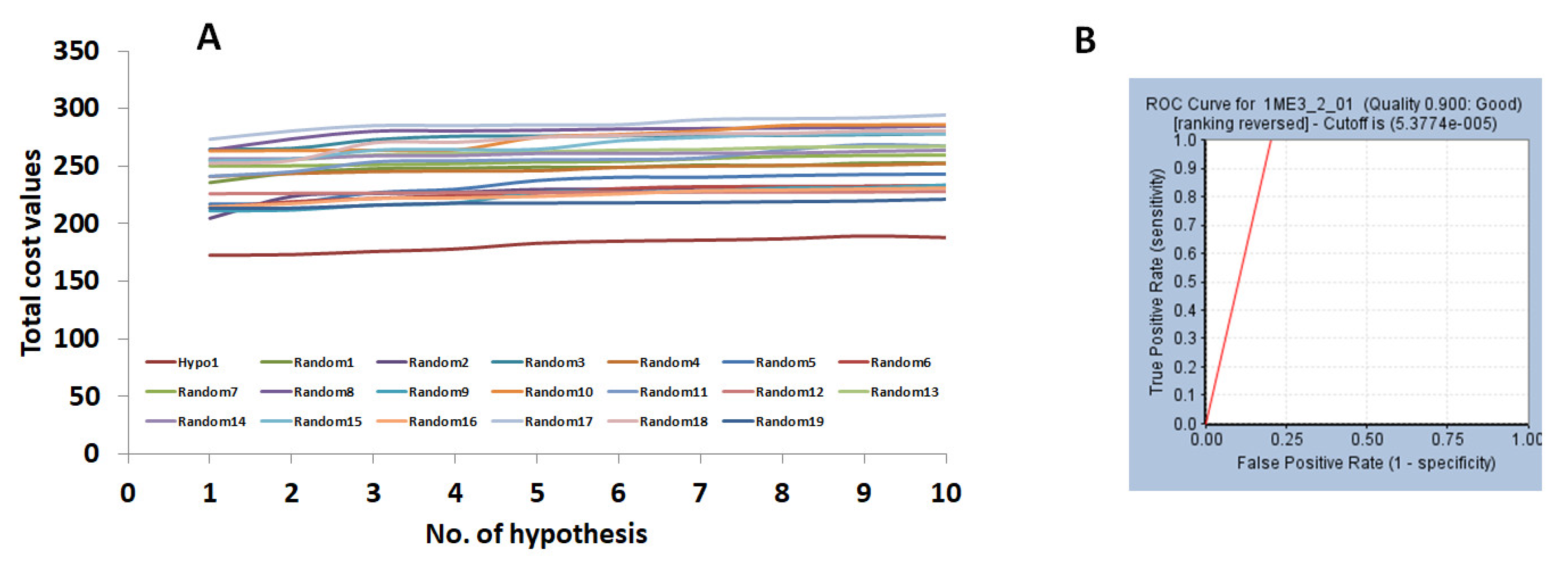
2.1. Generation of the Pharmacophore Model
2.1.1. Ligand-Based Pharmacophore Model Generation (Pharm 1)
2.1.2. Receptor-Based Pharmacophore Model Generation (Pharm 2)
2.2. Validation of the Pharmacophore Model
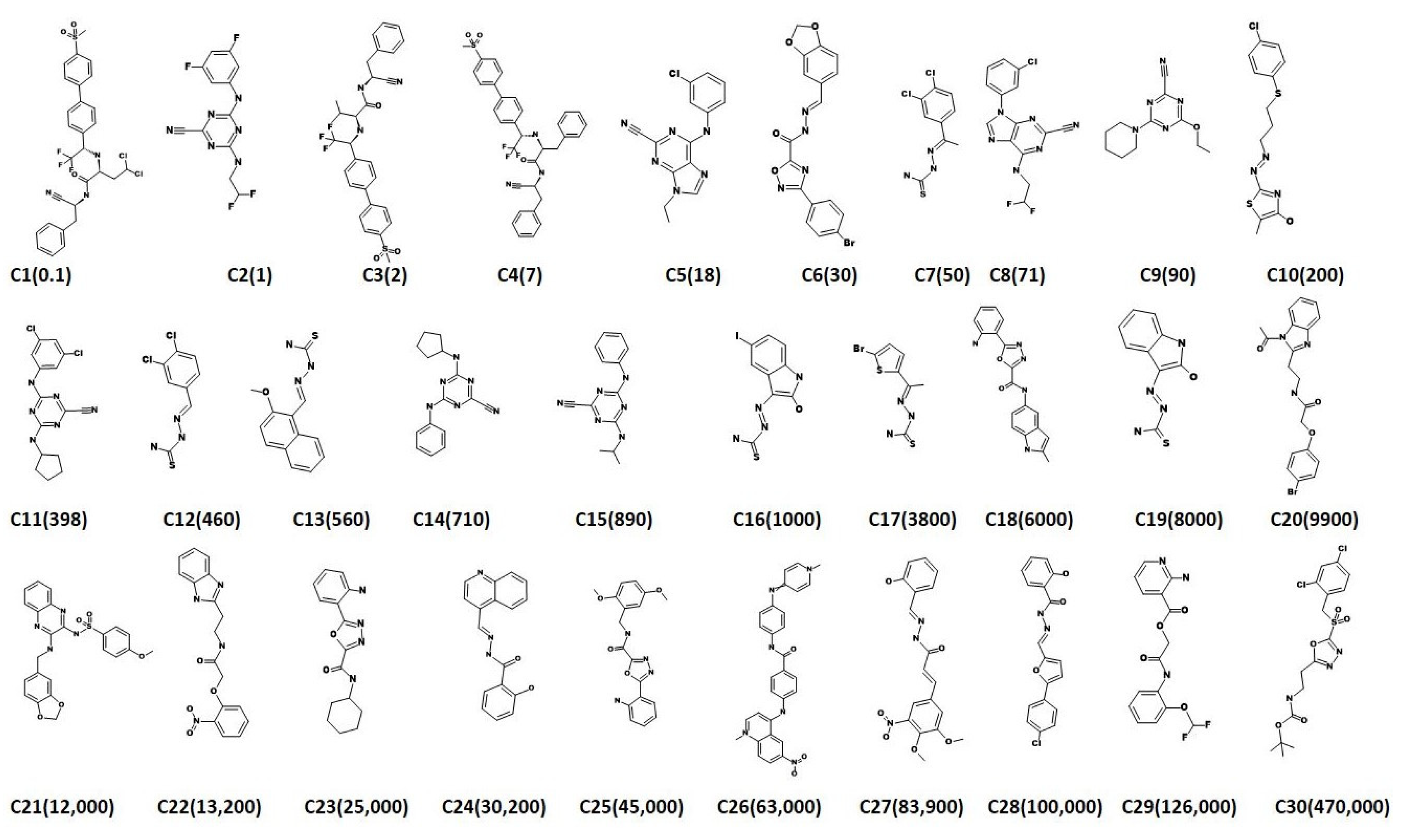
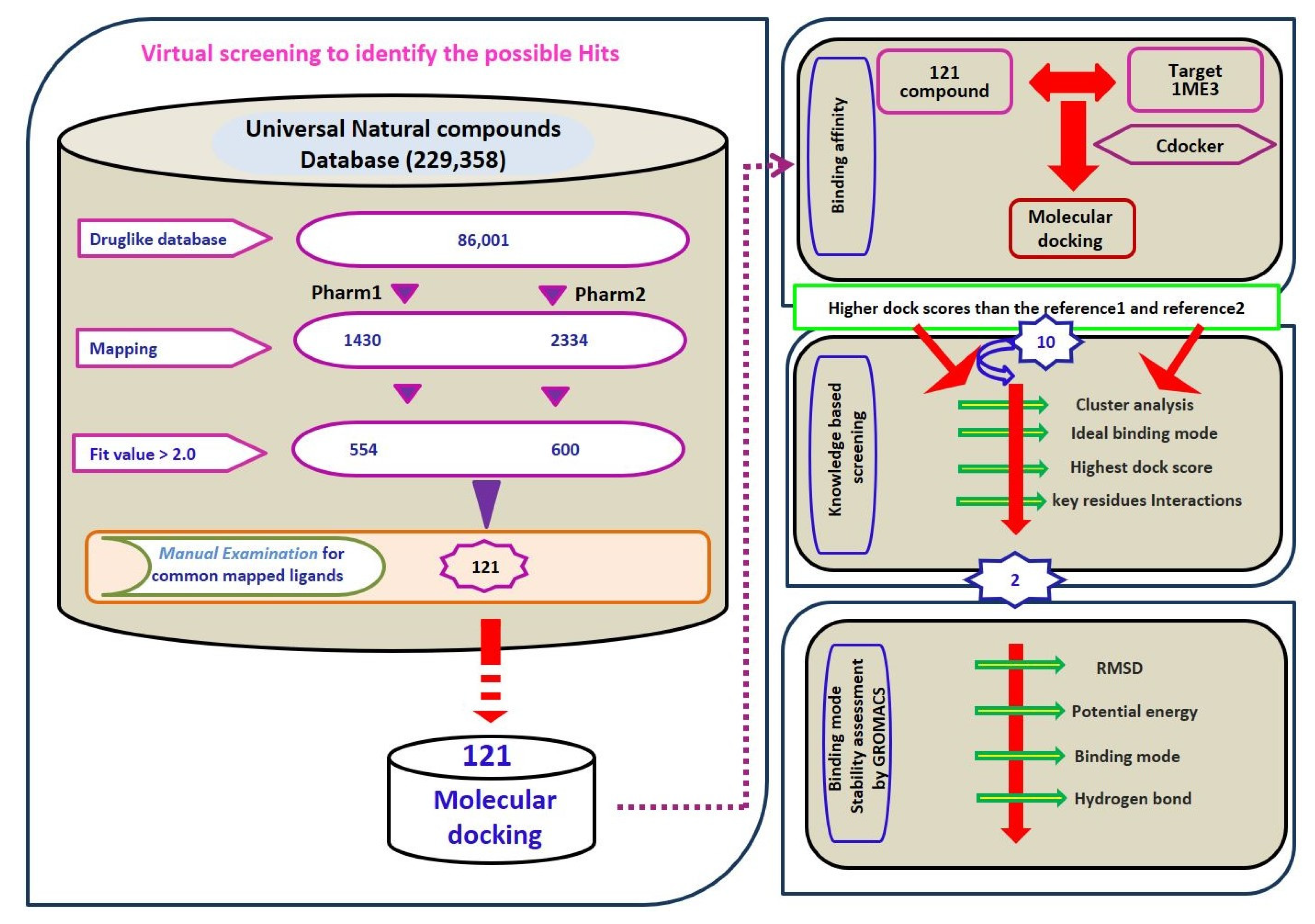
2.3. Virtual Screening for Redeeming the Lead-Like Candidates
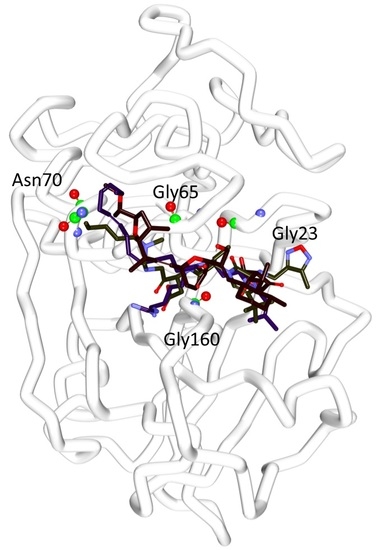
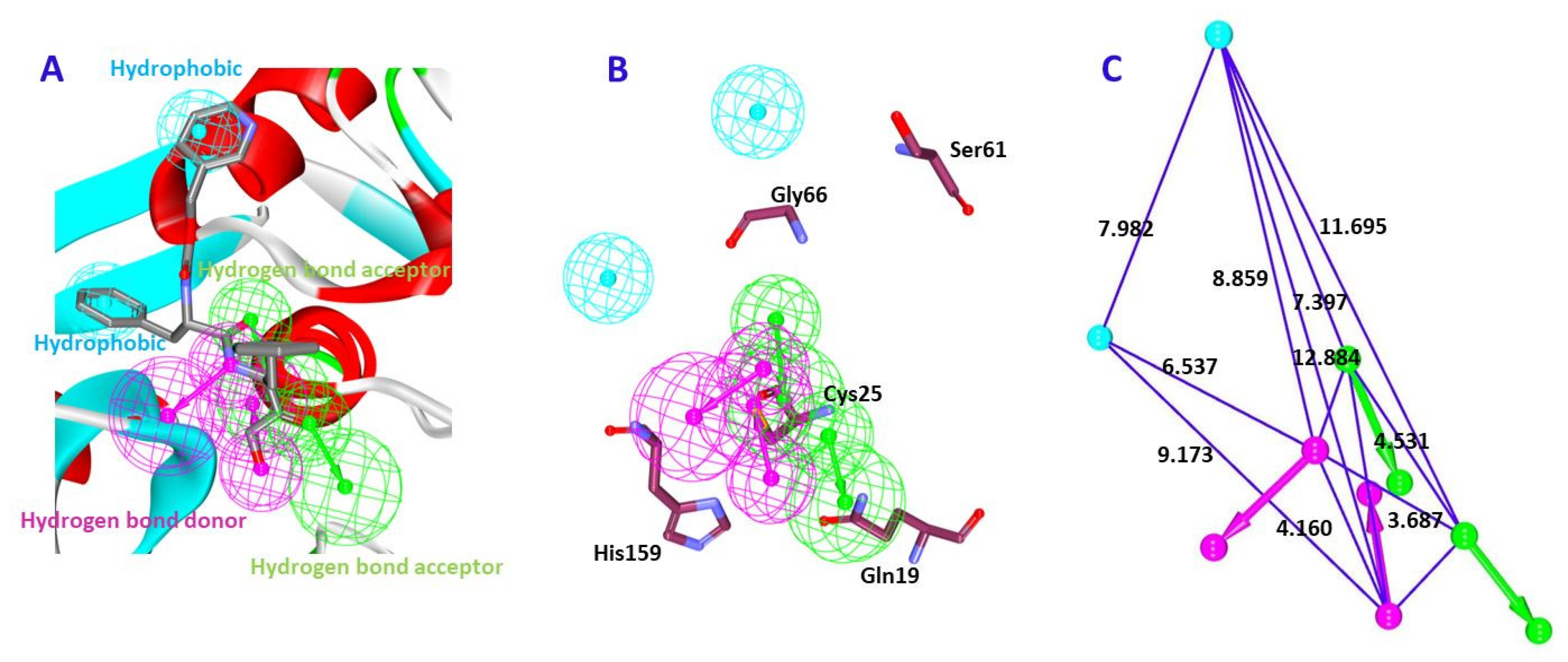
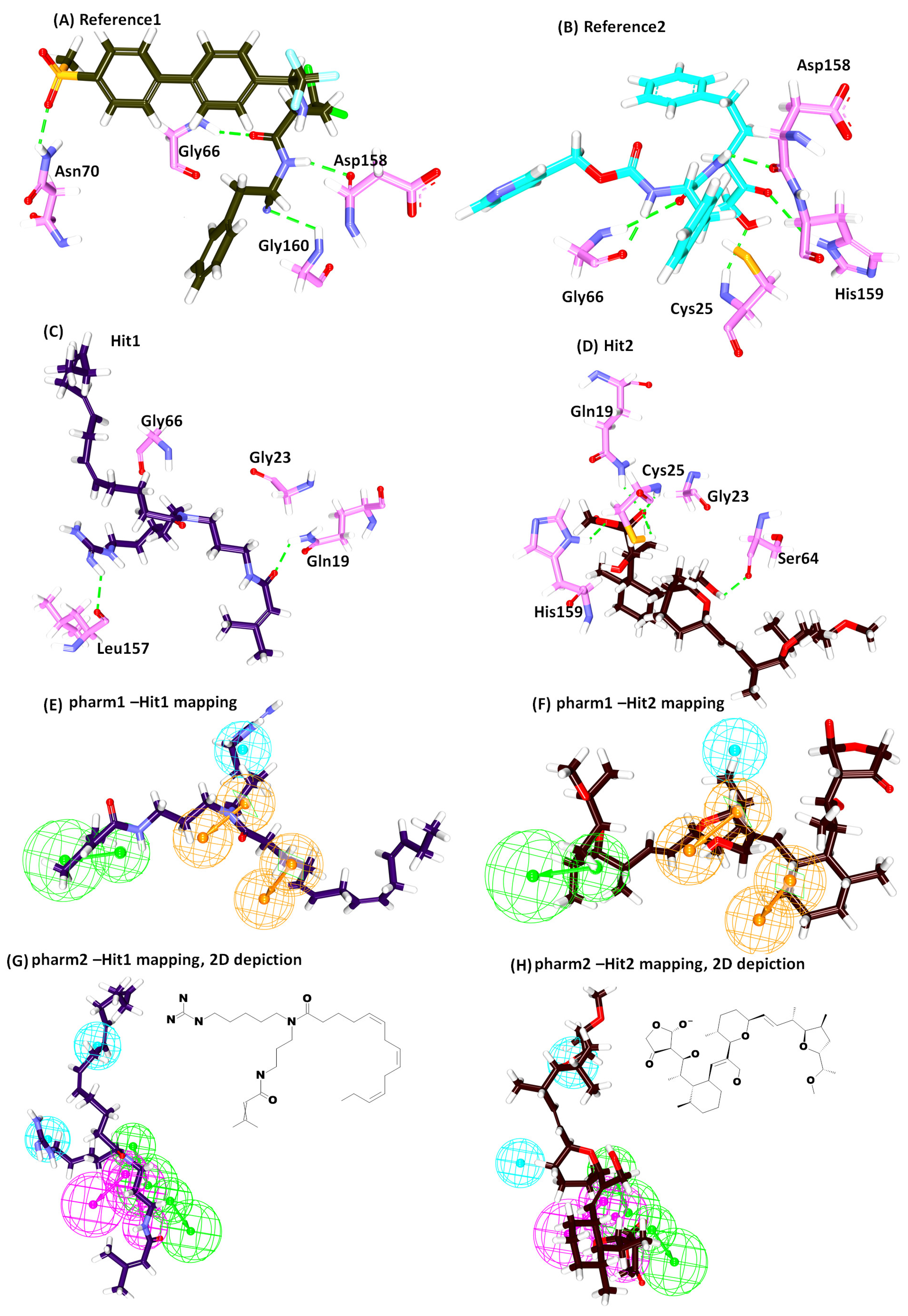
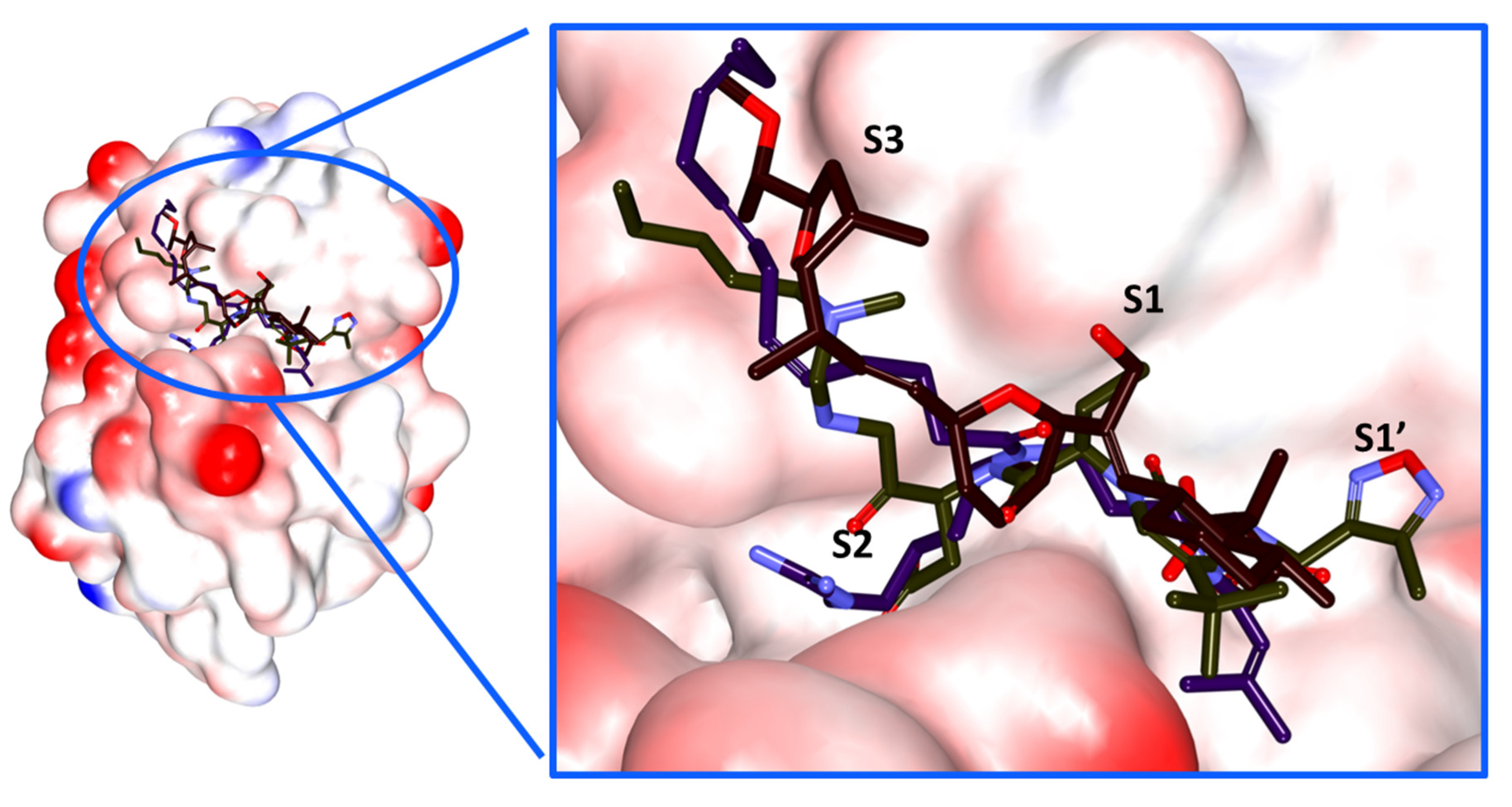
2.4. Molecular Docking Studies
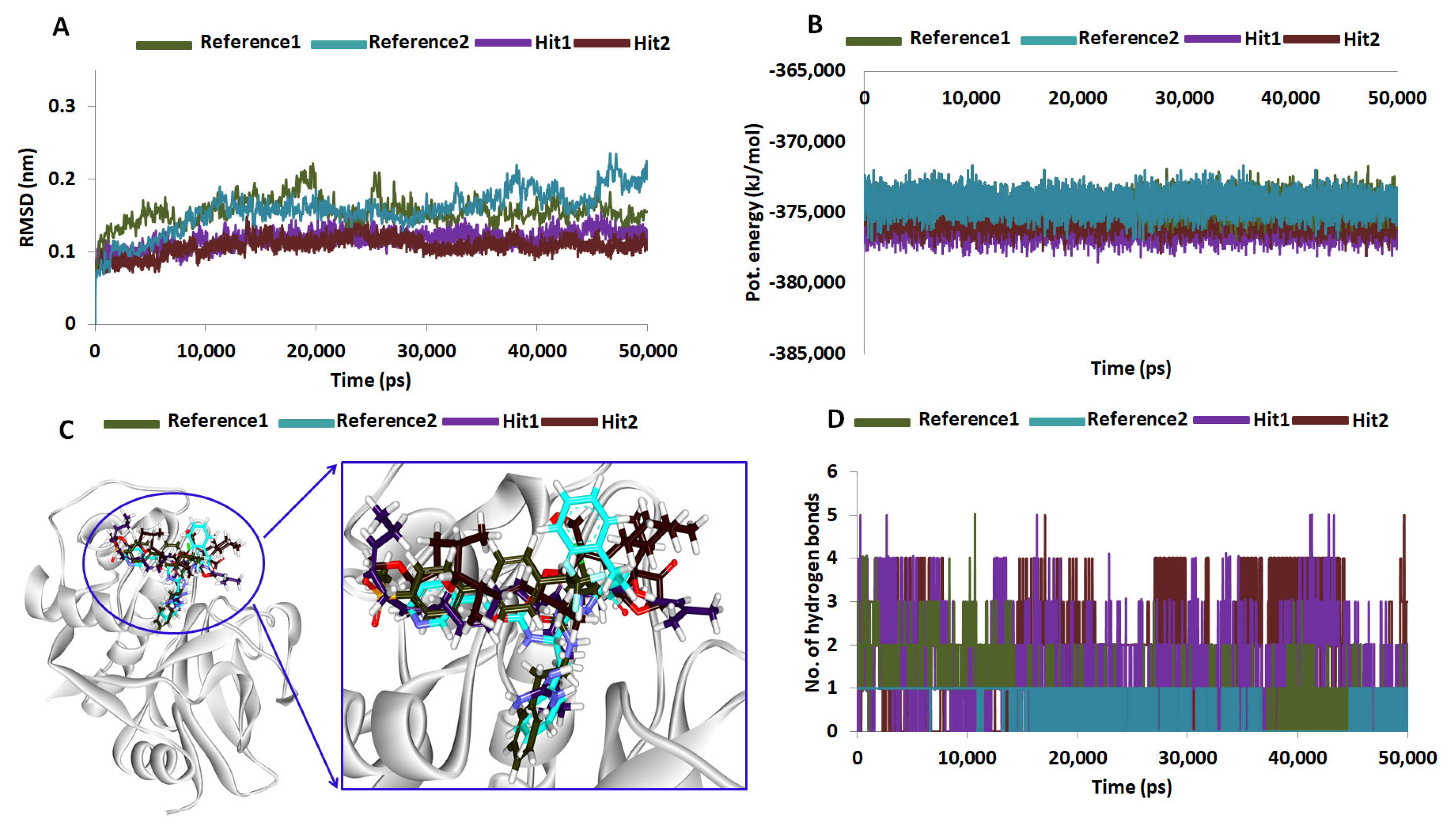
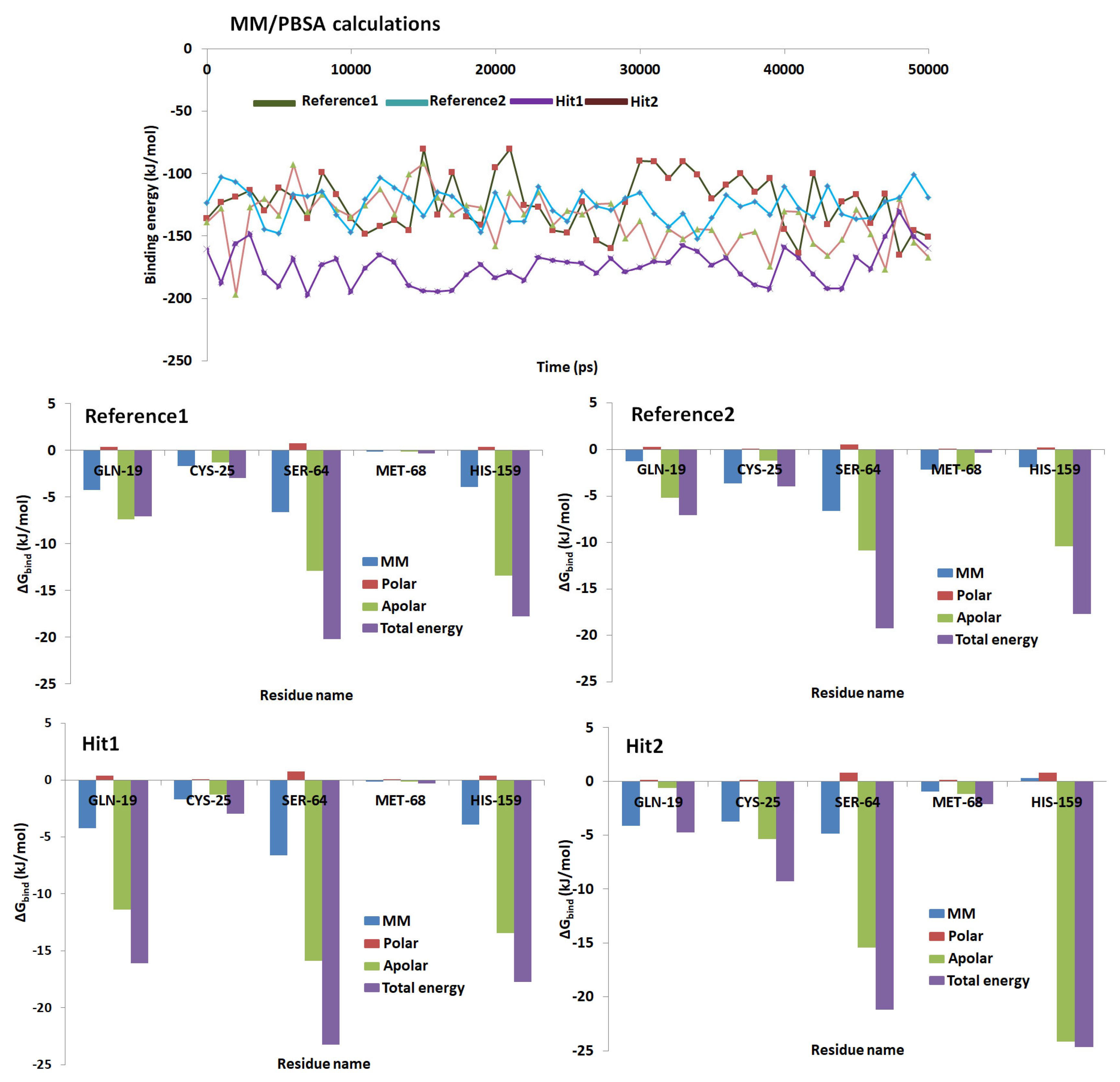
2.5. Molecular Dynamics Simulations
2.6. Binding Free Energy Calculations
3. Discussion
4. Materials and Methods
4.1. Preparation of the Dataset
4.2. Generation of the Pharmacophore Model
4.2.1. Ligand-Based Pharmacophore Model Generation (Pharm 1)
4.2.2. Receptor-Based Pharmacophore Model Generation (Pharm 2)
4.3. Validation of the Generated Pharmacophore
4.4. Virtual Database Screening and Drug Like Assessment
4.5. Molecular Docking Studies
4.6. Molecular Dynamics Simulation Studies
4.7. Binding Free Energy Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rassi, A.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Salas-Sarduy, E.; Landaburu, L.U.; Karpiak, J.X.; Madauss, K.P.; Cazzulo, J.J.; Agüero, F.; Alvarez, V.E. Novel scaffolds for inhibition of Cruzipain identified from high-throughput screening of anti-kinetoplastid chemical boxes. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; Keränen, H.; Wilson, B.A.; McCammon, J.A. Computational identification of uncharacterized cruzain binding sites. PLoS Negl. Trop. Dis. 2010, 4. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.E.; Keränen, H.; Durrant, J.D.; Ratnam, J.; Doak, A.; Arkin, M.R.; Mccammon, J.A. Novel Cruzain Inhibitors for the Treatment of Chagas’ Disease. Chem. Biol. Drug Des. 2012, 80, 398–405. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H.; Caffrey, C.; Kelly, B.; Loke, P.; Sajid, M. Proteases in parasitic diseases. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 497–536. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Guo, C.; Hansell, E.; Doyle, P.S.; Caffrey, C.R.; Holler, T.P.; McKerrow, J.H.; Cohen, F.E. Synthesis and structure-activity relationship study of potent trypanocidal thio semicarbazone inhibitors of the trypanosomal cysteine protease cruzain. J. Med. Chem. 2002, 45, 2695–2707. [Google Scholar] [CrossRef] [PubMed]
- Branquinha, M.H.; Oliveira, S.S.C.; Sangenito, L.S.; Sodre, C.L.; Kneipp, L.F.; d’Avila-Levy, C.M.; Santos, A.L.S. Cruzipain: An Update on its Potential as Chemotherapy Target against the Human Pathogen Trypanosoma cruzi. Curr. Med. Chem. 2015. [Google Scholar] [CrossRef]
- Pan, A.A.; McMahon-Pratt, D. Amastigote and epimastigote stage-specific components of Trypanosoma cruzi characterized by using monoclonal antibodies. Purification and molecular characterization of an 83-kilodalton amastigote protein. J. Immunol. 1989, 143, 1001–1008. [Google Scholar] [PubMed]
- Li, Y.; Shah-Simpson, S.; Okrah, K.; Belew, A.T.; Choi, J.; Caradonna, K.L.; Padmanabhan, P.; Ndegwa, D.M.; Temanni, M.R.; Corrada Bravo, H.; et al. Transcriptome Remodeling in Trypanosoma cruzi and Human Cells during Intracellular Infection. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.F.; Capettini, L.S.A.; da Silva, J.F.P.; Sales-Junior, P.; Cruz, J.S.; Cortes, S.F.; Lemos, V.S. Mechanisms of vascular dysfunction in acute phase of Trypanosoma cruzi infection in mice. Vascul. Pharmacol. 2016, 82, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Wiggers, H.J.; Rocha, J.R.; Fernandes, W.B.; Sesti-Costa, R.; Carneiro, Z.A.; Cheleski, J.; da Silva, A.B.F.; Juliano, L.; Cezari, M.H.S.; Silva, J.S.; et al. Non-peptidic Cruzain Inhibitors with Trypanocidal Activity Discovered by Virtual Screening and In Vitro Assay. PLoS Negl. Trop. Dis. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Machado, F.S.; Dutra, W.O.; Esper, L.; Gollob, K.J.; Teixeira, M.M.; Factor, S.M.; Weiss, L.M.; Nagajyothi, F.; Tanowitz, H.B.; Garg, N.J. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin. Immunopathol. 2012, 34, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Andrade, Z.A.; Andrade, S.G.; Correa, R.; Sadigursky, M.; Ferrans, V.J. Myocardial changes in acute Trypanosoma cruzi infection. Ultrastructural evidence of immune damage and the role of microangiopathy. Am. J. Pathol. 1994, 144, 1403–1411. [Google Scholar] [PubMed]
- Sharma, J.; Eickhoff, C.S.; Hoft, D.F.; Marentette, J.O.; Turk, J.; McHowat, J. Absence of calcium-independent phospholipase A2β impairs platelet-activating factor production and inflammatory cell recruitment in Trypanosoma cruzi-infected endothelial cells. Physiol. Rep. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Coura, J.R.; De Castro, S.L. A critical review on chagas disease chemotherapy. Mem. Inst. Oswaldo Cruz 2002, 97, 3–24. [Google Scholar] [CrossRef]
- de Castro, S.L. The challenge of Chagas’ disease chemotherapy: An update of drugs assayed against Trypanosoma cruzi. Acta Trop. 1993, 53, 83–98. [Google Scholar] [CrossRef]
- Mckerrow, J.H.; Doyle, P.S.; Engel, J.C.; Podust, L.M.; Robertson, S.A.; Ferreira, R.; Saxton, T.; Arkin, M.; Kerr, I.D.; Brinen, L.S.; et al. Two approaches to discovering and developing new drugs for Chagas disease. Mem. Inst. Oswaldo Cruz 2009, 104, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.R.; Taylor, M.C.; Horn, D.; Kelly, J.M.; Cheeseman, I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 5022–5027. [Google Scholar] [CrossRef] [PubMed]
- Lauria-Pires, L.; Braga, M.S.; Vexenat, A.C.; Nitz, N.; Simões-Barbosa, A.; Tinoco, D.L.; Teixeira, A.R.L. Progressive chronic chagas heart disease ten years after treatment with anti-Trypanosoma cruzi nitroderivatives. Am. J. Trop. Med. Hyg. 2000, 63, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A. Specific chemotherapy of Chagas disease: Relevance, current limitations and new approaches. Acta Trop. 2010, 115, 55–68. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.E.; Eakin, A.E.; Engel, J.C.; McKerrow, J.H.; Craik, C.S.; Fletterick, R.J. The crystal structure of Cruzain: A therapeutic target for Chagas’ disease. J. Mol. Biol. 1995, 247, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Robertson, S.A.; Brinen, L.S.; McKerrow, J.H. Cruzain: The path from target validation to the clinic. Adv. Exp. Med. Biol. 2011, 712, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Duschak, V.G.; Couto, A.S. Cruzipain, the major cysteine protease of Trypanosoma cruzi: A sulfated glycoprotein antigen as relevant candidate for vaccine development and drug target. A review. Curr. Med. Chem. 2009, 16, 3174–3202. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.P.C.A.; Dos Reis, F.C.G.; Serveau, C.; Lalmanach, G.; Juliano, L.; Ménard, R.; Vernet, T.; Thomas, D.Y.; Storer, A.C.; Scharfstein, J. Cysteine protease isoforms from Trypanosoma cruzi, cruzipain 2 and cruzain, present different substrate preference and susceptibility to inhibitors. Mol. Biochem. Parasitol. 2001, 114, 41–52. [Google Scholar] [CrossRef]
- Freitas, P.G.; Castilho, T.E.; De Almeida, L.; Maciel-Rezende, C.M.; Costa, L.T.; Viegas, C.; Marques, M.J.; Dos Santos, M.H.; Da Silveira, N.J.F. An in silico study of benzophenone derivatives as potential non-competitive inhibitors of trypanosoma cruzi and Leishmania amazonensis cysteine proteinases. J. Braz. Chem. Soc. 2018, 29, 515–527. [Google Scholar] [CrossRef]
- Tomas, A.M.; Miles, M.A.; Kelly, J.M. Overexpression of cruzipain, the major cysteine proteinase of Trypanosoma cruzi, is associated with enhanced metacyclogenesis. Eur. J. Biochem. 1997, 244, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Doyle, P.S.; Zhou, Y.M.; Hsieh, I.; Greenbaum, D.C.; McKerrow, J.H.; Engel, J.C. The trypanosoma cruzi protease cruzain mediates immune evasion. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Polticelli, F.; Zaini, G.; Bolli, A.; Antonini, G.; Gradoni, L.; Ascenzi, P. Probing the cruzain S2 recognition subsite: A kinetic and binding energy calculation study. Biochemistry 2005, 44, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Harth, G.; Andrews, N.; Mills, A.A.; Engel, J.C.; Smith, R.; McKerrow, J.H. Peptide-fluoromethyl ketones arrest intracellular replication and intercellular transmission of Trypanosoma cruzi. Mol. Biochem. Parasitol. 1993, 58, 17–24. [Google Scholar] [CrossRef]
- Ascenzi, P.; Salvati, L.; Bolognesi, M.; Colasanti, M.; Polticelli, F.; Venturini, G. Inhibition of cysteine protease activity by NO-donors. Curr. Protein Pept. Sci. 2001, 2, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Cazzulo, J.J. Proteinases of Trypanosoma cruzi: Patential targets for the chemotherapy of Changas desease. Curr. Top. Med. Chem. 2002, 2, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Barr, S.C.; Warner, K.L.; Kornreic, B.G.; Piscitelli, J.; Wolfe, A.; Benet, L.; Mckerrow, J.H. A Cysteine Protease Inhibitor Protects Dogs from Cardiac Damage during Infection by Trypanosoma cruzi A Cysteine Protease Inhibitor Protects Dogs from Cardiac Damage during Infection by Trypanosoma cruzi. Antimicrob. Agents Chemother. 2005, 49, 5160–5162. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H.; Rosenthal, P.J.; Swenerton, R.; Doyle, P. Development of protease inhibitors for protozoan infections. Curr. Opin. Infect. Dis. 2008, 21, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Turk, D.; Gunčar, G.; Podobnik, M.; Turk, B. Revised Definition of Substrate Binding Sites of Papain-Like Cysteine Proteases. Biol. Chem. 1998, 379. [Google Scholar] [CrossRef]
- Sakkiah, S.; Lee, K.W. Pharmacophore-based virtual screening and density functional theory approach to identifying novel butyrylcholinesterase inhibitors. Acta Pharmacol. Sin. 2012, 33, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.Z.; Xu, X. Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Brinen, L.S.; Hansell, E.; Cheng, J.; Roush, W.R.; McKerrow, J.H.; Fletterick, R.J. A target within the target: Probing cruzain’s P1’ site to define structural determinants for the Chagas’ disease protease. Structure 2000, 8, 831–840. [Google Scholar] [CrossRef]
- Huang, L.; Brinen, L.S.; Ellman, J.A. Crystal structures of reversible ketone-Based inhibitors of the cysteine protease cruzain. Bioorgan. Med. Chem. 2003, 11, 21–29. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wei, H.-Y.; Zhang, Y.-Z.; Jin, W.-Y.; Li, H.-L.; Zhou, H.; Cheng, X.-C.; Wang, R.-L. Synthesis, bioactivity, 3D-QSAR studies of novel dibenzofuran derivatives as PTP-MEG2 inhibitors. Oncotarget 2017, 8, 38466–38481. [Google Scholar] [CrossRef] [PubMed]
- Kandakatla, N.; Ramakrishnan, G. Ligand Based Pharmacophore Modeling and Virtual Screening Studies to Design Novel HDAC2 Inhibitors. Adv. Bioinform. 2014, 2014, 812148. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Baek, A.; Son, M.; Zeb, A.; Park, C.; Kumar, R.; Lee, G.; Kim, D.; Choi, Y.; Cho, Y.; et al. Computational Exploration for Lead Compounds That Can Reverse the Nuclear Morphology in Progeria. Biomed. Res. Int. 2017, 2017, 5270940. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Rampogu Lemuel, M. Network Based Approach in the Establishment of the Relationship between Type 2 Diabetes Mellitus and Its Complications at the Molecular Level Coupled with Molecular Docking Mechanism. Biomed. Res. Int. 2016, 2016, 6068437. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Baek, A.; Zeb, A.; Lee, K.W. Exploration for novel inhibitors showing back-to-front approach against VEGFR-2 kinase domain (4AG8) employing molecular docking mechanism and molecular dynamics simulations. BMC Cancer 2018, 18, 264. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Park, C.; Son, M.; Baek, A.; Zeb, A.; Lee, G.; Lee, K.W. Modulation of aromatase by natural compounds—A pharmacophore guided molecular modelling simulations. S. Afr. J. Bot. 2018. [Google Scholar] [CrossRef]
- Rampogu, S.; Baek, A.; Gajula, R.G.; Zeb, A.; Bavi, R.S.; Kumar, R.; Kim, Y.; Kwon, Y.J.; Lee, K.W. Ginger (Zingiber officinale) phytochemicals—Gingerenone-A and shogaol inhibit SaHPPK: Molecular docking, molecular dynamics simulations and in vitro approaches. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lopes, P.E.M.; Mackerell, A.D. Recent developments and applications of the CHARMM force fields. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for smallorganic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Senior, K. Chagas disease: Moving towards global elimination. Lancet Infect. Dis. 2007, 7, 572. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypo No | Total Cost | Cost Difference a | RMSD | Correlation | Features b | Maximum Fit |
|---|---|---|---|---|---|---|
| Hypo 1 | 214.15 | 66.93 | 2.42 | 0.71 | HBL, HyP, RA, RA | 8.24 |
| Hypo 2 | 216.71 | 66.82 | 2.45 | 0.70 | HBL, HyP, RA, RA | 7.79 |
| Hypo 3 | 216.72 | 63.81 | 2.45 | 0.70 | HBL, HyP, RA, RA | 7.92 |
| Hypo 4 | 218.92 | 61.61 | 2.48 | 0.66 | HBL, HyP, RA, RA | 7.23 |
| Hypo 5 | 219.39 | 61.14 | 2.49 | 0.59 | HBL, HyP, RA, RA | 7.97 |
| Hypo 6 | 220.94 | 59.59 | 2.51 | 0.66 | HyP, RA, RA, RA | 7.69 |
| Hypo 7 | 221.84 | 58.70 | 2.52 | 0.68 | HyP, RA, RA, RA | 7.70 |
| Hypo 8 | 222.14 | 58.39 | 2.53 | 0.68 | HyP, RA, RA, RA | 7.62 |
| Hypo 9 | 222.55 | 57.98 | 2.53 | 0.68 | HyP, RA, RA, RA | 7.01 |
| Hypo 10 | 224.35 | 56.16 | 2.55 | 0.67 | HBL, HyP, RA, RA | 7.92 |
| Pharmacophore Model | Number of Features | Feature Set * | Selectivity Score |
|---|---|---|---|
| Model1 | 6 | HyP, HyP, HBD, HBD, HBA, HBA | 11.63 |
| Model2 | 6 | HyP, HBD, HBA, HBA, HBA, HBA | 10.72 |
| Model3 | 6 | HyP, HBD, HBA, HBA, HBA, HBA | 10.72 |
| Model4 | 6 | HyP, HyP, HBD, HBD, HBA, HBA | 10.72 |
| Model5 | 6 | HyP, HyP, HBD, HBD, HBA, HBA | 10.72 |
| Model6 | 6 | HyP, HyP, HBD, HBD, HBA, HBA | 10.72 |
| Model7 | 6 | HyP, HyP, HBD, HBD, HBA, HBA | 10.72 |
| Model8 | 6 | HyP, HyP, HBD, HBD, HBA, HBA | 10.72 |
| Model9 | 5 | HyP, HyP, HBD, HBD, HBA | 10.12 |
| Model10 | 5 | HyP, HBD, HBD, HBA, HBA | 10.12 |
| Parameters | Pharm 1 | Pharm 2 |
|---|---|---|
| Total number of molecules in database (D) | 1000 | 1000 |
| Total number of actives in database (A) | 25 | 25 |
| Total number of hit molecules from the database (Ht) | 27 | 31 |
| Total number of active molecules in hit list (Ha) | 22 | 24 |
| % Yield of active (Ha/Ht) | 81.4 | 77.4 |
| Enrichment Factor (EF) | 32.59 | 30.96 |
| False negatives (A − Ha) | 3 | 1 |
| False positives (Ht – Ha) | 5 | 7 |
| Goodness of fit score (GF) | 0.80 | 0.73 |
| Name | -Cdocker Interaction Energy (kcal/mol) | Hydrogen Bond (<3 Å) | Alkyl/π-alkyl | van der Waals Interactions |
|---|---|---|---|---|
| * Ref1 | 47.79 | Gly66:NH-O23 (2.2) Asn70:HD22-O16 (2.1) Asp158:O-H56(2.0) Gly160:HN-N26 (2.7) | Leu67, Ala133 | Gln19, Gly23, Trp26, Cys25, Thr59, Ser61, Ser64, Asp60, Gly65, Met68, Asn69,Leu157, Leu159, Glu205 |
| * Ref2 | 50.69 | Cys25:HN-O3 (2.3) Gly66:HN-O16 (2.8) Gly66:HN-H58 (1.9) Asp158:O-H49 (2.0) His159:HD1-O4 (2.7) | Leu67, Asp60, Ala133 | Gln19, Gly23, Ser24, Thr59, Ser61, Ser64, Met68 Asn70, Leu157, Gly160, Glu205, |
| Hit1 | 53.57 | Gln19:HE22-O20 (2.1) Leu157:O-H74 (2.3) | Ala136, His159, Trp177 | Gly23, Cys25, Trp26, Thr50, Asp60, Ser61, Ser64, Gly65, Gly66, Leu67, Met68, Asn70, Ala133, Val134, Ser139, Asp158, Gly160 |
| Hit2 | 51.84 | Gln19:HE22-O21 (2.0) Cys25:HN-O21 (2.0) Cys25:HG-O21 (2.3) Ser64:O-H78 (2.1) His159:HD1-O14 (2.1) | Cys25, Trp26, Leu67 | Gly23, Ser24, Thr59, Asn60, Ser61, Cys63, Gly65, Asn70, Asp158, Gly160, Trp177 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rampogu, S.; Lee, G.; Baek, A.; Son, M.; Park, C.; Zeb, A.; Yoon, S.H.; Park, S.; Lee, K.W. Discovery of Non-Peptidic Compounds against Chagas Disease Applying Pharmacophore Guided Molecular Modelling Approaches. Molecules 2018, 23, 3054. https://doi.org/10.3390/molecules23123054
Rampogu S, Lee G, Baek A, Son M, Park C, Zeb A, Yoon SH, Park S, Lee KW. Discovery of Non-Peptidic Compounds against Chagas Disease Applying Pharmacophore Guided Molecular Modelling Approaches. Molecules. 2018; 23(12):3054. https://doi.org/10.3390/molecules23123054
Chicago/Turabian StyleRampogu, Shailima, Gihwan Lee, Ayoung Baek, Minky Son, Chanin Park, Amir Zeb, Sang Hwa Yoon, Suhyeon Park, and Keun Woo Lee. 2018. "Discovery of Non-Peptidic Compounds against Chagas Disease Applying Pharmacophore Guided Molecular Modelling Approaches" Molecules 23, no. 12: 3054. https://doi.org/10.3390/molecules23123054
APA StyleRampogu, S., Lee, G., Baek, A., Son, M., Park, C., Zeb, A., Yoon, S. H., Park, S., & Lee, K. W. (2018). Discovery of Non-Peptidic Compounds against Chagas Disease Applying Pharmacophore Guided Molecular Modelling Approaches. Molecules, 23(12), 3054. https://doi.org/10.3390/molecules23123054