
Prediction of GluN2B-CT1290-1310/DAPK1 Interaction by Protein–Peptide Docking and Molecular Dynamics Simulation


Abstract
1. Introduction
2. Results and Discussion
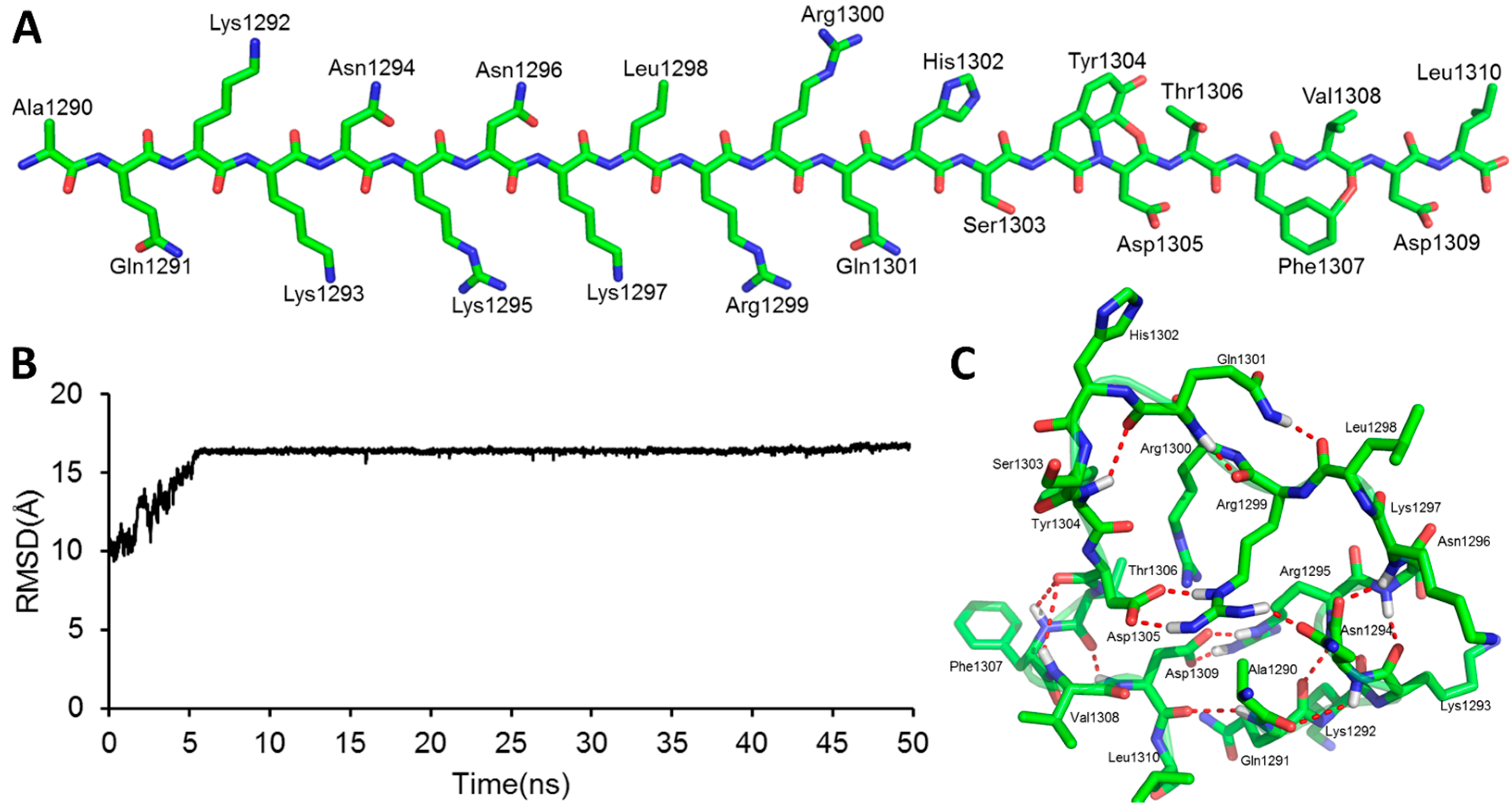
2.1. The Modeled Structure of the GluN2B C-Terminal Peptide
2.2. Docking of GluN2B-CT1290-1310 into the DAPK1 Active Site
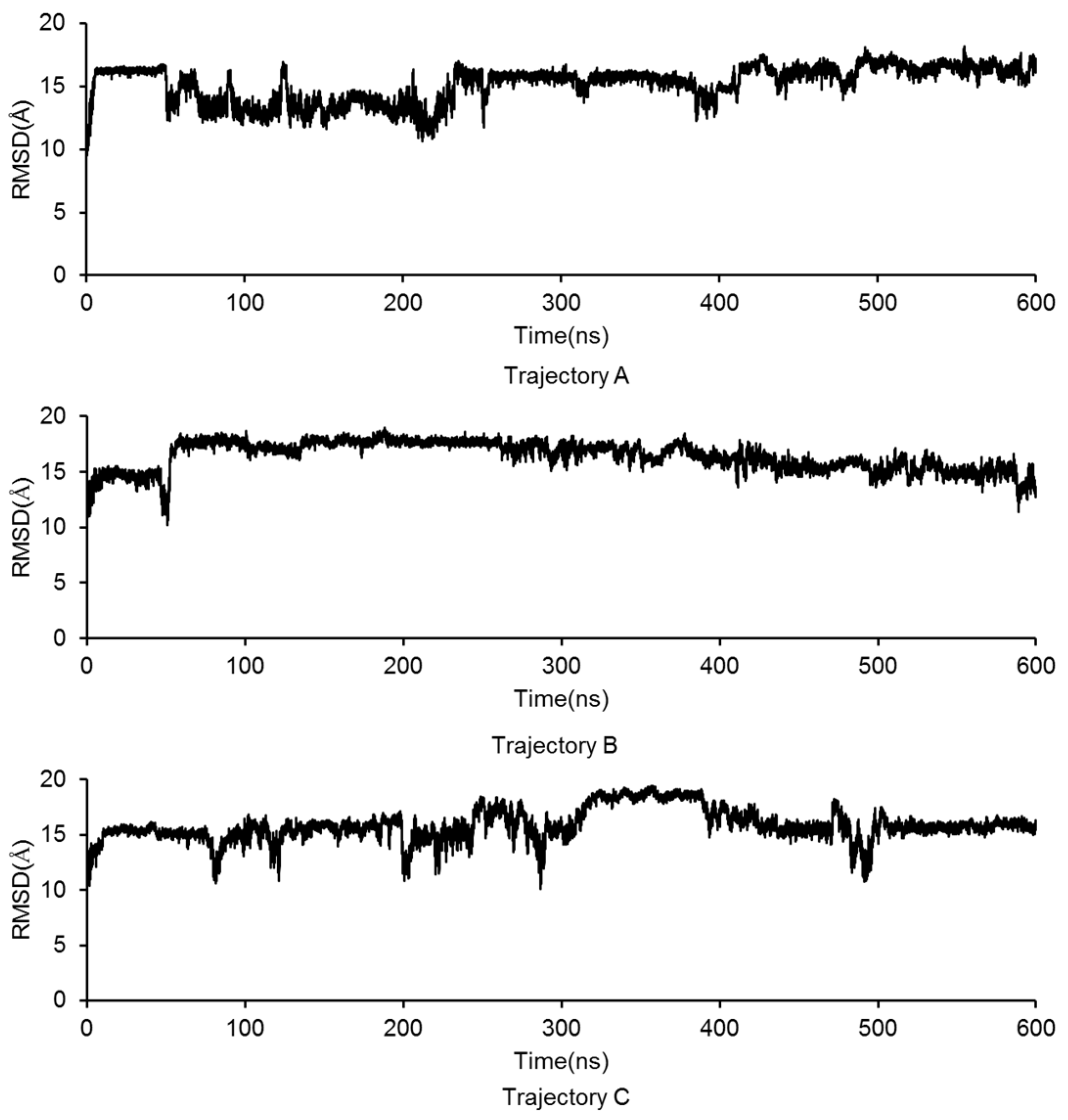
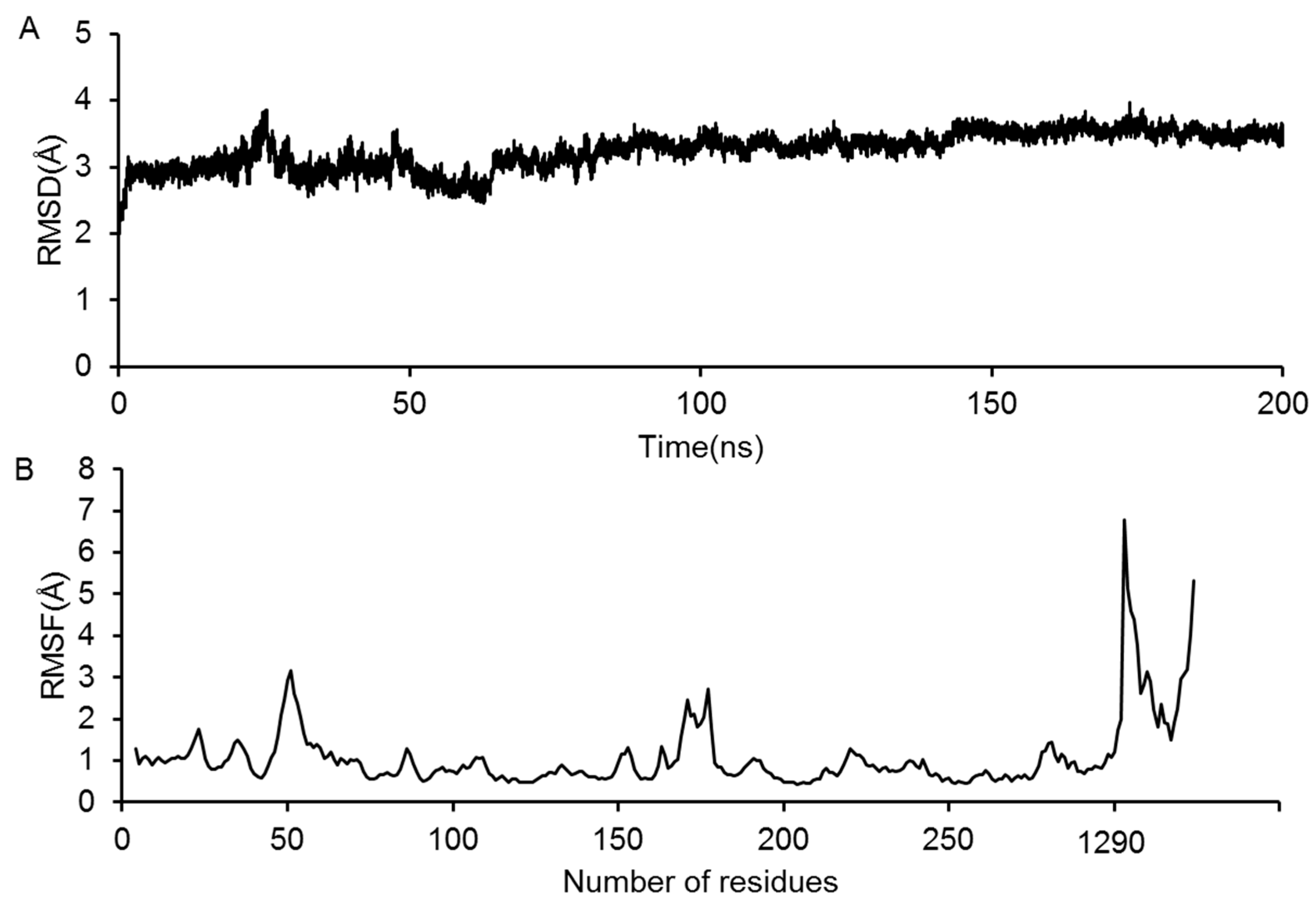
2.3. Molecular Dynamics Simulation of the GluN2B-CT1290-1310/DAPK1 Complex
2.4. Calculation of GluN2B-CT1290-1310/DAPK1 Binding Free Energy
2.5. Analysis of Interactions between GluN2B-CT1290-1310 and DAPK1
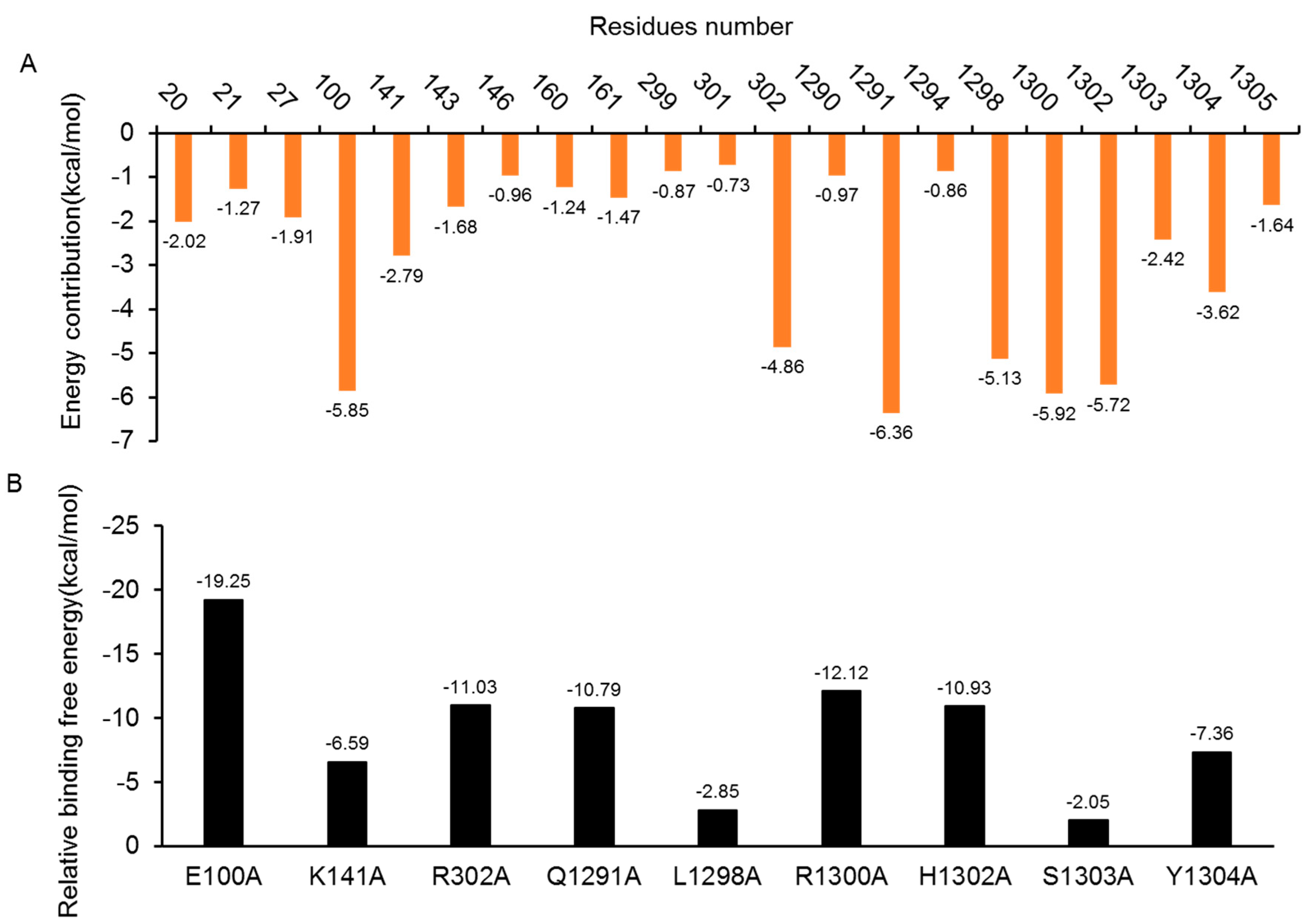
2.5.1. Insight from Free Energy Decomposition Analysis
2.5.2. Insights from the In-Silico Alanine Scanning Analysis
2.5.3. Insights from the Hydrogen Bond Interactions Network Analysis
2.6. Identification of the Interface Profile of the GluN2B-CT1290-1310/DAPK1 Complex
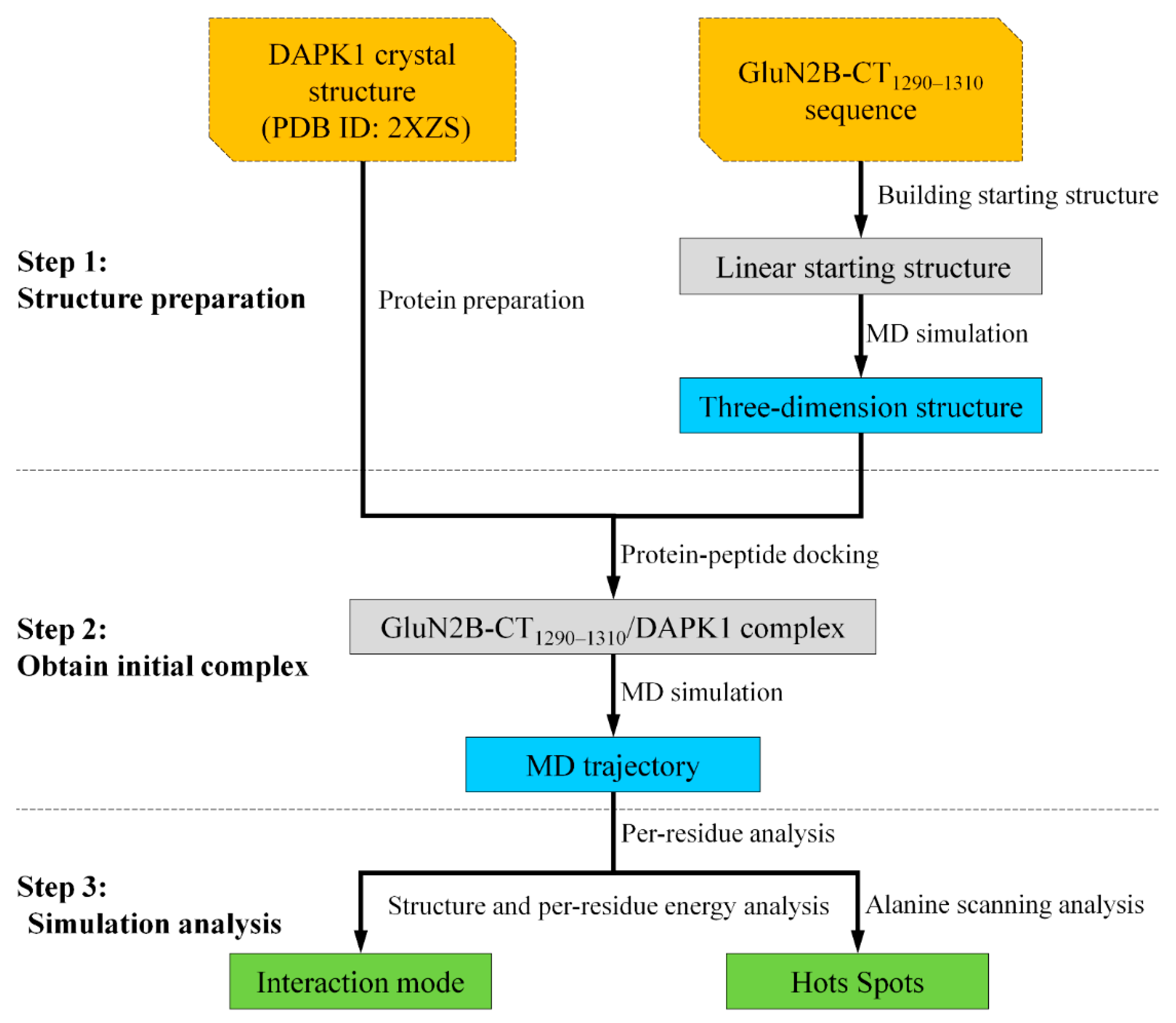
3. Materials and Methods
3.1. Structure Preparation
3.2. Protein–Peptide Docking
3.3. Molecular Dynamics Simulations
3.4. Binding Free Energy Calculation and Per-Residue Energy Decomposition Analysis
3.5. In-Silico Alanine Scanning Analysis
3.6. Hydrogen Bond Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 743–800. [Google Scholar] [CrossRef]
- Zheng, G.; Xue, W.; Yang, F.; Zhang, Y.; Chen, Y.; Yao, X.; Zhu, F. Revealing vilazodone’s binding mechanism underlying its partial agonism to the 5-HT1A receptor in the treatment of major depressive disorder. Phys. Chem. Chem. Phys. 2017, 19, 28885–28896. [Google Scholar] [CrossRef] [PubMed]
- Margret, A.A.; Arumugam, G.K. A therapeutic paradigm to appraise the competence of chitosan oligosaccharide lactate targeting monoamine oxidase-A and p-glycoprotein to contest depression by channeling the blood brain barrier. Curr. Bioinform. 2018, 13, 273–279. [Google Scholar] [CrossRef]
- Thomas, S.J.; Shin, M.; McInnis, M.G.; Bostwick, J.R. Combination therapy with monoamine oxidase inhibitors and other antidepressants or stimulants: Strategies for the management of treatment-resistant depression. Pharmacotherapy 2015, 35, 433–449. [Google Scholar] [CrossRef] [PubMed]
- De Berardis, D.; Marini, S.; Fornaro, M.; Srinivasan, V.; Iasevoli, F.; Tomasetti, C.; Valchera, A.; Perna, G.; Quera-Salva, M.A.; Martinotti, G.; et al. The melatonergic system in mood and anxiety disorders and the role of agomelatine: Implications for clinical practice. Int. J. Mol. Sci. 2013, 14, 12458–12483. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Yu, C.Y.; Li, X.X.; Zhang, P.; Tang, J.; Yang, Q.; Fu, T.; Zhang, X.; Cui, X.; Tu, G.; et al. Therapeutic target database update 2018: Enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2018, 46, D1121–D1127. [Google Scholar] [PubMed]
- Xue, W.; Fu, T.; Zheng, G.; Tu, G.; Zhang, Y.; Yang, F.; Tao, L.; Yao, L.; Zhu, F. Recent advances and challenges of the drugs acting on monoamine transporters. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ramaker, M.J.; Dulawa, S.C. Identifying fast-onset antidepressants using rodent models. Mol. Psychiatry 2017, 22, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Wang, P.; Li, B.; Li, Y.; Xu, X.; Yang, F.; Yao, X.; Chen, Y.Z.; Xu, F.; Zhu, F. Identification of the inhibitory mechanism of FDA approved selective serotonin reuptake inhibitors: An insight from molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2016, 18, 3260–3271. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Wang, P.; Tu, G.; Yang, F.; Zheng, G.; Li, X.; Li, X.; Chen, Y.; Yao, X.; Zhu, F. Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder. Phys. Chem. Chem. Phys. 2018, 20, 6606–6616. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, F.; Yang, H.; Xu, X.; Liu, D.; Xue, W.; Zhu, F. Identification of dual active agents targeting 5-HT1A and SERT by combinatorial virtual screening methods. Biomed. Mater. Eng. 2015, 26, S2233–S2239. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, X.X.; Yang, S.Y.; Chen, Y.Z. Clinical success of drug targets prospectively predicted by in silico study. Trends Pharmacol. Sci. 2018, 39, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Shyu, W.C.; Wang, Y.T. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol. Med. 2011, 17, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Del Rosario, J.S.; Feldmann, K.G.; Ahmed, T.; Amjad, U.; Ko, B.; An, J.; Mahmud, T.; Salama, M.; Mei, S.; Asemota, D.; et al. Death associated protein kinase (DAPK) -mediated neurodegenerative mechanisms in nematode excitotoxicity. BMC Neurosci. 2015, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Xu, J.Y.; Tao, L.; Li, X.F.; Li, S.; Zeng, X.; Chen, S.Y.; Zhang, P.; Qin, C.; Zhang, C.; et al. SVM-Prot 2016: A web-server for machine learning prediction of protein functional families from sequence irrespective of similarity. PLoS ONE 2016, 11, e0155290. [Google Scholar] [CrossRef] [PubMed]
- Li, S.X.; Han, Y.; Xu, L.Z.; Yuan, K.; Zhang, R.X.; Sun, C.Y.; Xu, D.F.; Yuan, M.; Deng, J.H.; Meng, S.Q.; et al. Uncoupling DAPK1 from NMDA receptor GluN2B subunit exerts rapid antidepressant-like effects. Mol. Psychiatry 2018, 23, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Huart, A.S.; Temmerman, K.; Vahokoski, J.; Mertens, H.D.; Komadina, D.; Hoffmann, J.E.; Yumerefendi, H.; Svergun, D.I.; Kursula, P.; et al. Death-associated protein kinase activity is regulated by coupled calcium/calmodulin binding to two distinct sites. Structure 2016, 24, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Kimchi, A. DAP-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Semin. Cancer Biol. 2004, 14, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.; Hupp, T.R. Novel insights into DAPK autophagic signalling using peptide aptamer combinatorial protein-interaction screens. Autophagy 2008, 4, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Shi, Z.; Qin, C.; Tao, L.; Liu, X.; Xu, F.; Zhang, L.; Song, Y.; Liu, X.; Zhang, J.; et al. Therapeutic target database update 2012: A resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2012, 40, D1128–D1136. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 2018, 34, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Hu, Q.; Guo, M.; Wang, G. HAlign: Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 2015, 31, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Hagberg, H.; Krishnamurthy, R.; Thornton, C.; Mallard, C. Death associated protein kinases: Molecular structure and brain injury. Int. J. Mol. Sci. 2013, 14, 13858–13872. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ding, Q.; Chen, Z.; Yun, H.; Wang, H. Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J. Biol. Chem. 2013, 288, 24151–24159. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ravanan, P.; Talwar, P. Death associated protein kinase 1 (DAPK1): A regulator of apoptosis and autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Zuo, Y.; Zou, Q. O-GlcNAcPRED-II: An integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 2018, 34, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Qin, C.; Li, Y.H.; Tao, L.; Zhou, J.; Yu, C.Y.; Xu, F.; Chen, Z.; Zhu, F.; Chen, Y.Z. Therapeutic target database update 2016: Enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res. 2016, 44, D1069–D1074. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Han, B.; Kumar, P.; Liu, X.; Ma, X.; Wei, X.; Huang, L.; Guo, Y.; Han, L.; Zheng, C.; et al. Update of TTD: Therapeutic Target Database. Nucleic Acids Res. 2010, 38, D787–D791. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zheng, C.J.; Han, L.Y.; Xie, B.; Jia, J.; Liu, X.; Tammi, M.T.; Yang, S.Y.; Wei, Y.Q.; Chen, Y.Z. Trends in the exploration of anticancer targets and strategies in enhancing the efficacy of drug targeting. Curr. Mol. Pharmacol. 2008, 1, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Han, L.; Zheng, C.; Xie, B.; Tammi, M.T.; Yang, S.; Wei, Y.; Chen, Y. What are next generation innovative therapeutic targets? Clues from genetic, structural, physicochemical, and systems profiles of successful targets. J. Pharmacol. Exp. Ther. 2009, 330, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Karakas, E.; Simorowski, N.; Furukawa, H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009, 28, 3910–3920. [Google Scholar] [CrossRef] [PubMed]
- Karakas, E.; Simorowski, N.; Furukawa, H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 2011, 475, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lu, W.; Michel, J.C.; Goehring, A.; Du, J.; Song, X.; Gouaux, E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 2014, 511, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Zheng, G.; Tu, G.; Yang, F.; Chen, Y.; Yao, X.; Li, X.; Xue, W.; Zhu, F. Exploring the binding mechanism of metabotropic glutamate receptor 5 negative allosteric modulators in clinical trials by molecular dynamics simulations. ACS Chem. Neurosci. 2018, 9, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Torchala, M.; Moal, I.H.; Chaleil, R.A.; Fernandez-Recio, J.; Bates, P.A. SwarmDock: A server for flexible protein-protein docking. Bioinformatics 2013, 29, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.A.; Harvey, J.A.; Spampinato, U.; Clarke, W.P. Physiological relevance of constitutive activity of 5-HT2A and 5-HT2C receptors. Trends Pharmacol. Sci. 2005, 26, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Temmerman, K.; Simon, B.; Wilmanns, M. Structural and functional diversity in the activity and regulation of DAPK-related protein kinases. FEBS J. 2013, 280, 5533–5550. [Google Scholar] [CrossRef] [PubMed]
- Wilbek, T.S.; Skovgaard, T.; Sorrell, F.J.; Knapp, S.; Berthelsen, J.; Stromgaard, K. Identification and characterization of a small-molecule inhibitor of death-associated protein kinase 1. Chembiochem 2015, 16, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Zhang, S. Computational prediction of protein hot spot residues. Curr. Pharm. Des. 2012, 18, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, S.; Fernandez-Recio, J. Identification of hot-spot residues in protein-protein interactions by computational docking. BMC Bioinformatics 2008, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Zerbe, B.S.; Hall, D.R.; Vajda, S.; Whitty, A.; Kozakov, D. Relationship between hot spot residues and ligand binding hot spots in protein-protein interfaces. J. Chem. Inf. Model. 2012, 52, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Li, X.X.; Yang, H.; Li, Y.H.; Xue, W.W.; Chen, Y.Z.; Tao, L.; Zhu, F. Assessing the performances of protein function prediction algorithms from the perspectives of identification accuracy and false discovery rate. Int. J. Mol. Sci. 2018, 19, 183. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.Y.; Fu, T.T.; Zhang, X.Y.; Hu, J.; Xue, W.W.; Zheng, G.X.; Li, B.; Li, Y.H.; Yao, X.J.; Zhu, F. Comparison of computational model and X-ray crystal structure of human serotonin transporter: Potential application for the pharmacology of human monoamine transporters. Mol. Simul. 2017, 43, 1089–1098. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, X.; Fu, T.; Li, S.; Li, B.; Xue, W.; Yao, X.; Chen, Y.; Zhu, F. Differentiating physicochemical properties between addictive and nonaddictive ADHD drugs revealed by molecular dynamics simulation studies. ACS Chem. Neurosci. 2017, 8, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Li, Y.; Yu, C.; Xue, W.; Hu, J.; Li, B.; Wang, P.; Zhu, F. What makes species productive of anti-cancer drugs? Clues from drugs’ species origin, druglikeness, target and pathway. Anticancer Agents Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Merrill, M.A.; Chen, Y.; Strack, S.; Hell, J.W. Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol. Sci. 2005, 26, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Parida, P.; Yadav, R.N.; Sarma, K. Insight into structural organization and protein-protein interaction of non structural 3 (NS3) proteins from dengue serotypes. Curr. Pharm. Biotechnol. 2014, 15, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Vakser, I.A. Evaluation of GRAMM low-resolution docking methodology on the hemagglutinin-antibody complex. Proteins 1997, 29, 226–230. [Google Scholar] [CrossRef]
- Tovchigrechko, A.; Vakser, I.A. Development and testing of an automated approach to protein docking. Proteins 2005, 60, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, L.; Weng, Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins 2003, 52, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Weng, Z. A novel shape complementarity scoring function for protein-protein docking. Proteins 2003, 51, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Ma, X.H.; Qin, C.; Tao, L.; Liu, X.; Shi, Z.; Zhang, C.L.; Tan, C.Y.; Chen, Y.Z.; Jiang, Y.Y. Drug discovery prospect from untapped species: Indications from approved natural product drugs. PLoS ONE 2012, 7, e39782. [Google Scholar] [CrossRef] [PubMed]
- Moal, I.H.; Bates, P.A. SwarmDock and the use of normal modes in protein-protein docking. Int. J. Mol. Sci. 2010, 11, 3623–3648. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Moal, I.H.; Bates, P.A. Detection and refinement of encounter complexes for protein-protein docking: Taking account of macromolecular crowding. Proteins 2010, 78, 3189–3196. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, P.; Yang, H.; Zhou, J.; Li, Y.; Li, X.; Xue, W.; Yu, C.; Tian, Y.; Zhu, F. Comparison of FDA approved kinase targets to clinical trial ones: Insights from their system profiles and drug-target interaction networks. Biomed. Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Han, L.Y.; Chen, X.; Lin, H.H.; Ong, S.; Xie, B.; Zhang, H.L.; Chen, Y.Z. Homology-free prediction of functional class of proteins and peptides by support vector machines. Curr. Protein Pept. Sci. 2008, 9, 70–95. [Google Scholar] [PubMed]
- Hara, Y.; Murayama, S. Effects of analgesic-antipyretics on the spinal reflex potentials in cats: An analysis of the excitatory action of aminopyrine. Nihon Yakurigaku Zasshi 1992, 100, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Springborg, M.; Kirtman, B. Efficient vector potential method for calculating electronic and nuclear response of infinite periodic systems to finite electric fields. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Chong, L.T.; Pitera, J.W.; Swope, W.C.; Pande, V.S. Comparison of computational approaches for predicting the effects of missense mutations on p53 function. J. Mol. Graph. Model. 2009, 27, 978–982. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Xue, W.; Wang, P.; Yang, F.; Li, B.; Li, X.; Li, Y.; Yao, X.; Zhu, F. Exploring the inhibitory mechanism of approved selective norepinephrine reuptake inhibitors and reboxetine enantiomers by molecular dynamics study. Sci. Rep. 2016, 6, 26883. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Fu, T.; Zhang, X.; Yang, F.; Zheng, G.; Xue, W.; Chen, Y.; Yao, X.; Zhu, F. Differentiating physicochemical properties between NDRIs and sNRIs clinically important for the treatment of ADHD. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2766–2777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, J.; Huang, L.; Tian, J.; Yao, X.; Liu, H. The molecular mechanism of two coreceptor binding site antibodies X5 and 17b neutralizing HIV-1: Insights from molecular dynamics simulation. Chem. Biol. Drug Des. 2018, 92, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Zhou, S.; Liu, X.; Zhao, C.; Liu, H.; Yao, X. Understanding the structural and energetic basis of PD-1 and monoclonal antibodies bound to PD-L1: A molecular modeling perspective. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Wang, M.; Jin, X.; Liu, H.; Yao, X. Understanding the structural and energetic basis of inhibitor and substrate bound to the full-length NS3/4A: Insights from molecular dynamics simulation, binding free energy calculation and network analysis. Mol. Biosyst. 2012, 8, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.R.; Wang, Z.J.; Zheng, Q.C.; Zhang, H.X. Exploring the inhibition mechanism on HIF-2 by inhibitor PT2399 and 0X3 using molecular dynamics simulations. J. Mol. Recognit. 2018, 31, e2730. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Qiu, M.; Guo, L.; Yao, X. Computational study of the binding mechanism between farnesoid X receptor alpha and antagonist N-benzyl-N-(3-(tertbutyl)-4-hydroxyphenyl)-2,6-dichloro-4-(dimethylamino) benzamide. J. Biomol. Struct. Dyn. 2018. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, X.; Nie, Q.; Yang, J.; Yao, X. Computational study of the binding mechanism of medium chain acyl-CoA synthetase with substrate in Methanosarcina acetivorans. J. Biotechnol. 2017, 259, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Yang, F.; Wang, P.; Zheng, G.; Chen, Y.; Yao, X.; Zhu, F. What contributes to serotonin-norepinephrine reuptake inhibitors’ dual-targeting mechanism? The key role of transmembrane domain 6 in human serotonin and norepinephrine transporters revealed by molecular dynamics simulation. ACS Chem. Neurosci. 2018, 9, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tang, J.; Yang, Q.; Li, S.; Cui, X.; Li, Y.; Chen, Y.; Xue, W.; Li, X.; Zhu, F. NOREVA: Normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017, 45, W162–W170. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Computational alanine scanning to probe protein-protein interactions: a novel approach to evaluate binding free energies. J. Am. Chem. Soc. 1999, 121, 8133–8143. [Google Scholar] [CrossRef]
- Liu, H.; Yao, X. Molecular basis of the interaction for an essential subunit PA-PB1 in influenza virus RNA polymerase: Insights from molecular dynamics simulation and free energy calculation. Mol. Pharm. 2010, 7, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Liu, L.; Lu, L.; Zou, Q. Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 2018, 34, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.Y.; Ma, K.; Zheng, Q.C.; Yang, G.H.; Zhang, H.X. Exploring the interactional details between aldose reductase (AKR1B1) and 3-Mercapto-5H-1,2,4-triazino[5,6-b]indole-5-acetic acid through molecular dynamics simulations. J. Biomol. Struct. Dyn. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.B.; Hu, S.C.; Wang, H.; Zou, Q.; Ji, Z.L. PaGeFinder: Quantitative identification of spatiotemporal pattern genes. Bioinformatics 2012, 28, 1544–1545. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Wang, P.P.; Li, X.X.; Yu, C.Y.; Yang, H.; Zhou, J.; Xue, W.W.; Tan, J.; Zhu, F. The human kinome targeted by FDA approved multi-target drugs and combination products: A comparative study from the drug-target interaction network perspective. PLoS ONE 2016, 11, e0165737. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Tang, J.; Wang, Y.; Cui, X.; Yang, Q.; Hong, J.; Li, X.; Li, S.; Chen, Y.; Xue, W.; et al. Discovery of the consistently well-performed analysis chain for SWATH-MS based pharmacoproteomic quantification. Front. Pharmacol. 2018, 9, 681. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GluN2B-CT1290-1310/DAPK1 Complex | Structure of GluN2B-CT1290-1310 | DAPK1 PDB Code a | Docking Program | Ranking Number in Docking b | Number of Water Molecules | Total Number of Atoms | Simulation Time |
|---|---|---|---|---|---|---|---|
| 1 | Trajectory A 50 ns | 2XZS | GRAMM-X | 2 | 15570 | 51941 | 200 ns |
| 2 | Trajectory A 600 ns | 2XZS | GRAMM-X | 5 | 12968 | 44135 | 200 ns |
| 3 | Trajectory C 600 ns | 2XZS | GRAMM-X | 4 | 11877 | 40862 | 200 ns |
| 4 | Trajectory A 600 ns | 2XZS | ZDOCK | 3 | 12013 | 41270 | 200 ns |
| 5 | Trajectory A 600 ns | 2XZS | ZDOCK | 9 | 12613 | 43070 | 200 ns |
| 6 | Trajectory C 600 ns | 2XZS | ZDOCK | 1 | 11897 | 40922 | 200 ns |
| 7 | Trajectory A 600 ns | 2XZS | SwarmDock | 1 | 12003 | 41240 | 200 ns |
| 8 | Trajectory B 600 ns | 2XZS | SwarmDock | 5 | 12748 | 43475 | 200 ns |
| Energy Contribution | GluN2B-CT1290–1310/DAPK1 | DAPK1 | GluN2B | Delta | ||||
|---|---|---|---|---|---|---|---|---|
| Mean | σ j | Mean | σ j | Mean | σ j | Mean | σ j | |
| ELE a | −10147.50 | 108.22 | −8860.67 | 108.82 | −805.70 | 18.87 | −481.09 | 34.11 |
| VDW b | −1371.34 | 28.80 | −1279.68 | 27.00 | −17.06 | 7.32 | −74.59 | 7.85 |
| INT c | 7827.43 | 54.97 | 7276.88 | 52.29 | 550.55 | 14.92 | 0.00 | 0.00 |
| GAS d | −3691.36 | 119.20 | −2863.47 | 115.65 | −272.21 | 21.94 | −555.68 | 35.79 |
| GBSUR e | 113.59 | 3.03 | 107.98 | 2.43 | 16.66 | 0.51 | −11.05 | 1.10 |
| GB f | −4397.01 | 98.49 | −4284.63 | 99.70 | −622.19 | 14.48 | 509.81 | 33.16 |
| GBSOLg | −4283.42 | 96.91 | −4176.65 | 98.54 | −605.53 | 14.30 | 498.76 | 32.86 |
| GBELEh | −14544.50 | 29.69 | −13145.30 | 27.41 | −1427.89 | 8.83 | 28.72 | 5.81 |
| GBTOT i | −7974.78 | 53.64 | −7040.12 | 50.22 | −877.75 | 15.02 | −56.92 | 8.73 |
| Acceptor | DonorH | Donor | Frames | Occupancy a | AvgDist b | AvgAng c |
|---|---|---|---|---|---|---|
| 100@OE1 | 1300@HH11 | 1300@NH1 | 13754 | 68.77% | 2.92 | 149.35 |
| 100@OE2 | 1300@HH11 | 1300@NH1 | 13009 | 65.04% | 2.92 | 150.07 |
| 100@OE1 | 1300@HE | 1300@NE | 11175 | 55.87% | 2.99 | 148.35 |
| 100@OE2 | 1300@HE | 1300@NE | 10038 | 50.19% | 3.00 | 148.24 |
| 161@OD2 | 1302@HD1 | 1302@ND1 | 9403 | 47.02% | 2.88 | 158.51 |
| 161@OD2 | 1303@HG | 1303@OG | 9091 | 45.46% | 2.78 | 157.89 |
| 143@OE2 | 302@HH22 | 302@NH2 | 11794 | 58.97% | 2.90 | 152.66 |
| 143@OE1 | 302@HH22 | 302@NH2 | 10289 | 51.44% | 3.02 | 146.13 |
| 143@OE1 | 302@HH12 | 302@NH1 | 9107 | 45.53% | 2.87 | 157.70 |
| 1298@O | 302@HH11 | 302@NH1 | 13591 | 67.95% | 2.86 | 157.30 |
| GluN2B-CT1290–1310/DAPK1 Complex | Identified Interface Residues |
|---|---|
| 1 | Val27, Glu100, Glu143, Met146, Ile160, Asp161, Arg302, Asp1305, Tyr1304, His1302, Arg1300, Leu1298 |
| 3 | Val27, Glu100, Glu143, Asn144, Met146, Asp161, Arg1295, Asp1309, His1302, Ser1303, Phe1307 |
| 4 | Gln23, Val27, Glu143, Met146, Leu164, Phe178, Phe183, Arg1300, Asp1305, Arg1299, His1302, Tyr1304 |
| 5 | Gln23, Glu143, Asp139, Asp161, Leu164, Thr180, Glu182, Arg1300, Arg1295, His1302, Ser1303, Tyr1304, Thr1306, Phe1307 |
| 7 | Leu19, Val27, Val96, Ala97, Glu107, Met146, Lie160, Arg1295, Phe1307, Arg1309 |
| 8 | Glu18, Leu19, Val27, Leu95, Asp103, Met146, Lys1297, Asp1305, Thr1306, Phe1307 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, G.; Fu, T.; Yang, F.; Yao, L.; Xue, W.; Zhu, F. Prediction of GluN2B-CT1290-1310/DAPK1 Interaction by Protein–Peptide Docking and Molecular Dynamics Simulation. Molecules 2018, 23, 3018. https://doi.org/10.3390/molecules23113018
Tu G, Fu T, Yang F, Yao L, Xue W, Zhu F. Prediction of GluN2B-CT1290-1310/DAPK1 Interaction by Protein–Peptide Docking and Molecular Dynamics Simulation. Molecules. 2018; 23(11):3018. https://doi.org/10.3390/molecules23113018
Chicago/Turabian StyleTu, Gao, Tingting Fu, Fengyuan Yang, Lixia Yao, Weiwei Xue, and Feng Zhu. 2018. "Prediction of GluN2B-CT1290-1310/DAPK1 Interaction by Protein–Peptide Docking and Molecular Dynamics Simulation" Molecules 23, no. 11: 3018. https://doi.org/10.3390/molecules23113018
APA StyleTu, G., Fu, T., Yang, F., Yao, L., Xue, W., & Zhu, F. (2018). Prediction of GluN2B-CT1290-1310/DAPK1 Interaction by Protein–Peptide Docking and Molecular Dynamics Simulation. Molecules, 23(11), 3018. https://doi.org/10.3390/molecules23113018