3.2. Chemistry
3.2.1. Methyl 4-Bromobutanoate (11)
4-Bromobutanoic acid (7.5 g, 45.2 mmole) was dissolved in methanol (50 mL), and then CH3SiCl (11.4 mL, 90.4 mmole) was added at 0 °C; the mixture was stirred at this temperature for 30 min and for 72 h at 25 °C. The solvent was removed in vacuuo. The resultant residue was purified by column chromatography (silica-gel) eluting with hexanes:ethyl acetate 90:10 to afford the title compound as a colorless oil (6.78 g, 83.4%). 1H-NMR (CDCl3): δ 3.68 (s, 3H), 3.46 (t, J = 6.4 Hz, 2H), 2.50 (t, J = 7.2 Hz, 2H), 2.22–2.13 (m, 2H). 13C-NMR (CDCl3): δ 173.11, 51.85, 32.80, 32.34, 27.87. HRMS (FAB+): m/z [M]+ calcd for C5H9BrO2: 179.9786, found (M + 1): 180.9786.
3.2.2. General Methodology for the Synthesis of Esters 12a–f
The corresponding amine 10a–f (1.0 equiv) was added dropwise to methyl 4-bromobutanoate (2, 1.0 equiv) at 25 °C, and the mixture was allowed to warm to 65–70 °C for 2 h. When the reaction was complete, aqueous NaHCO3 was added, and the mixture was extracted with EtOAc (3 × 10 mL), dried over Na2SO4, filtered and removed in vacuuo. The crude product was purified by column chromatography (silica-gel), eluting with hexanes: ethyl acetate 90:10.
Methyl 4-(thiazolidin-3-yl)butanoate (12a). 0.445 g (37%) as a yellow oil. 1H-NMR (200 MHz, CDCl3): δ 4.00 (s, 2H), 3.63 (s, 1H), 3.02 (dd, J = 9.5, 3.3 Hz, 2H), 2.82 (dd, J = 9.3, 3.6 Hz, 1H), 2.37 (t, J = 7.4, 2H), 2.35 (t, J = 7 Hz, 2H), 1.77 (q, J = 7.2 Hz, 1H, H-4). 13C-NMR (50 MHz, CDCl3) δ 173.94, 60.44, 58.01, 52.07, 51.62, 31.71, 29.6, 24.28. HRMS (FAB+): m/z [M]+ calcd for C8H15NO2S: 189.0823, found (M + 1): 190.0904.
Methyl 4-(piperidin-1-yl) butanoate (12b). 0.37 g (67%) as a yellow oil. 1H-NMR (CDCl3) δ 3.62 (s, 3H), 2.33 (sa, 4H), 2.28 (t, J = 8 Hz, 2H), 2.27 (t, J = 8 Hz, 2H), 1.77 (q, J = 8 Hz, 2H), 1.55–1.49 (m, 4H), 1.4–1.37 (m, 2H). 13C-NMR (CDCl3) δ 174.13, 58.49, 54.54, 51.56, 32.23, 25.98, 24.48, 22.24. HRMS (FAB+): m/z calcd for C10H19NO2: 185.1416, found (M + 1): 186.1532.
Methyl 4-(3-methylpiperidin-1-yl) butanoate (12c). 0.537 g (79%) as a yellow oil. 1H-NMR (CDCl3): δ 3.67 (s, 3H), 2.84–2.78 (m, 2H), 2.35–2.29 (m, 4H), 1.87–1.77 (m, 3H), 1.71–1.49 (m, 6H), 0.85 (d, J = 6.4 Hz, 3H). 13C-NMR (CDCl3) δ 174.13, 62.08, 58.25, 54.02, 51.54, 33.14, 32.22, 31.18, 25.62, 22.30, 19.83. HRMS (FAB+): m/z calcd for C11H21NO2: 199.1572, found (M + 1): 200.1636.
Methyl 4-(4-methylpiperidin-1-yl)butanoate (12d). 0.45 g (67%) as a yellow oil. 1H-NMR (CDCl3): δ 3.67 (s, 3H), 2.87–2.84 (m, 2H), 2.35–2.3 (m, 4H), 1.92–1.86 (m, 2H), 1.85–1.77 (m, 2H), 1.62–1.58 (m, 2H), 1.40–1.28 (m, 1H), 1.28 (m, 2H), 0.91 (d, J = 6.4 Hz, 3H). 13C-NMR (CDCl3) δ 174.18, 58.21, 54.05, 51.61, 34.43, 32.30, 30.93, 22.47, 22.02. HRMS (FAB+): m/z calcd for C11H21NO2, 199.1572, found (M + 1): 200.1656.
Methyl 4-morpholinobutanoate (12e). 0.445 g (52%) as a yellow oil. 1H-NMR (CDCl3): δ 3.7 (dd, J = 4, 4 Hz, 4H), 3.68 (s, 3H), 2.43 (dd, J = 4, 4 Hz, 4H), 2.36 (t, J = 8 Hz, 4H), 1.82 (q, J = 8 Hz, 1H). 13C-NMR (CDCl3) δ 173.92, 66.92, 57.97, 53.55, 51.48, 31.87, 21.73. HRMS (FAB+): m/z calcd for C9H17NO3: 187.1208, found (M + 1): 188.1308.
Methyl 4-thiomorpholinobutanoate (12f). 0.47 g (58%) as a yellow oil. 1H-NMR (CDCl3): δ 3.60 (s, 3H), 2.64–2.62 (m, 4H), 2.6–2.58 (m, 4H), 2.31 (t, J = 8 Hz), 2.26 (t, J = 8Hz), 1.73 (q, J = 8 Hz). 13C-NMR (CDCl3) δ 173.93, 58.24, 54.92, 51.47, 31.88, 27.93 (2C), 21.78. HRMS (FAB+): m/z calcd for C9H17NO2S: 203.098, found (M + 1): 204.1057.
3.2.3. General Methodology for Hydrolysis of Methyl Esters 12a–f
The corresponding methyl ester 12a–f (1.0 equiv) was dissolved in MeOH (6 mL) and NaOH (1.1 equiv) dissolved in water (2 mL) was added dropwise. The reaction was monitored until the consumption of the starting material. At the end of the reaction, the MeOH was evaporated. Then HCl was added to pH = 5, the mixture was extracted with EtOAc (3 × 5 mL), dried over Na2SO4, filtered and the solvent removed in vacuo.
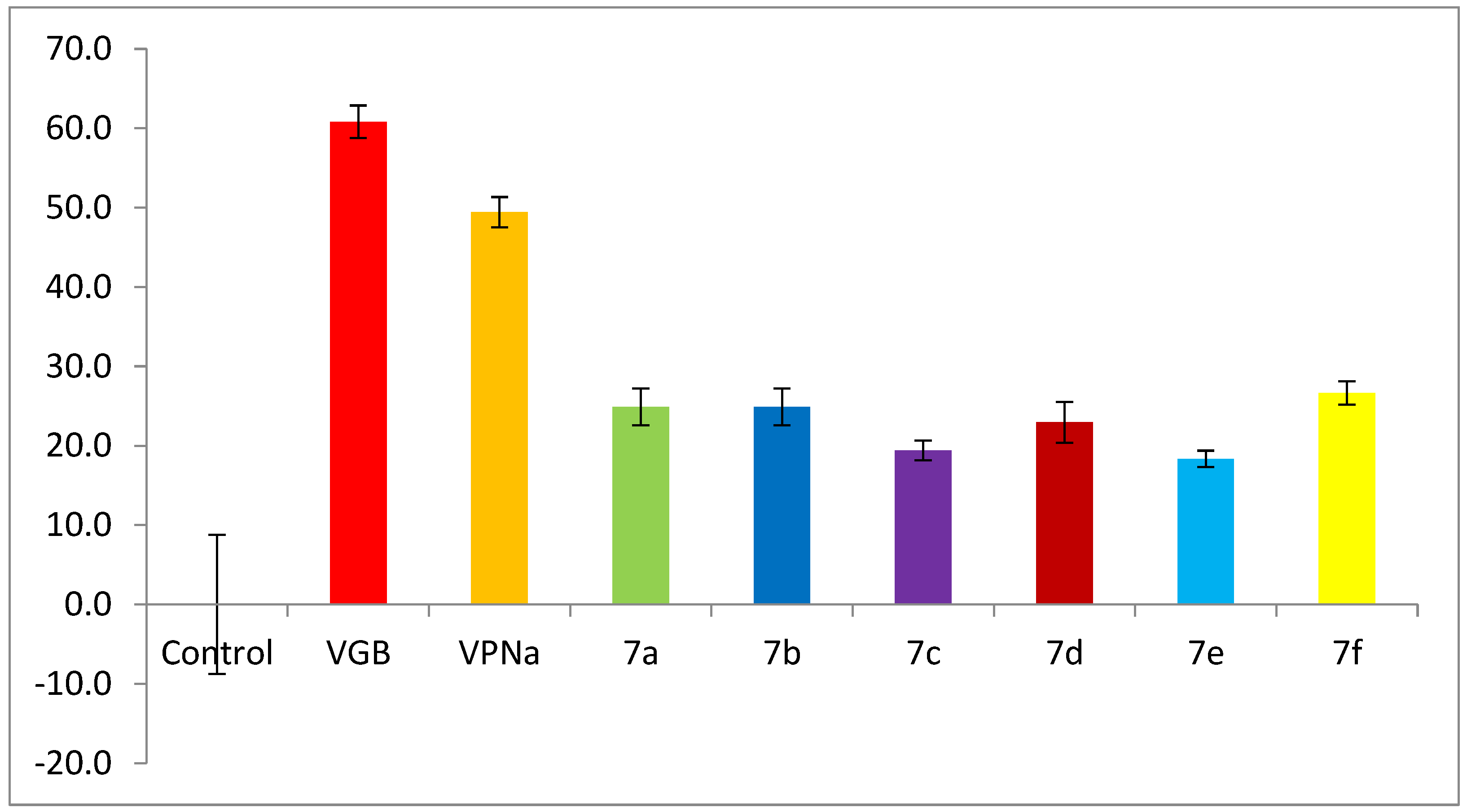
4-(Thiazolidin-3-yl)butanoic acid (7a). 0.165 g (81%) as a yellow oil. 1H-NMR (200 MHz, CD3OD) δ 4.02 (s, 2H), 3.08 (t, J= 5.9 Hz, 2H), 2.88 (t, J = 6.2 Hz, 2H), 2.54–2.41 (m, 2H), 2.29 (t, J= 7.3 Hz, 2H), 1.90–1.68 (m, 2H). 13C-NMR (50 MHz, CD3OD) δ 180.16, 60.29, 58.68, 53.73, 35.05, 29.94, 26.13. HRMS (FAB+): m/z calcd for C7H13NO2S, 175.0667, experimental (M + 1): 176.0769.
4-(Piperidin-1-yl)butanoic acid (7b). 0.22 g (95%) as a yellow oil. 1H-NMR (CD3OD): δ 3.3–3.12 (sa, 2H), 3.08 (t, J = 7.2 Hz, 2H), 2.74 (ddd, J = 15.5, 12, 12 Hz, 2H), 2.41 (t, J = 6.6 Hz, 2H), 2.01–1.92 (m, 2H), 1.86–1.84 (m, 4H), 1.66 (sa, 2H). 13C-NMR (CD3OD) δ 181.22, 59.77, 54.39, 37.50, 24.73, 23.15, 21.54HRMS (FAB+): m/z calcd for C9H17NO2: 171.1259, found (M + 1): 172.1324.
4-(3-Methylpiperidin-1-yl)butanoic acid (7c). 0.21 g (93%) as a colorless oil. 1H-NMR (D2O): δ 2.92 (m, 2H), 2.46–2.41 (m, 2H), 2.09 (td, J = 12.4, 2.6 Hz, 1H), 2.02 (t, J = 7.3 Hz, 2H), 1.81 (t, J = 11.4 Hz, 1H), 1.68–1.47 (m, 5H), 1.46–1.34 (m, 1H), 0.86–0.75 (m, 1H), 0.71 (d, J = 6.5 Hz, 3H). 13C-NMR (D2O) δ 182.34, 59.72, 57.37, 52.70, 35.20, 31.10, 29.65, 23.62, 21.53, 18.45. HRMS (FAB+): m/z calcd for C10H19NO2: 185.1416, found (M + 1): 186.1532.
4-(4-Methylpiperidin-1-yl)butanoic acid (7d). 0.14 g (80%) as a colorless oil. 1H-NMR (D2O): δ 2.81 (d, J = 11.6 Hz, 2H), 2.30–2.22 (m, 2H), 2.02–1.96 (m, 4H), 1.63–1.46 (m, 4H), 1.28–1.21 (m, 1H), 1.05–0.95 (m, 2H), 0.71 (d, J = 6.5 Hz). 13C-NMR (D2O) δ 182.67, 57.36, 52.78, 35.41, 32.57, 29.41, 22.13, 20.84. HRMS (FAB+): m/z calcd for C10H19NO2: 185.1416, found (M + 1): 186.1532.
4-Morpholinobutanoic acid (7e). 0.22 g (83%) of a yellow solid, m.p = 73–73.5 °C. 1H-NMR (CD3OD): δ 3.73–3.66 (m, 4H), 2.46 (s, 4H), 2.36 (m, 2H), 2.17 (t, J = 7.5 Hz), 1.79 (q, J = 7.5 Hz). 13C-NMR (CD3OD) δ 180.87, 66.28 (2C), 58.7, 53.41 (2C), 35.61, 22.89. HRMS (FAB+): m/z calcd for C8H15NO3: 173.11, found (M + 1): 174.1171.
4-Thiomorpholinobutanoic acid (7f). 0.22 g (83%) of a yellow solid, m.p = 82–83 °C. 1H-NMR (CDCl3): δ 2.77–2.70 (m, 4H), 2.66–2.65 (m, 4H), 2.41–2.37 (m, 2H), 2.14 (t, J = 7.5 Hz, 2H), 1.78 (q, J = 7.6 Hz). 13C-NMR (CDCl3) δ 182.32, 60.37, 56.20 (2C), 36.95, 28.37 (2C), 24.14. HRMS (FAB+): m/z calcd for C8H15NO2S: 189.08, found (M + 1): 190.1053.
3.2.4. General Procedure for the Synthesis of Esters 14a–f
Amines 10a–f (1.0 equiv), N,N-diisopropylethylamine (1.5 equiv), ethyl (E)-4-bromobut-2-enoate (5, 1.3 equiv) and CH2Cl2 (25 mL) were placed in a round bottom flask and stirred at −20 °C for 1 h, under nitrogen. After completion of the reaction (TLC) the reaction mixture was extracted with NaHCO3 (3 × 30 mL) and water (1 × 30 mL). The organic phase was dried over Na2SO4 and the solvent removed under reduced pressure to yield the crude product. The crude product was purified by silica gel column chromatography (9:1 hexanes:EtOAc).
Ethyl (E)-4-(thiazolidin-3-yl)but-2-enoate (14a). 1.21 g (63 %) as a yellow oil. 1H-NMR (CDCl3) δ 6.93 (dt, J = 16, 8 Hz, 1H), 6.01 (dt, J = 16, 1.8 Hz, 1H), 4.18 (q, J = 7 Hz, 2H), 4.03 (s, 2H), 3.15 (dd, J = 8, 1.8 Hz, 2H), 3.07 (t, J = 6.4 Hz, 2H), 2.87 (t, J = 6.4 Hz, 2H), 1.27 (t, J = 7 Hz, 3H). 13C-NMR (CDCl3) δ 166.24, 144.94, 123.42, 60.59 (2C), 57.92, 53.90, 29.62, 14.34. HRMS (FAB+): m/z calcd for C8H15NO2S: 201.284, found (M + 1): 202.0912.
Ethyl (E)-4-(piperidin-1-yl)but-2-enoate (14b). 0.9 g (90 %) as a yellow oil. 1H-NMR (CDCl3): δ 6.95 (dt, J = 15.7, 6.3 Hz, 1H), 5.92 (dt, J = 15.7, 1.6 Hz, 1H), 4.16 (q, J = 7.1 Hz, 2H), 3.07 (dd, J = 6.3, 1.6 Hz, 2H), 2.37 (sa, 4H), 1.56 (q, J = 5.6 Hz, 4H), 1.44–1.38 (m, 2H), 1.25 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3) δ 166.39, 145.69, 123.34, 60.45, 60.25, 54.84 (2C), 26.05 (2C), 24.24, 14.39. HRMS (FAB+): m/z calcd for C11H19NO2: 197.1416, found (M + 1): 198.1506.
Ethyl (E)-4-(3-methylpiperidin-1-yl)but-2-enoate (14c). 0.83 g (77%) as a yellow oil. 1H-NMR (CDCl3): δ 6.96 (dt, J = 15.7, 6.3 Hz, 1H), 5.94 (dt, J = 15.7, 1.6 Hz, 1H), 4.17 (q, J = 7.2 Hz, 2H), 3.09 (dd, J = 6.3, 1.6 Hz, 2H), 2.85–2.74 (m, 2H), 1.87 (td, J = 11.3, 3.2 Hz), 1.71–1.54 (m, 5H), 1.27 (t, J = 7.2 Hz, 3H), 0.84 (d, J = 6.4 Hz, 4H). 13C-NMR (CDCl3) δ 166.44, 145.63, 123.43, 62.22, 60.50, 60.01, 54.32, 32.90, 31.31, 25.65, 19.83, 14.42. HRMS (FAB+): m/z calcd for C12H21NO2: 211.1572, found (M + 1): 212.1651.
Ethyl (E)-4-(4-methylpiperidin-1-yl)but-2-enoate (14d). 0.85 g (80%) as a yellow oil. 1H-NMR (CDCl3) δ 6.94 (dt, J = 15.6, 6.4 Hz, 1H), 5.92 (dt, J = 15.5, 1.6 Hz, 1H), 4.15 (q, J = 7.1 Hz, 2H), 3.08 (dd, J = 6.4, 1.6 Hz, 2H), 2.82 (d, J = 11.8 Hz, 4H), 2.02–1.80 (m, 2H), 1.65–1.55 (m, 2H), 1.38–1.31 (m, 1H), 1.25 (t, J = 7.1 Hz, 3H), 0.89 (d, J = 5.7 Hz, 3H). 13C-NMR (CDCl3) δ 166.46, 145.77, 123.42, 60.53, 59.96, 54.35 (2C), 34.45 (2C), 30.73, 22.06, 14.46. HRMS (FAB+): m/z calcd for C12H21NO2: 211.1572, found (M + 1): 212.1651.
Ethyl (E)-4-morpholinobut-2-enoate (14e). 1.02 g (89%) as a yellow oil. 1H-NMR (200 MHz, CDCl3) δ 6.91 (dt, J = 15.7, 6.2 Hz, 1H), 5.96 (dt, J = 15.7, 1.6 Hz, 1H), 4.17 (q, J = 7.1 Hz, 2H), 3.72–3.67 (m, 4H), 3.1 (dd, J = 6.2, 1.6 Hz, 2H), 2.49–2.39 (m, 4H), 1.26 (t, J = 7.1 Hz, 3H). 13C-NMR (50 MHz, CDCl3) δ 166.05, 144.38, 123.65, 66.83 (2C), 60.38, 59.55, 53.64 (2C), 14.20. HRMS (FAB+): m/z calcd for C10H17NO3: 199.1208, found (M + 1): 200.1291.
Ethyl (E)-4-thiomorpholinobut-2-enoate (14f). 0.85 g (80%) as a yellow oil. 1H-NMR (CDCl3) δ 6.88 (dt, J = 15.7, 6.1 Hz, 1H), 5.94 (dt, J = 15.7, 1.7 Hz, 1H), 4.16 (q, J = 7.1 Hz, 2H), 3.11 (dd, J = 6.1, 1.7 Hz, 2H), 2.67 (m, 8H), 1.25 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3) δ 166.00, 144.72, 123.43, 60.30, 59.91, 54.97 (2C), 27.88 (2C), 14.15. HRMS (FAB+): m/z calcd for C12H21NO2: 215.098, found (M + 1): 216.1044.
3.2.5. General Methodology for the Conjugate Additions of 15, 17
Most of the reactions were carried out using the following procedure: CuI (2.00 equiv) was placed in a one-neck 50 mL round-bottom flask equipped with a stirring bar and sealed with a septum. The flask was evacuated with a vacuum pump and then purged with nitrogen, this process was repeated three times to assure a complete nitrogen atmosphere. Anhydrous ethyl ether (25.00 mL) was injected and the mixture stirred for 15 min and then cooled to 0 °C. Simultaneously, a Grignard solution was prepared mixing finely divided Mg turnings (4.1 equiv) and catalytic amounts of I2, with 1-bromo-2-methylpropane (4.0 equiv) or 1-bromo-4-chlorobenzene (4.0 equiv) at room temperature. The mixture was stirred for 30 min and then transferred via syringe dropwise to the copper solution at 0 °C, and stirred at this temperature for another 30 min period. Then, the unsaturated substrate 14a–f (1.0 equiv) were added dropwise. The reaction was allowed to proceed for 24 h before being quenched with a saturated aqueous solution of NH4Cl. The mixture was extracted with ether (3 × 25 mL) dryed over Na2SO4 and the solvent removed in vacuo to afford the crude products which were purified by flash chromatography eluting with hexane:EtOAc (95:5). Some modifications done to this procedure are mentioned in the specific examples below.
Ethyl 5-methyl-3-(thiazolidin-3-ylmethyl)hexanoate (16a). 0.023 g (5%) as a yellow oil. 1H-NMR (CDCl3): δ 4.16–4.07 (m, 2H), 4.02 (q, J = 9.2 Hz, 2H), 3.04–3.0 (m, 2H), 2.90–2.82 (m, 2H), 2.44–2.36 (m, 2H), 2.27 (dd, J = 15.1, 5.6 Hz, 1H), 2.12–2.06 (m, 2H), 1.64 (m, 1H), 1.25 (t, J = 7.1 Hz, 3H), 1.22–1.08 (m, 2H), 0.89 (t, J = 6.4 Hz, 6H). 13C-NMR (CDCl3) δ 173.54, 61.44, 60.25, 58.65, 58.14, 42.40, 38.58, 33.28, 29.88, 25.40, 23.02, 22.77, 14.42. HRMS (FAB+): m/z calcd for C13H25NO2S: 259.1606, found (M + 1): 260.1699.
Ethyl 5-methyl-3-(piperidin-1-ylmethyl)hexanoate (16b). 0.17 g (52%) as a yellow oil. 1H-NMR (CDCl3): δ 4.05 (q, J = 7.2 Hz, 2H), 2.44–2.32 (m, 3H), 2.24–2.11 (m, 5H), 2.05–1.99 (m, 1H), 1.68–1.57 (m, 1H), 1.52–1.46 (m, 4H), 1.39–1.32 (m, 2H), 1.19 (t, J = 7.2 Hz, 3H), 1.11–0.97 (m, 2H), 0.81 (dd, J = 6.5, 5.3 Hz, 6H). 13C-NMR (CDCl3) δ 173.85, 64.54, 60.04, 55.21 (2C), 43.00, 39.25, 30.97, 26.23 (2C), 25.52, 24.69, 22.95 (2C), 14.40. HRMS (FAB+): m/z calcd for C15H29NO2: 255.2198, found (M + 1): 256.2267.
Ethyl 5-methyl-3-((3-methylpiperidin-1-yl)methyl)hexanoate (16c). 0.09 g (20%) as an amber oil. 1H-NMR (CDCl3): δ 4.09 (q, J = 7.1 Hz, 2H), 2.84–2.76 (m, 1H), 2.68–2.61 (m, 1H), 2.38–2.32 (m, 1H), 2.21–2.09 (m, 3H), 2.08–1.99 (m, 1H), 1.92–1.85 (m, 1H), 1.71–1.36 (m, 6H), 1.24 (t, J = 7.1 Hz, 3H), 1.17–1.02 (m, 2H), 0.88–0.81 (m, 10H). 13C-NMR (CDCl3): δ 173.79 (C-3), 64.31 (C-6), 64.27 (C-6), 63.69 (C-7), 62.03 (C-7), 60.01 (C-2), 55.52 (C-12), 53.90 (C-12), 43.10 (C-13), 43.07 (C-13), 39.21 (C-4), 33.33 (C-10), 31.41 (C-8), 31.13 (C-5), 25.80 (C-11), 25.70 (C-11), 25.54 (C-14), 23.03(C-15), 22.97 (C-16), 19.83 (C-9), 19.81 (C-9), 14.40 (C-1). HRMS (FAB+): m/z calcd for C16H31NO2: 269.2355, found (M + 1): 270.2411.
Ethyl 5-methyl-3-((4-methylpiperidin-1-yl)methyl)hexanoate (16d). 0.164 g (37%) as a yellow oil. 1H-NMR (CDCl3): δ 4.12 (q, J = 7.2 Hz, 2H), 2.87 (d, J = 11.3 Hz, 1H), 2.71 (d, J = 11.8 Hz, 1H), 2.36 (dd, J = 5.7 Hz, 1H), 2.18–2.11 (m, 2H), 2.07 (m, 1H), 1.96 (ddd, J = 11.5,, 11.5, 2.5 Hz), 1.74 (ddd, J = 11.6, 2.4 Hz, 1H), 1.64 (m, J = 6.4 Hz, 1H), 1.57–1.53 (m, 2H), 1.26 (t, J = 7.1 Hz, 4H), 1.20–1.03 (m, 4H), 0.91–0.85 (m, 9H). 13C-NMR (CDCl3) δ 173.89, 64.13, 60.06, 55.56, 53.67, 42.98, 39.27, 34.78, 34.52, 31.05, 31.03, 25.44, 22.95 (2C), 22.13, 14.41. HRMS (FAB+): m/z calcd for C16H31NO2: 269.2355, found (M + 1): 270.2429.
Ethyl 5-methyl-3-(morpholinomethyl)hexanoate (16e). 0.166 g (37%) as a yellow oil. 1H-NMR (CDCl3): δ 4.13 (q, J = 7.2 Hz, 2H), 3.71–3.59 (m, 4H), 2.50 (m, 2H), 2.34 (m, 3H), 2.26–2.16 (m, 3H), 2.13–2.06 (m, 1H), 1.71–1.59 (m, 1H), 1.26 (t, J = 7.1 Hz, 3H), 1.20–1.06 (m, 2H), 0.89 (d, J = 6.3 Hz, 6H). 13C-NMR (CDCl3): δ 173.71, 67.23 (2C), 64.25, 60.21, 54.16 (2C), 42.82, 39.09, 30.42, 25.41, 22.96, 22.86, 14.40. HRMS (FAB+): m/z calcd for C14H27NO3: 257.1991, found (M + 1): 258. 2077.
Ethyl 5-methyl-3-(thiomorpholinomethyl)hexanoate (16f). 0.11 g (25%) as a yellow oil. 1H-NMR (CDCl3): δ 4.17–4.07 (m, 2H), 2.78–2.71 (m, 2H), 2.63–2.53 (m, 6H), 2.32–2.24 (m, 2H), 2.2–2.13 (m, 2H), 2.04 (dd, J = 12.2, 9.0 Hz, 1H), 1.69–1.57 (m, 1H), 1.27 (t, J = 7.2 Hz, 3H), 1.16–1.03 (m, 2H), 0.88 (d, J = 6.2 Hz, 6H). 13C-NMR (CDCl3): δ 173.76, 64.60, 60.22, 55.75, 42.82, 39.15, 30.87, 28.18, 25.47, 23.03, 22.91, 14.46. HRMS (FAB+): m/z calcd for C14H27NO3S: 273.1762, found (M + 1): 274.1858.
Ethyl 3-(4-chlorophenyl)-4-(piperidin-1-yl)butanoate (18b). 0.2 g (52%) as a yellow oil. 1H-NMR (CDCl3): δ 7.25 (d, J = 8.5 Hz, 2H) 7.13 (d, J = 8.5 Hz, 2H), 4.03 (q, J = 7.1 Hz, 2H), 3.35 (ddd, J = 11.9, 9.1, 6.0 Hz, 1H), 2.87 (dd, J = 15.5, 6.3 Hz, 1H), 2.49–2.41 (m, 3H), 2.41–2.32 (m, 2H), 2.31–2.23 (m, 2H), 1.55–1.46 (m, 4H), 1.42–1.38 (m, 2H), 1.16 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3) δ 172.57, 141.72, 132.11, 128.90 (2C), 128.47 (2C), 65.17, 60.19, 54.87 (2C), 39.25 (2C), 26.00, 24.34, 14.12. HRMS (FAB+): m/z calcd for C17H24ClNO2: 309.1496, found (M + 1): 310.1556.
Ethyl 3-(4-chlorophenyl)-4-(3-methylpiperidin-1-yl)butanoate (18c). 0.22 g (49%) as a yellow oil. 1H-NMR (CDCl3): δ 7.24 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 8.5 Hz, 2H), 4.02 (q, J = 7.1 Hz, 2H), 3.42–3.32 (m, 1H), 2.86 (dd, J = 15.5, 6.3 Hz, 2H), 2.69 (d, J= 8.8 Hz, 1H), 2.49–2.39 (m, 2H), 2.35 (dd, J = 12.6, 5.7 Hz, 1H), 1.96 (ddd, J = 11.2, 11.2, 3.0 Hz, 1H), 1.75 (ddd, J = 11.4, 11.4, 2.7 Hz, 1H), 1.71–1.39 (m, 5H), 1.15 (t, J = 7.1 Hz, 3H), 0.83 (t, J = 6.2 Hz, 4H). 13C-NMR (CDCl3): δ 172.66 (C-3), 141.66 (C-13), 132.14 (C-16), 128.90 (C-15), 128.48 (C-14), 64.93 (C-6), 64.90 (C-6), 63.35 (C-7), 61.42 (C-7), 60.25 (C-2), 55.28 (C-12), 53.40 (C-12), 39.32 (C-5), 39.24 (C-4), 32.92 (C-10), 31.14 (C-8), 30.87 (C-8), 25.52 (C-11), 25.38 (11), 19.67 (C-9), 19.63 (C-9), 14.10 (C-1). HRMS (FAB+): m/z calculado para C18H26ClNO2: 323.1652, experimental (M + 1): 324.1734.
Ethyl 3-(4-chlorophenyl)-4-(4-methylpiperidin-1-yl) butanoate (18d). 0.75 g (49%) as a yellow oil. 1H-NMR (CDCl3): δ 7.24 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 8.5 Hz, 2H), 4.03 (q, J = 7.1 Hz, 2H), 3.39–3.31 (m, 1H), 2.94–2.89 (d, J = 11.1 Hz, 1H), 2.85 (dd, J = 15.5, 6.3 Hz, 1H), 2.74 (d, J = 11.1 Hz, 1H), 2.50–2.41 (m, 2H), 2.36 (dd, J = 12.6, 5.7 Hz, 1H), 2.03 (ddd, J = 11.1, 11.1, 2.5 Hz, 1H), 1.83 (ddd, J = 11.1, 11.1, 2.5 Hz, 1H), 1.59–1.52 (m, 2H), 1.35–1.24 (m, 1H), 1.15 (t, J = 7.1 Hz, 6H), 0.89 (d, J = 6.4 Hz, 3H). 13C-NMR (CDCl3) δ 172.36, 141.75, 132.06, 128.88 (2C), 128.42 (2C), 64.76, 60.04, 55.34, 53.18, 39.38, 39.18, 34.55, 34.28, 30.71, 21.90, 14.11. HRMS (FAB+): m/z calcd for C18H26ClNO2: 323.1652, found (M + 1): 324.1772.
Ethyl 3-(4-chlorophenyl)-4-morpholinobutanoate (18e). 0.645 g (55 %) as a yellow oil. 1H-NMR (CDCl3): δ 7.25 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H), 4.05 (q, J = 7.1 Hz, 2H), 3.67–3.59 (m, 4H), 3.43–3.32 (m, 1H), 2.85 (dd, J = 15.5, 6.6 Hz, 1H), 2.58–2.30 (m, 7H), 1.16 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3) δ 172.25, 141.22, 132.21, 128.82 (2C), 128.50 (2C), 66.89 (2C), 64.71, 60.20, 53.79 (2C), 39.12, 38.67, 14.09. HRMS (FAB+): m/z calcd for C16H22ClNO3: 311.1288, found (M + 1): 312.1299.
Ethyl 3-(4-chlorophenyl)-4-thiomorpholinobutanoate (18f). 0.66 g (58 %) as a yellow oil. 1H-NMR (CDCl3): δ 7.25 (d, J = 8.5 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 4.05 (q, J = 7.1 Hz, 2H), 3.40–3.30 (m, 1H, H-5), 2.83–2.77 (m, 3H), 2.66–2.56 (m, 6H), 2.5–2.39 (m, 3H), 1.18 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3): δ 172.42, 141.23, 132.29, 128.86 (2C), 128.55 (2C), 65.06, 60.34, 55.39 (2C), 39.13 (2C), 27.92 (2C), 14.14. HRMS (FAB+): m/z calcd for C16H22ClNO2S: 327.106, found (M + 1): 328.1084.
3.2.6. General Procedure for the Hydrolysis of Esters 16a–f and 18b–f
The corresponding ethyl ester 16a–f or 18b–f (1.0 equiv) were dissolved in MeOH (6.0 mL), and NaOH (1.1 equiv) dissolved in water (2 mL) was added dropwise. The reaction was monitored until the consumption of the starting materials (tlc). When the reaction was complete, the solvent was evaporated and aqueous HCl was added until the mixture reached a pH = 5. The mixture was extracted with ethyl acetate (3 × 5 mL), dried over Na2SO4 and the solvent removed in vacuo.
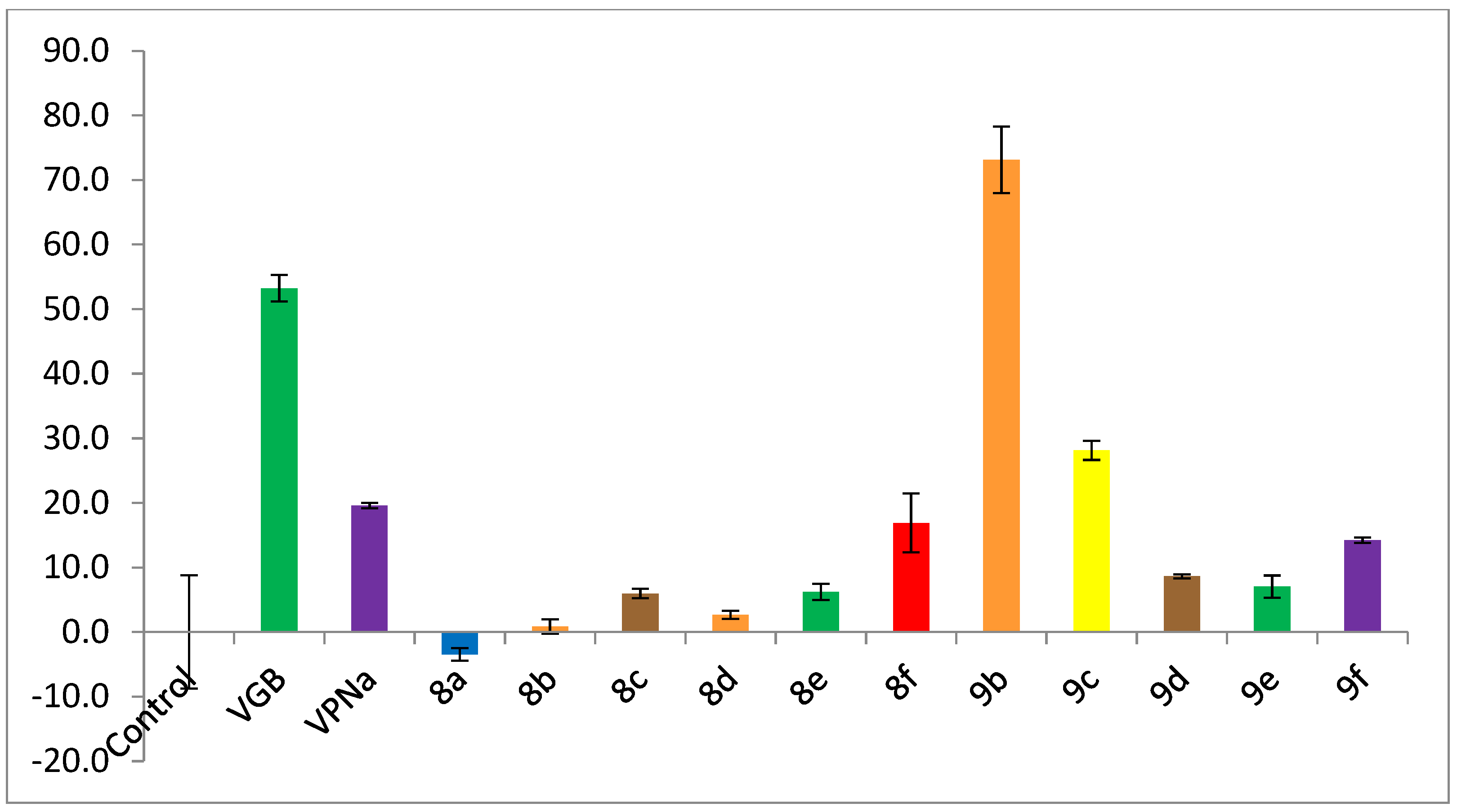
5-Methyl-3-(thiazolidin-3-ylmethyl)hexanoic acid (8a). 0.055 g (77%) as a yellow oil. 1H-NMR (CD3OD): δ 3.83 (s, 2H), 3.51–3.42 (m, 1H), 3.23 (m, 2H), 3.10–2.68 (m, 3H), 2.56–2.44 (m, 1H), 2.37–2.18 (m, 2H), 1.74–1.63 (m, 1H), 1.22–1.12 (m, 2H), 0.94–0.91 (m, 6H). 13C-NMR (CD3OD): δ 180.44, 60.81, 59.41, 51.43, 43.92, 39.09, 31.45, 30.39, 26.31, 23.36, 22.65.
5-Methyl-3-(piperidin-1-ylmethyl)hexanoic acid (8b). 0.55 g (90%) as a yellow oil. 1H-NMR (CD3OD): δ 3.04–2.99 (m, 1H), 2.85 (dd, J = 13.2, 10.4 Hz, 1H), 2.52 (dt, J = 16.6, 1.8 Hz, 1H), 2.35 (dd, J = 16.6, 10.3 Hz, 1H), 2.20 (m, 1H), 1.86 (s, 5H), 1.74–1.63 (m, 2H), 1.14 (dd, J = 7.2, 7.2 Hz, 2H), 0.935 (d, J = 4 Hz, 3H), 0.925 (d, J = 4 Hz, 3H). 13C-NMR (CD3OD) δ 181.03, 65.16 (3C), 44.96, 44.16, 30.36, 26.11, 24.76 (2C), 23.00, 22.96, 22.77. HRMS (FAB+): m/z calcd for C13H25NO2: 227.1885, found (M + 1): 228.1988.
5-Methyl-3-((3-methylpiperidin-1-yl)methyl)hexanoic acid (8c). 0.04 g (70%) as a yellow oil. 1H-NMR (CD3OD): δ 3.61–3.47 (m, 2H), 3.28–3.26 (m, 1H), 3.04 (d, J = 13.2 Hz, 1H), 2.94–2.85 (m, 2H), 2.74–2.59 (m, 1H), 2.53 (dd, J = 16.7 Hz, 1H), 2.36 (dd, J = 16.6, 10.2 Hz, 1H), 2.27–2.17 (m, 1H), 1.94–1.88 (m, 2H), 1.88–1.77 (m, 2H), 1.73–1.64 (m, 1H), 1.15 (dd, J = 7.2, 7.2 Hz, 2H), 1.02–0.99 (m, 3H), 0.95–0.92 (m, 6H). 13C-NMR (CD3OD): δ 180.92 (C-1), 65.05 (C-4), 59.38 (C-5), 53.18 (C-10), 44.79 (C-2), 44.70 (C-2), 44.15 (C-11), 44.13 (C-11), 31.54 (C-8), 30.37 (C-6, C-9), 30.26 (C-3), 26.11 (C-12), 23.05 (C-15), 22.99 (C-15), 22.80 (C-16), 22.77 (C-16), 18.95 (C-7). δ HRMS (FAB+): m/z calcd for C14H27NO2: 241.2042, found (M + 1): 242.2106.
5-Methyl-3-((4-methylpiperidin-1-yl)methyl)hexanoic acid (8d). 0.1 g (90%) as a yellow oil. 1H-NMR (CD3OD): δ 3.64–3.56 (m, 2H), 3.04–3.00 (m, 1H), 2.88 (dd, J = 13.4, 10.3 Hz, 1H), 2.57–2.48 (m, 1H), 2.35 (dd, J = 16.8, 10.3 Hz, 1H), 2.25–2.14 (m, 1H), 1.91–1.82 (m, 2H), 1.68 (m, 1H), 1.53–1.48 (m, 3H), 1.14 (ddd, J = 7.3 Hz, 2H), 1.01 (d, J = 6.7 Hz, 3H), 0.93 (d, J = 6.7 Hz, 6H). 13C-NMR (CD3OD) δ 180.86, 64.63, 53.21 (2C), 44.75, 44.07, 32.56 (2C), 30.37, 28.50, 26.05, 22.99, 22.76, 21.24. HRMS (FAB+): m/z calcd for C14H27NO2: 241.2042, found (M + 1): 242.2121.
5-Methyl-3-(morpholinomethyl)hexanoic acid (8e). 0.1 g (85%) as a yellow oil. 1H-NMR (CD3OD): δ 3.89–3.87 (m, 4H), 3.29–3.24 (m, 2H), 3.12–3.06 (m, 2H), 3.05–3.00 (m, 1H), 2.88 (dd, J = 13.1, 10.2 Hz), 2.54–2.48 (m, 1H), 2.41 (dd, J = 16.6, 9.6 Hz, 1H), 2.29–2.18 (m, 1H), 1.75–1.62 (m, 1H), 1.16 (dd, J = 3.9, 3.9 Hz, 2H), 0.93 (d, J =4.8 Hz, 3H), 0.92 (d, J =4.8 Hz, 3H). 13C-NMR (CD3OD) δ 180.28, 65.80 (2C), 64.92, 53.58 (2C), 43.95 (2C), 29.94, 26.12, 23.03, 22.79. HRMS (FAB+): m/z calcd for C14H27NO2: 229.1678, found (M + 1): 230.1743.
5-Methyl-3-(thiomorpholinomethyl)hexanoic acid (8f). 0.067 g (90%) as a yellow oil. 1H-NMR (CD3OD): δ 3.51–3.44 (m, 2H), 3.35–3.29 (m, 2H), 3.11–3.02 (m, 1H), 2.95–2.92 (m, 4H), 2.88–2.80 (m, 1H), 2.50 (d, J = 16.6 Hz, 1H), 2.39 (dd, J = 16.6, 10.0 Hz, 1H), 2.25–2.16 (m, 1H), 1.68 (sept, J = 8 Hz, 1H), 1.14 (dd, J = 7.2, 7.2 Hz, 2H), 0.92 (d, J = 5.2 Hz, 3H), 0.9 (d, J = 5.2 Hz, 3H). 13C-NMR (CD3OD) δ 179.55, 64.35, 54.31 (2C), 43.16, 42.58, 28.83, 25.24 (2C), 24.71, 21.72, 21.41. HRMS (FAB+): m/z calcd for C14H27NO2: 245.1449, found (M + 1): 246.1524.
3-(4-Chlorophenyl)-4-(piperidin-1-yl)butanoic acid (9b). 0.3 g (85%) of a white solid, m.p = 146–149 °C. 1H-NMR (CD3OD): δ 7.34 (d, J = 8.6 Hz, 2H), 7.27 (d, J = 8.6 Hz, 2H), 3.52 (dddd, J = 8 Hz, 8 Hz, 4 Hz, 4 Hz, 1H), 3.37 (dd, J = 14, 8 Hz, 3H), 3.11 (ddd, J = 13.0, 2.0, 1.2 Hz, 3H), 2.91 (dd, J = 16.7, 5.2 Hz, 1H), 2.63 (ddd, J = 13.6, 13, 1.2 Hz, 1H), 1.89–1.82 (m, 4H), 1.64 (sa, 2H). 13C-NMR (CD3OD) δ 179.28, 142.37, 133.06, 130.11 (2C), 129.81 (2C), 64.71, 54.68, 45.63, 38.12, 24.62 (2C), 22.93. HRMS (FAB+): m/z calcd for C15H20ClNO2: 281.1183, found (M + 1): 282.1287.
3-(4-Chlorophenyl)-4-(3-methylpiperidin-1-yl)butanoic acid (9c). 0.2 g (52%) as a yellow oil. 1H-NMR (CD3OD): δ 7.37–7.26 (m, 4H), 3.76 (d, J = 11.5 Hz, 1H), 3.68 (d, J = 11.8 Hz, 1H), 3.58–3.47 (m, 1H), 3.43–3.36 (m, 2H), 3.08 (d, J = 13.0 Hz, 1H), 2.93 (dd, J = 16.7, 10.8 Hz, 1H), 2.70–2.55 (m, 2H), 2.41 (t, J = 11.7 Hz, 1H), 1.95–1.79 (m, 4H), 1.21–1.09 (m, 1H), 1.01 (d, J = 6.7 Hz, 1H), 0.99 (d, J = 6.7 Hz, 2H). 13C-NMR (CD3OD): δ 179.60 (C-1), 179.57 (C-1), 142.62(C-11), 134.13 (C-14), 130.23 (C-13), 129.97 (C-12), 64.91(C-4), 64.81 (C-4), 61.19 (C-5), 59.66 (C-5), 54.89 (C-10), 53.48 (C-10), 45.93 (C-2), 38.34 (C-3), 38.19 (C-3), 31.67 (C-8), 31.21 (C-6), 30.85 (C-6), 24.39 (C-9), 24.27 (C-9), 19.13 (C-7), 19.11 (C-7). HRMS (FAB+): m/z calcd for C16H22ClNO2: 295.1339, found (M + 1): 296.1445.
3-(4-Chlorophenyl)-4-(4-methylpiperidin-1-yl)butanoic acid (9d). 0.26 g (68%) of a yellow solid, m.p = 142–144 °C. 1H-NMR (CD3OD): δ 7.34 (d, J = 8.6 Hz, 1H), 7.27 (d, J = 8.6 Hz, 1H), 3.77 (d, J = 11.7 Hz, 1H), 3.54–3.49 (m, 1H), 3.47–3.34 (m, 2H), 3.08 (d, J = 12.9 Hz, 1H), 3.01–2.95 (m, 1H), 2.93 (dd, J = 16, 16 Hz, 1H), 2.78 (dd, J = 11.4, 11.4 Hz, 1H), 2.62 (dd, J = 16, 4 Hz,1H), 1.94–1.85 (m, 2H), 1.69 (m, 1H), 1.59–1.45 (m, 2H), 1.01 (d, J = 6.5 Hz, 3H). 13C-NMR (CD3OD) δ 179.75, 142.61, 134.00, 130.10 (2C), 129.74 (2C), 64.59, 54.86, 53.36, 46.09, 38.25, 32.84, 32.57, 29.94, 21.22. HRMS (FAB+): m/z calcd for C16H22ClNO2: 295.1339, found (M + 1): 296.1445
3-(4-Chlorophenyl)-4-morpholinobutanoic acid (9e). 0.28 g (69%) of a yellow solid, m.p = 166–168 °C. 1H-NMR (CD3OD): δ 7.32 (d, J = 8.5 Hz, 2H), 7.25 (d, J = 8.5 Hz, 2H), 3.81–3.78 (m, 4H), 3.5–3.43 (dddd, J = 4.8, 4.8, 4, 4 Hz, 1H), 3.08–2.98 (m, 3H), 2.94–2.81 (m, 4H), 2.57 (dd, J = 16.2, 4.5 Hz, 1H). 13C-NMR (CD3OD) δ 178.35, 142.88, 133.90, 130.13 (2C), 130.00 (2C), 66.77 (2C), 65.41, 54.26 (2C), 43.60 (1C), 38.88. HRMS (FAB+): m/z calcd for C16H22ClNO2: 283.0975, found (M + 1): 284.1009.
3-(4-Chlorophenyl)-4-thiomorpholinobutanoic acid (9f). 0.288 g (70%) of a white solid, m.p = 144–146 °C. 1H-NMR (CD3OD): δ 7.32 (d, J = 8 Hz, 2H), 7.25 (d, J = 8 Hz, 2H), 3.53–3.44 (m, 1H), 3.37–3.33 (m, 2H), 3.17–3.06 (m, 3H), 2.97–2.85 (m, 2H), 2.87–2.85 (m, 4H), 2.58 (dd, J = 16.3, 3.8 Hz, 1H). 13C-NMR (CD3OD) δ 178.90, 142.77, 133.79, 129.92 (2C), 129.89 (2C), 65.79, 55.95 (2C), 44.32, 38.78, 27.23 (2C). HRMS (FAB+): m/z calcd for C16H22ClNO2: 299.0747, found (M + 1): 300.0811.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



























