
Preparation of Enantiomeric β-(2′,5′-Dimethylphenyl)Bromolactones, Their Antiproliferative Activity and Effect on Biological Membranes

 ,
, 
 ,
,
Abstract
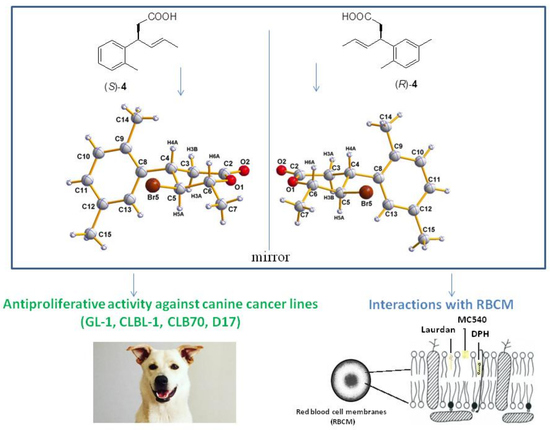
1. Introduction
2. Results and Discussion
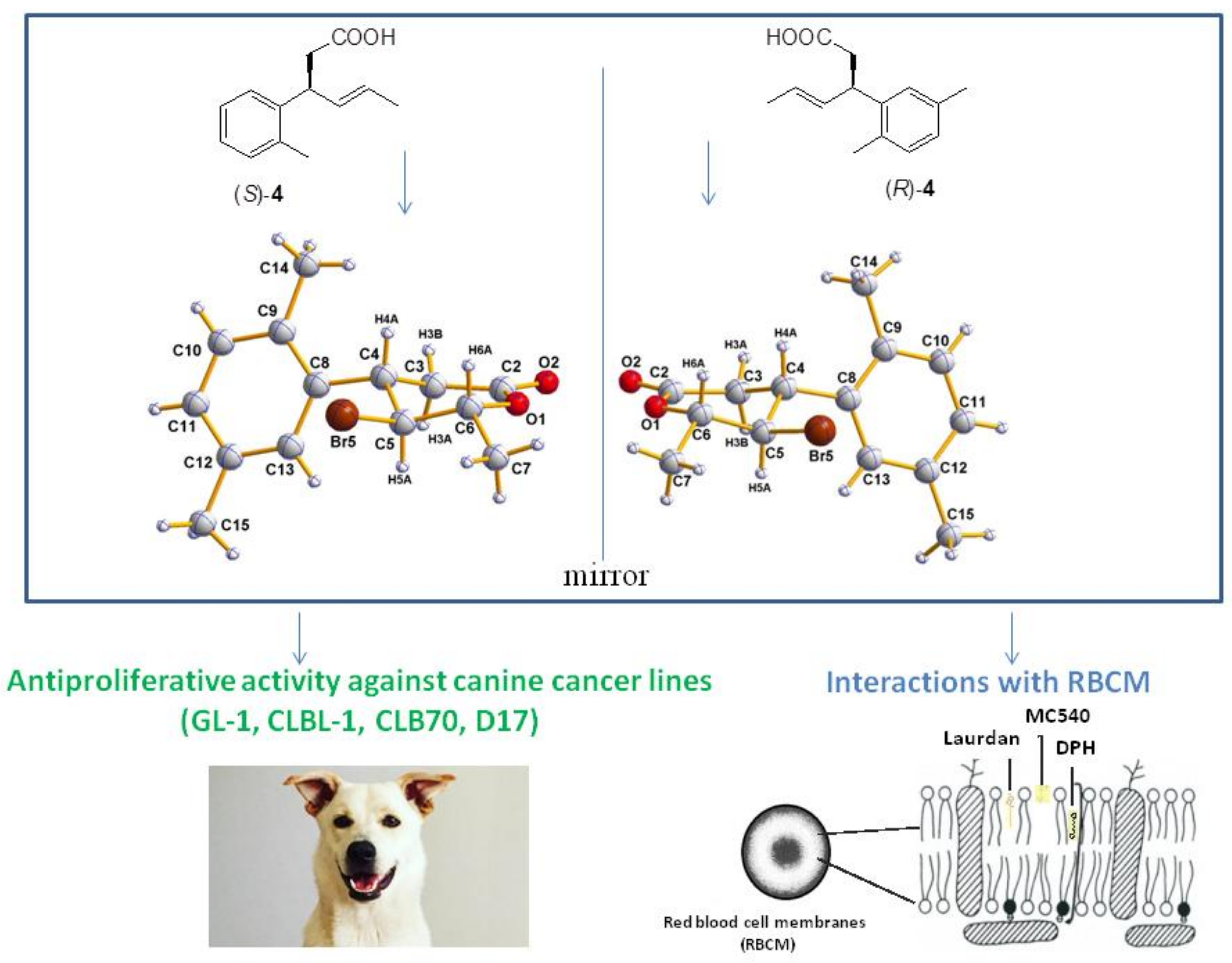
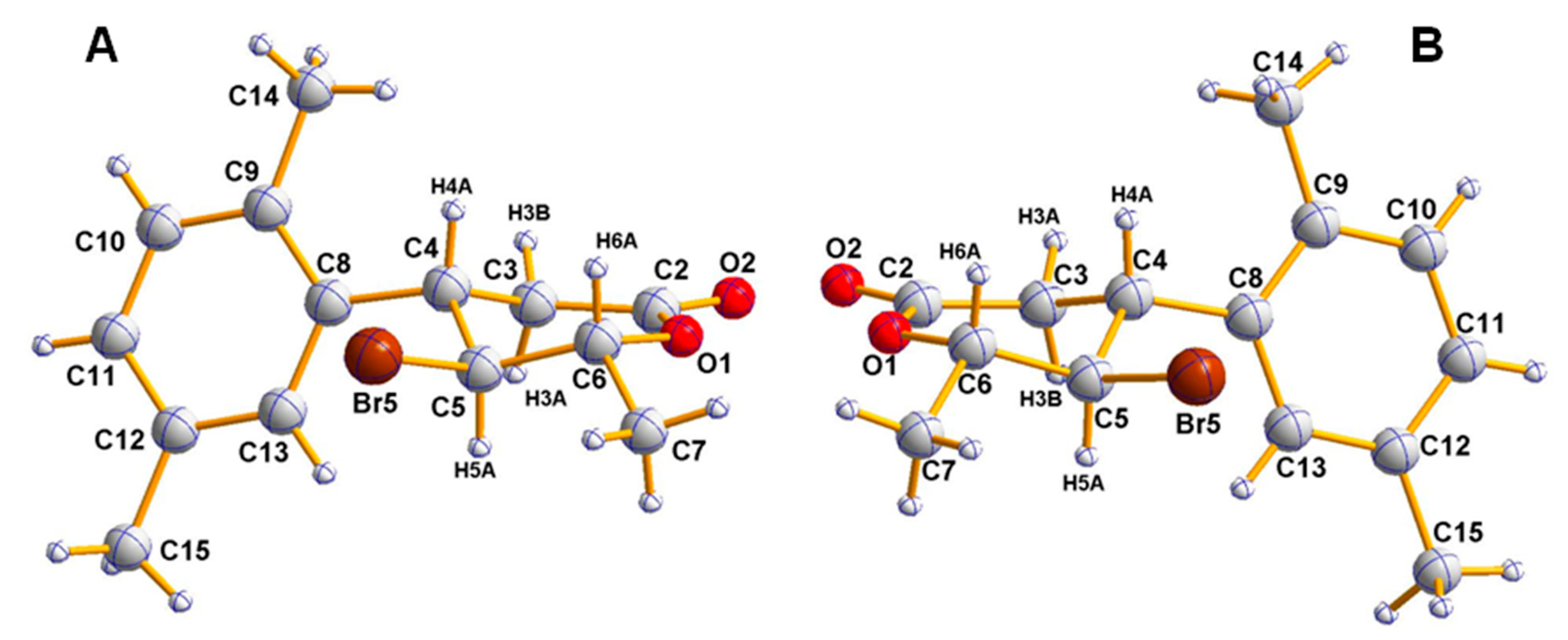
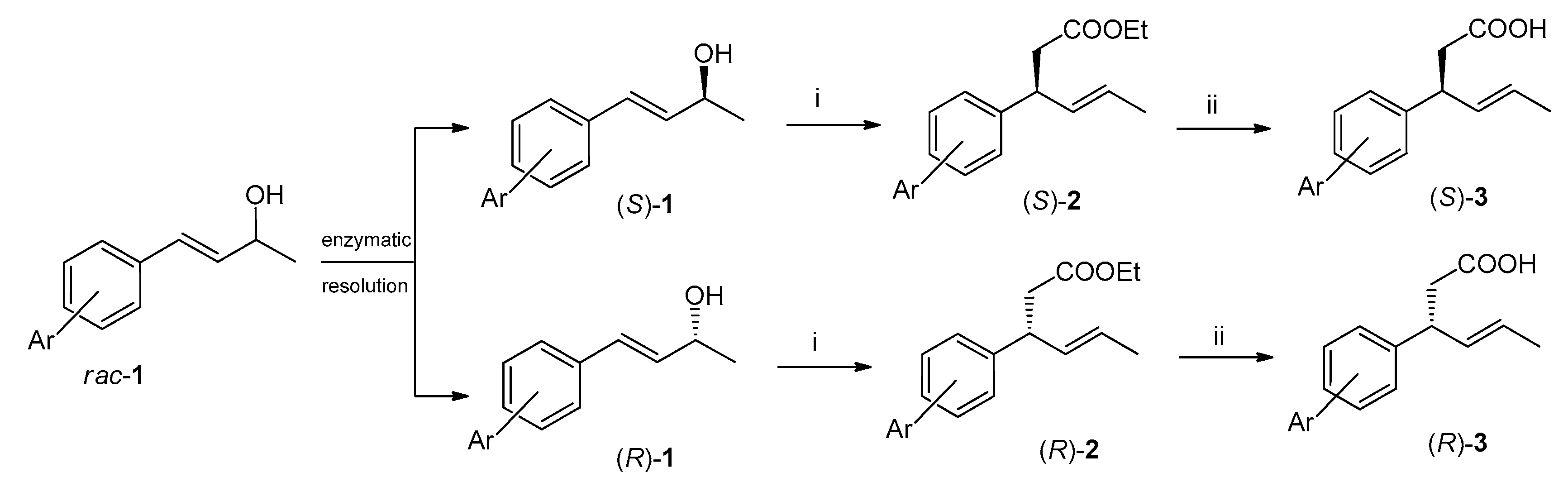
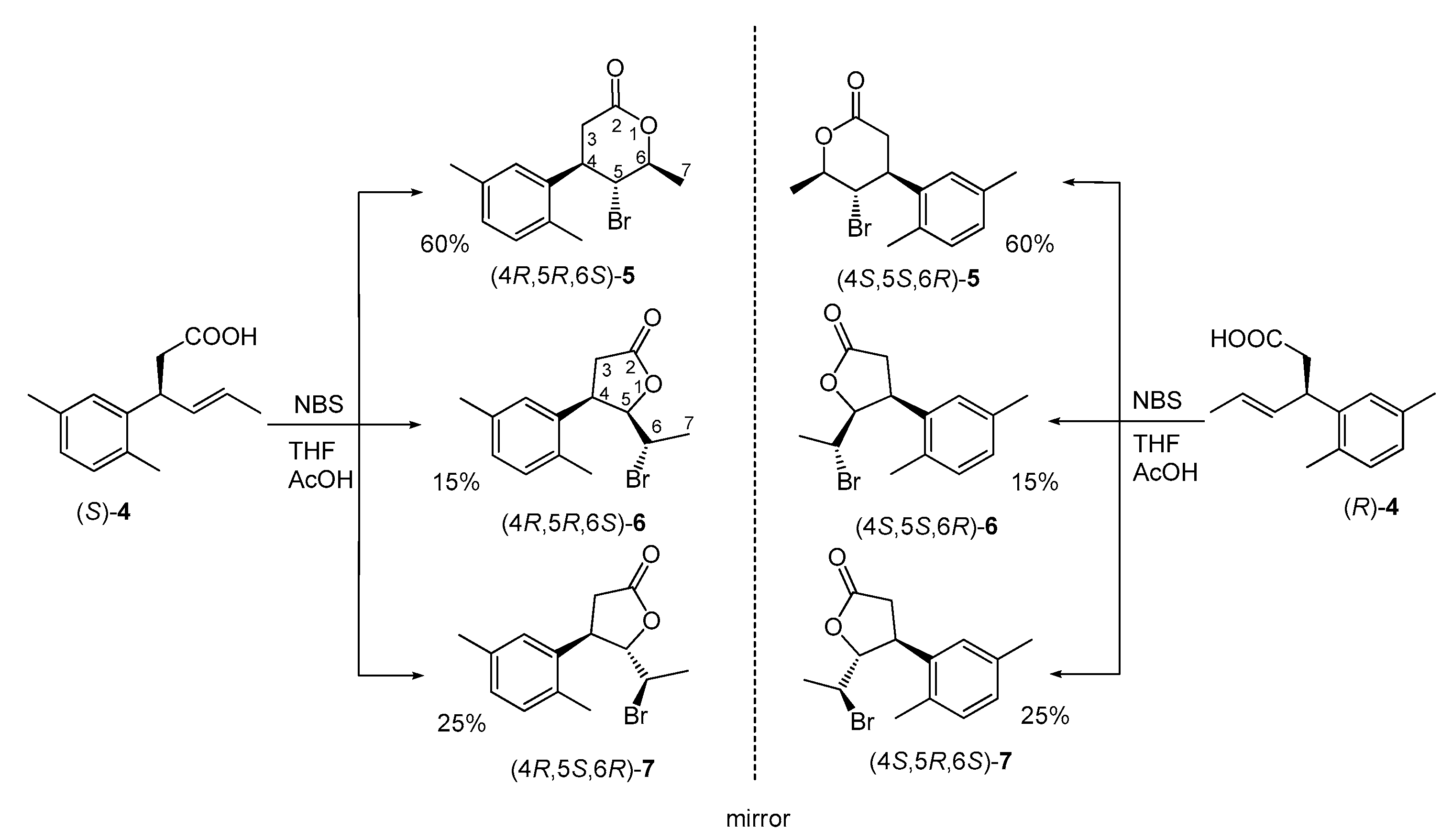
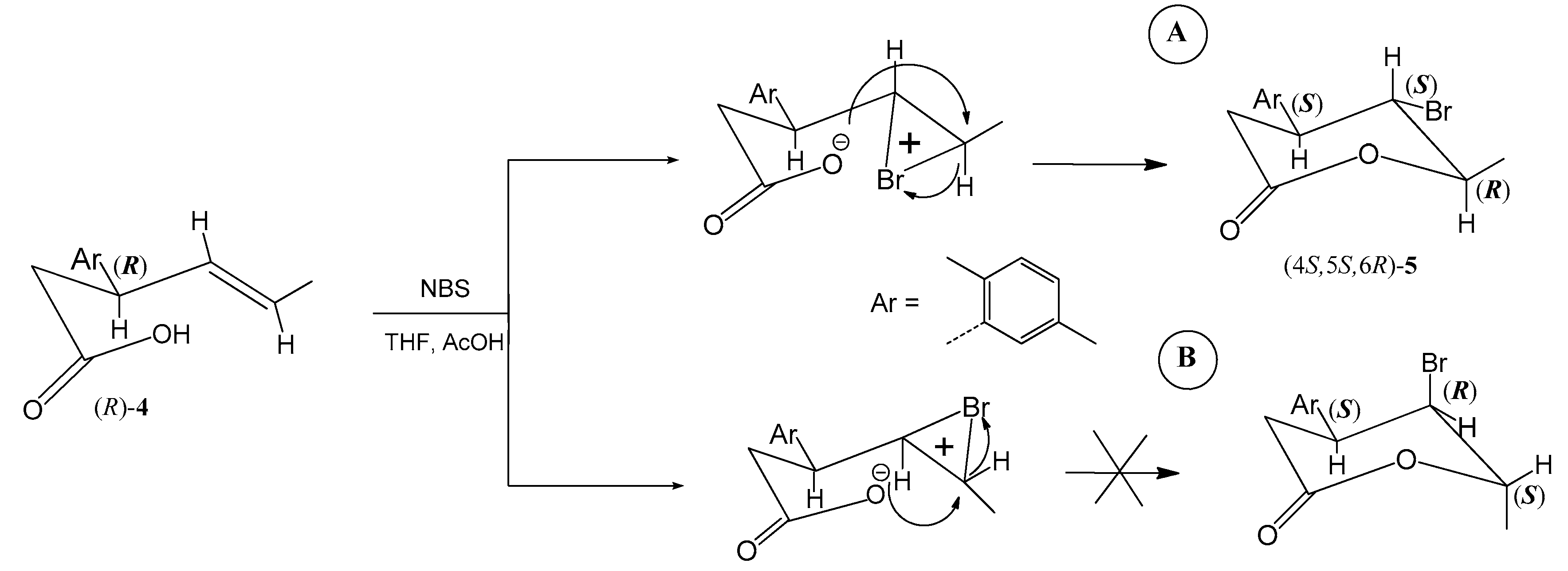
2.1. Synthesis of Enantiomeric Bromolactones (5–7)
2.2. Antiproliferative Activity
2.3. Hemolytic Activity
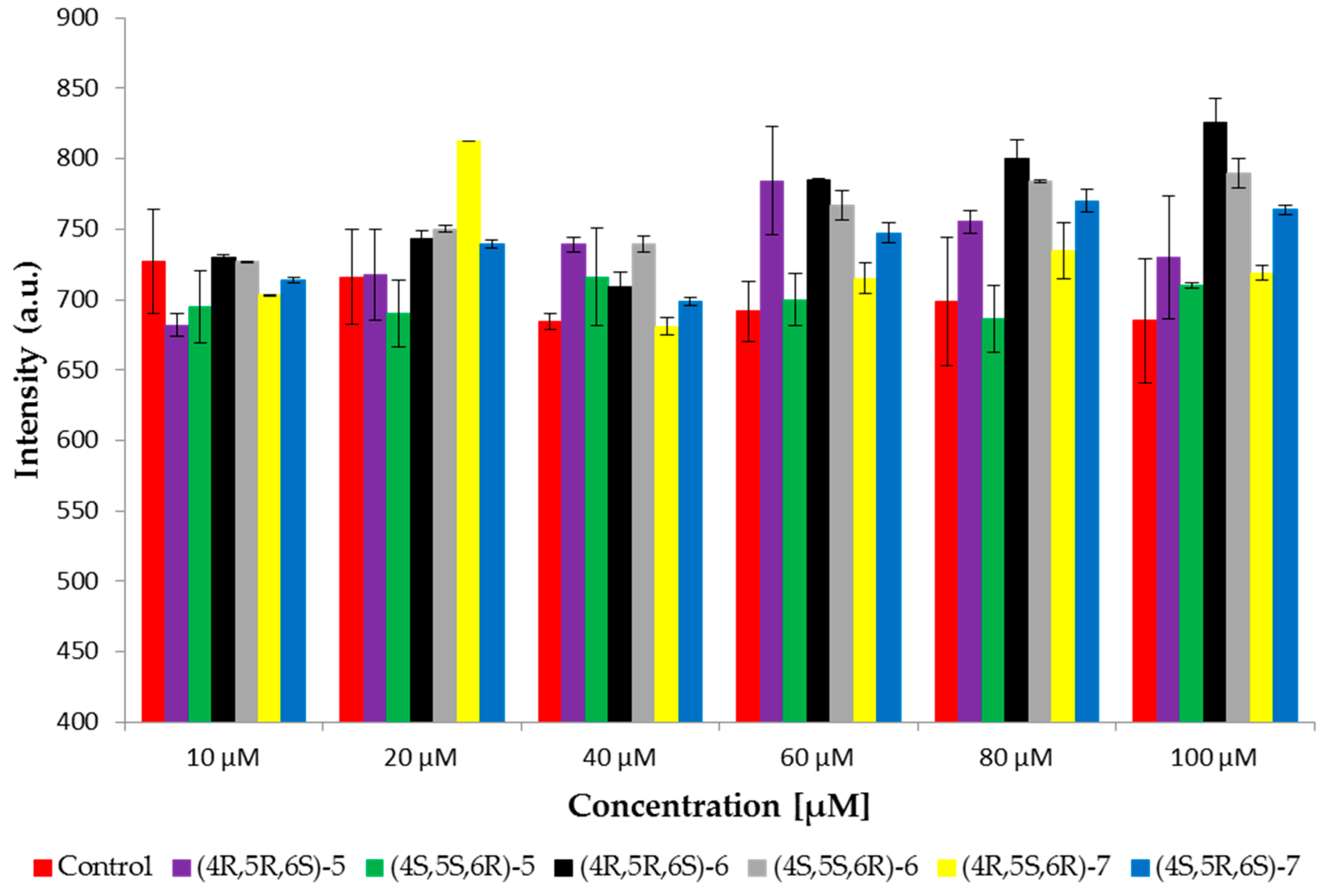
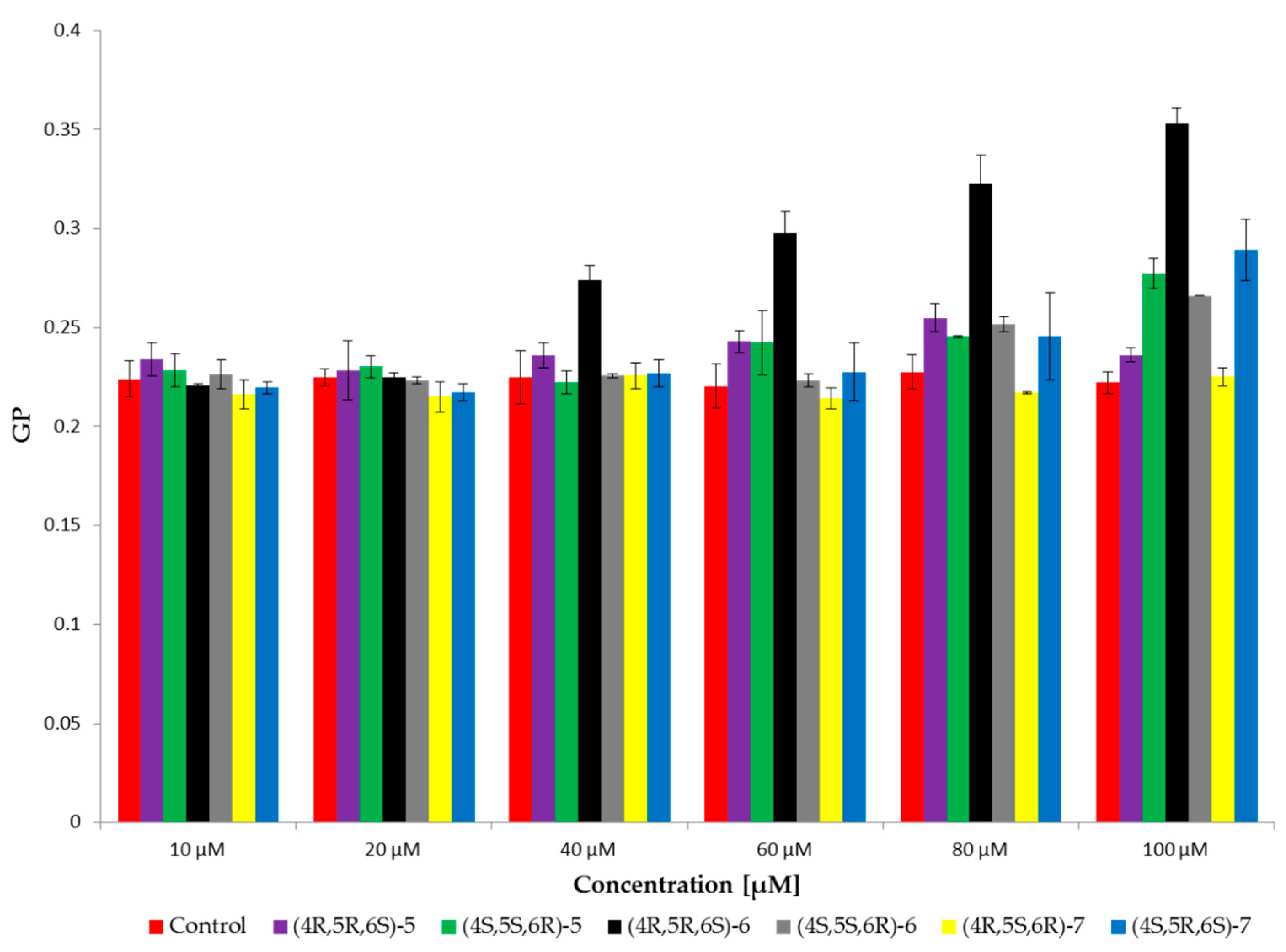
2.4. Fluorescence Spectroscopy
3. Materials and Methods
3.1. Chemicals
3.2. Analysis
3.3. Synthesis of Bromolactones 5–7—General Procedure
3.4. Cell Lines
3.5. Determination of Antiproliferative Activity
3.6. Hemolytic Activity
3.7. Fluorescence Spectroscopy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mirunalini, S.; Deepalakshmi, K.; Manimozhi, J. Antiproliferative effect of coumarin by modulating oxidant/antioxidant status and inducing apoptosis in Hep2 cells. Biomed. Aging Pathol. 2014, 4, 131–135. [Google Scholar] [CrossRef]
- Pizao, P.E.; Smitskamp-Wilms, E.; Van Ark-Otte, J.; Beijnen, J.H.; Peters, G.J.; Pinedo, H.M.; Giaccone, G. Antiproliferative activity of the topoisomerase I inhibitors topotecan and camptothecin, on sub- and postconfluent tumor cell cultures. Biochem. Pharmacol. 1994, 48, 1145–1154. [Google Scholar] [CrossRef]
- Benedeković, G.; Kovačević, I.; Popsavin, M.; Francuz, J.; Kojić, V.; Bogdanović, G.; Popsavin, V. New antitumour agents with α,β-unsaturated δ-lactone scaffold: Synthesis and antiproliferative activity of (−)-cleistenolide and analogs. Bioorganic Med. Chem. Lett. 2016, 26, 3318–3321. [Google Scholar] [CrossRef] [PubMed]
- Tuchinda, P.; Munyoo, B.; Pohmakotr, M.; Thinapong, P.; Sophasan, S.; Santisuk, T.; Reutrakul, V. Cytotoxic styryl-lactones from the leaves and twigs of Polyalthia crassa. J. Nat. Prod. 2006, 69, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Popsavin, V.; Benedeković, G.; Popsavin, M.; Kojić, V.; Bogdanović, G. Divergent synthesis of cytotoxic styryl lactones isolated from Polyalthia crassa. The first total synthesis of crassalactone B. Tetrahedron Lett. 2010, 51, 3426–3429. [Google Scholar] [CrossRef]
- Lan, Y.H.; Chang, F.R.; Yu, J.H.; Yang, Y.L.; Chang, Y.L.; Lee, S.J.; Wu, Y.C. Cytotoxic styrylpyrones from Goniothalamus amuyon. J. Nat. Prod. 2003, 66, 487–490. [Google Scholar] [CrossRef] [PubMed]
- De Fátima, Â.; Kohn, L.K.; De Carvalho, J.E.; Pilli, R.A. Cytotoxic activity of (S)-goniothalamin and analogs against human cancer cells. Bioorganic Med. Chem. 2006, 14, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Roman Junior, W.A.; Gomes, D.B.; Zanchet, B.; Schönell, A.P.; Diel, K.A.P.; Banzato, T.P.; Ruiz, A.L.T.G.; Carvalho, J.E.; Neppel, A.; Barison, A.; et al. Antiproliferative effects of pinostrobin and 5,6-dehydrokavain isolated from leaves of Alpinia zerumbet. Brazilian J. Pharmacogn. 2017, 27, 592–598. [Google Scholar] [CrossRef]
- Zhao, C.; Rakesh, K.P.; Mumtaz, S.; Moku, B.; Asiri, A.M.; Marwani, H.M.; Manukumar, H.M.; Qin, H.L. Arylnaphthalene lactone analogs: Synthesis and development as excellent biological candidates for future drug discovery. RSC Adv. 2018, 8, 9487–9502. [Google Scholar] [CrossRef]
- Chen, Y.L.; Lin, S.Z.; Chang, J.Y.; Cheng, Y.L.; Tsai, N.M.; Chen, S.P.; Chang, W.L.; Harn, H.J. In vitro and in vivo studies of a novel potential anticancer agent of isochaihulactone on human lung cancer A549 cells. Biochem. Pharmacol. 2006, 72, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Landete, J.M. Plant and mammalian lignans: A review of source, intake, metabolism, intestinal bacteria and health. Food Res. Int. 2012, 46, 410–424. [Google Scholar] [CrossRef]
- Bylund, A.; Sarrinen, N.; Zhang, J.; Bergh, A.; Widmark, A.; Johansson, A.; Lundin, E.; Adlercreutz, H. Anticancer effects of a plant lignan 7-hydroxymatairesinol on a prostate cancer model in vivo. Exp. Biol. Med. 2005, 230, 217–223. [Google Scholar] [CrossRef]
- Chen, L.-H.; Fang, J.; Li, H.; Demark-Wahnefried, W.; Lin, X. Enterolactone induces apoptosis in human prostate carcinoma LNCaP cells via a mitochondrial-mediated, caspase-dependent pathway. Mol. Cancer Ther. 2007, 6, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Q.; Huang, C.; Jia, Y.M.; Song, B.A.; Li, J.; Liu, X.H. Novel coumarin-dihydropyrazole thio-ethanone derivatives: Design, synthesis and anticancer activity. Eur. J. Med. Chem. 2014, 74, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Deredas, D.; Huben, K.; Janecka, A.; Długosz, A.; Pomorska, D.K.; Mirowski, M.; Krajewska, U.; Janecki, T.; Krawczyk, H. Synthesis and anticancer properties of 3-methylene-4-(2-oxoalkyl)-3,4-dihydrocoumarins. Medchemcomm 2016, 7, 1745–1758. [Google Scholar] [CrossRef]
- An, R.; Hou, Z.; Li, J.; Yu, H.; Mou, Y.; Guo, C. Design, synthesis and biological evaluation of novel 4-substituted coumarin derivatives as antitumor agents. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Y.; Zhang, Q.-Q.; Song, J.-L.; Zhang, L.; Jiang, C.-S.; Zhang, H. Design, synthesis, and antiproliferative evaluation of novel coumarin/2-cyanoacryloyl hybrids as apoptosis inducing agents by activation of caspase-dependent pathway. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, P.-Y.; Hsieh, K.-Y.; Hsu, P.-L.; Goto, M.; Morris-Natschke, S.L.; Harn, H.-J.; Lee, K.-H. Design, synthesis and structure–activity relationships of (±)-isochaihulactone derivatives. Med. Chem. Commun. 2017, 2, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, S.; Ma, L.; Chen, Y.; Lu, W. Design, synthesis and biological evaluation of novel homocamptothecin analogs as potent antitumor agents. Bioorganic Med. Chem. 2015, 23, 1950–1962. [Google Scholar] [CrossRef] [PubMed]
- Benedeković, G.; Popsavin, M.; Francuz, J.; Kovacević, I.; Kojić, V.; Bogdanovic, G.; Divjaković, V.; Popsavin, V. Design, synthesis and SAR analysis of antitumour styryl lactones related to (+)-crassalactones B and C. Eur. J. Med. Chem. 2014, 87, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Popsavin, V.; Francuz, J.; Srećo Zelenović, B.; Benedeković, G.; Popsavin, M.; Kojić, V.; Bogdanović, G. Heteroannelated and 7-deoxygenated goniofufurone mimics with antitumour activity: Design, synthesis and preliminary SAR studies. Bioorganic Med. Chem. Lett. 2013, 23, 5507–5510. [Google Scholar] [CrossRef] [PubMed]
- Wzorek, A.; Gawdzik, B.; Gładkowski, W.; Urbaniak, M.; Barańska, A.; Malińska, M.; Woźniak, K.; Kempinska, K.; Wietrzyk, J. Synthesis, characterization and antiproliferative activity of β-aryl-δ-iodo-γ-lactones. J. Mol. Struct. 2013, 1047, 160–168. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Białońska, A.; Poradowski, D.; Drynda, A.; Urbaniak, M. Synthesis and anticancer activity of novel halolactones with β-aryl substituents from simple aromatic aldehydes. Tetrahedron 2013, 69, 10414–10423. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Gliszczyńska, A.; Czarnecka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Maciejewska, G.; Białońska, A. Chiral δ-iodo-γ-lactones derived from cuminaldehyde, 2,5-dimethylbenzaldehyde and piperonal: Chemoenzymatic synthesis and antiproliferative activity. Tetrahedron Asymmetry 2016, 27, 227–237. [Google Scholar] [CrossRef]
- Pawlak, A.; Gładkowski, W.; Kutkowska, J.; Mazur, M.; Obmińska-Mrukowicz, B.; Rapak, A. Enantiomeric trans β-aryl-δ-iodo-γ-lactones derived from 2,5-dimethylbenzaldehyde induce apoptosis in canine lymphoma cell lines by downregulation of antiapoptotic Bcl-2 family members Bcl-xL and Bcl-2. Bioorganic Med. Chem. Lett. 2018, 28, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.C.; Ribeiro, D.; Nunes, C.; Reis, S. Biophysics in cancer: The relevance of drug-membrane interaction studies. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 2231–2244. [Google Scholar] [CrossRef] [PubMed]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Białońska, A. Convenient chemoenzymatic route to optically active β-aryl-δ-iodo-γ-lactones and β-aryl-γ-iodo-δ-lactones with the defined configurations of stereogenic centers. European J. Org. Chem. 2015, 605–615. [Google Scholar] [CrossRef]
- Gładkowski, W.; Gliszczyńska, A.; Siepka, M.; Czarnecka, M.; Maciejewska, G. Kinetic resolution of (E)-4-(2′,5′-dimethylphenyl)-but-3-en-2-ol and (E)-4-(benzo[d][1′,3′]dioxol-5′-yl)-but-3-en-2-ol through lipase-catalyzed transesterification. Tetrahedron Asymmetry 2015, 26, 702–709. [Google Scholar] [CrossRef]
- Jew, S.; Terashima, S.; Koga, K. Asymmetric halolactonisation reaction-1. Asymmetric synthesis of optically active α,α-disubstituted-α-hydroxy acids from α,β-unsaturated acids by the novel use of halolactonisation. Tetrahedron 1979, 35, 2337–2343. [Google Scholar] [CrossRef]
- Jew, S. Novel aspects of bromolactonization reaction using N-haloimides in an aprotic polar solvent. Arch. Pharm. Res. 1982, 5, 97–101. [Google Scholar] [CrossRef]
- Snider, B.B.; Johnston, M.I. Regioselectivity of the halolactonization of γ,δ-unsaturated acids. Tetrahedron Lett. 1985, 26, 5497–5500. [Google Scholar] [CrossRef]
- Williams, D.L.H.; Bienvenue-Goetz, E.; Dubois, J.E. Participation by neighbouring groups in addition reactions. Part 1. Hydroxy-group participation in the bromination and iodination of olefins. J. Chem. Soc. B Phys. Org. 1969, 517–522. [Google Scholar] [CrossRef]
- Holbert, G.W.; Weiss, L.B.; Ganem, B. Masked arenes; synthesis of substituted benzenes and benzene oxides. Tetrahedron Lett. 1976, 17, 4435–4438. [Google Scholar] [CrossRef]
- Pawlak, A.; Gładkowski, W.; Mazur, M.; Henklewska, M.; Obmińska-Mrukowicz, B.; Rapak, A. Optically active stereoisomers of 5-(1-iodoethyl)-4-(4′-isopropylphenyl)dihydrofuran-2-one: The effect of the configuration of stereocenters on apoptosis induction in canine cancer cell lines. Chem. Biol. Interact. 2017, 261, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Faggio, C. The use of erythrocyte fragility to assess xenobiotic cytotoxicity. Cell Biochem. Funct. 2015, 33, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Lelkes, P.I.; Miller, I.R. Perturbations of membrane structure by optical probes: I. Location and structural sensitivity of merocyanine 540 bound to phospholipid membranes. J. Membr. Biol. 1980, 52, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alay, M. Spectroscopic analysis of the interaction of a peptide sequence of Hepatitis G virus with bilayers. Talanta 2003, 60, 269–277. [Google Scholar] [CrossRef]
- Stillwell, W.; Wassall, S.R.; Dumaual, A.C.; Ehringer, W.D.; Browning, C.W.; Jenski, L.J. Use of merocyanine (MC540) in quantifying lipid domains and packing in phospholipid vesicles and tumor cells. BBA-Biomembr. 1993, 1146, 136–144. [Google Scholar] [CrossRef]
- Stott, B.M.; Vu, M.P.; McLemore, C.O.; Lund, M.S.; Gibbons, E.; Brueseke, T.J.; Wilson-Ashworth, H.A.; Bell, J.D. Use of fluorescence to determine the effects of cholesterol on lipid behavior in sphingomyelin liposomes and erythrocyte membranes. J. Lipid Res. 2008, 49, 1202–1215. [Google Scholar] [CrossRef] [PubMed]
- Langner, M.; Hui, S.W. Merocyanine 540 as a fluorescence indicator for molecular packing stress at the onset of lamellar-hexagonal transition of phosphatidylethanolamine bilayers. Biochim. Biophys. Acta-Biomembr. 1999, 1415, 323–330. [Google Scholar] [CrossRef]
- Parasassi, T.; De Stasio, G.; Ravagnan, G.; Rusch, R.M.; Gratton, E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys. J. 1991, 60, 179–189. [Google Scholar] [CrossRef]
- Parasassi, T.; Krasnowska, E.K.; Bagatolli, L.; Gratton, E. Laurdan and Prodan as polarity-sensitive fluorescent membrane probes. J. Fluoresc. 1998, 8, 365–373. [Google Scholar] [CrossRef]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr. Sect. A 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Rütgen, B.C.; Hammer, S.E.; Gerner, W.; Christian, M.; de Arespacochaga, A.G.; Willmann, M.; Kleiter, M.; Schwendenwein, I.; Saalmüller, A. Establishment and characterization of a novel canine B cell line derived from a spontaneously occurring diffuse large cell lymphoma. Leuk. Res. 2010, 34, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Nakaichi, M.; Taura, Y.; Kanki, M.; Mamba, K.; Momoi, Y.; Tsujimoto, H.; Nakama, S. Establishment and characterization of a new canine B cell leukemia cell line. J. Vet. Med. Sci. 1996, 58, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, A.; Ziolo, E.; Kutkowska, J.; Blazejczyk, A.; Wietrzyk, J.; Krupa, A.; Hildebrand, W.; Dziegiel, P.; Dzimira, S.; Obminska-Mrukowicz, B.; et al. A novel canine B cell leukaemia cell line. Establishment, characterisation and sensitivity to chemotherapeutics. Vet. Comp. Oncol. 2017, 15, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Łuczyński, J.; Frąckowiak, R.; Włoch, A.; Kleszczyńska, H.; Witek, S. Gemini ester quat surfactants and their biological activity. Cell. Mol. Biol. Lett. 2013, 18, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Włoch, A.; Strugała, P.; Pruchnik, H.; Żyłka, R.; Oszmiański, J.; Kleszczyńska, H. Physical effects of buckwheat extract on biological membrane in vitro and its protective properties. J. Membr. Biol. 2016, 249, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.T.; Mitchell, C.; Hanahan, D.J. The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch. Biochem. Biophys. 1963, 100, 119–130. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 5–7 are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | IC50 [μg/mL] 3 | ||||
|---|---|---|---|---|---|---|
| D17 | CLBL-1 | CLB70 | GL-1 | Jurkat | ||
| 1 | (4R,5R,6S)-5 | >50 | 45.86 ± 3.50 | 31.30 ± 4.20 | 31.10 ± 3.89 | 32.51 ± 2.1 |
| 2 | (4S,5S,6R)-5 | >50 | 33.35 ± 0.40 | 21.39 ± 5.40 | 15.65 ± 1.20 | 30.80 ± 4.90 |
| 3 | (4R,5R,6S)-6 | >50 | 33.27 ± 2.40 | 19.61 ± 3.70 | 14.74 ± 2.87 | 22.47 ± 2.30 |
| 4 | (4S,5S,6R)-6 | >50 | 30.98 ± 1.90 | 13.21 ± 2.30 | 8.51 ± 0.81 | 14.48 ± 5.20 |
| 5 | (4R,5S,6R)-7 | >50 | 18.09 ± 3.33 | 20.08 ± 0.32 | 14.92 ± 0.48 | 16.73 ± 3.40 |
| 6 | (4S,5R,6S)-7 | 34.31 ± 1.88 | 14.33 ± 2.53 | 15.80 ± 0.15 | 6.97 ± 0.45 | 9.80 ± 2.10 |
| 7 | (4R,5R,6S)-9 | 19.39 ± 2.60 | 8.07 ± 1.21 | n.i. 4 | 25.65 ± 4.3 | 29.40 ± 1.66 |
| 8 | (4S,5S,6R)-9 | 26.57 ± 3.54 | 8.01 ± 0.96 | n.i. | 20.48 ± 3.95 | 33.84 ± 6.85 |
| 9 | (4R,5S,6R)-10 | 14.81 ± 3.39 | 7.10 ± 0.65 | n.i. | 14.24 ± 6.06 | 14.30 ± 3.72 |
| 10 | (4S,5R,6S)-10 | 16.99 ± 4.88 | 4.76 ± 0.52 | n.i. | 16.30 ± 4.69 | 16.16 ± 4.73 |
| 11 | carboplatin | 13.10± 4.30 | n.i. | n.i. | 7.06± 1.15 | 21.37± 4.00 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gładkowski, W.; Włoch, A.; Pawlak, A.; Sysak, A.; Białońska, A.; Mazur, M.; Mituła, P.; Maciejewska, G.; Obmińska-Mrukowicz, B.; Kleszczyńska, H. Preparation of Enantiomeric β-(2′,5′-Dimethylphenyl)Bromolactones, Their Antiproliferative Activity and Effect on Biological Membranes. Molecules 2018, 23, 3035. https://doi.org/10.3390/molecules23113035
Gładkowski W, Włoch A, Pawlak A, Sysak A, Białońska A, Mazur M, Mituła P, Maciejewska G, Obmińska-Mrukowicz B, Kleszczyńska H. Preparation of Enantiomeric β-(2′,5′-Dimethylphenyl)Bromolactones, Their Antiproliferative Activity and Effect on Biological Membranes. Molecules. 2018; 23(11):3035. https://doi.org/10.3390/molecules23113035
Chicago/Turabian StyleGładkowski, Witold, Aleksandra Włoch, Aleksandra Pawlak, Angelika Sysak, Agata Białońska, Marcelina Mazur, Paweł Mituła, Gabriela Maciejewska, Bożena Obmińska-Mrukowicz, and Halina Kleszczyńska. 2018. "Preparation of Enantiomeric β-(2′,5′-Dimethylphenyl)Bromolactones, Their Antiproliferative Activity and Effect on Biological Membranes" Molecules 23, no. 11: 3035. https://doi.org/10.3390/molecules23113035
APA StyleGładkowski, W., Włoch, A., Pawlak, A., Sysak, A., Białońska, A., Mazur, M., Mituła, P., Maciejewska, G., Obmińska-Mrukowicz, B., & Kleszczyńska, H. (2018). Preparation of Enantiomeric β-(2′,5′-Dimethylphenyl)Bromolactones, Their Antiproliferative Activity and Effect on Biological Membranes. Molecules, 23(11), 3035. https://doi.org/10.3390/molecules23113035