Gas Phase Hydrogenation of Furaldehydes via Coupling with Alcohol Dehydrogenation over Ceria Supported Au-Cu


Abstract
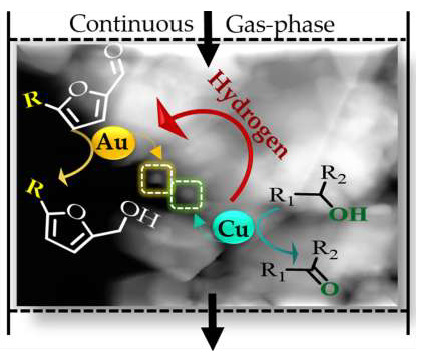
1. Introduction
2. Results and Discussion
2.1. Catalytic Conversion of 5-Hydroxymethyl-2-Furaldehyde (HMF): Coupling Dehydrogenation-Hydrogenation vs. Conventional Stand-Alone Hydrogenation Using an External H2 Supply
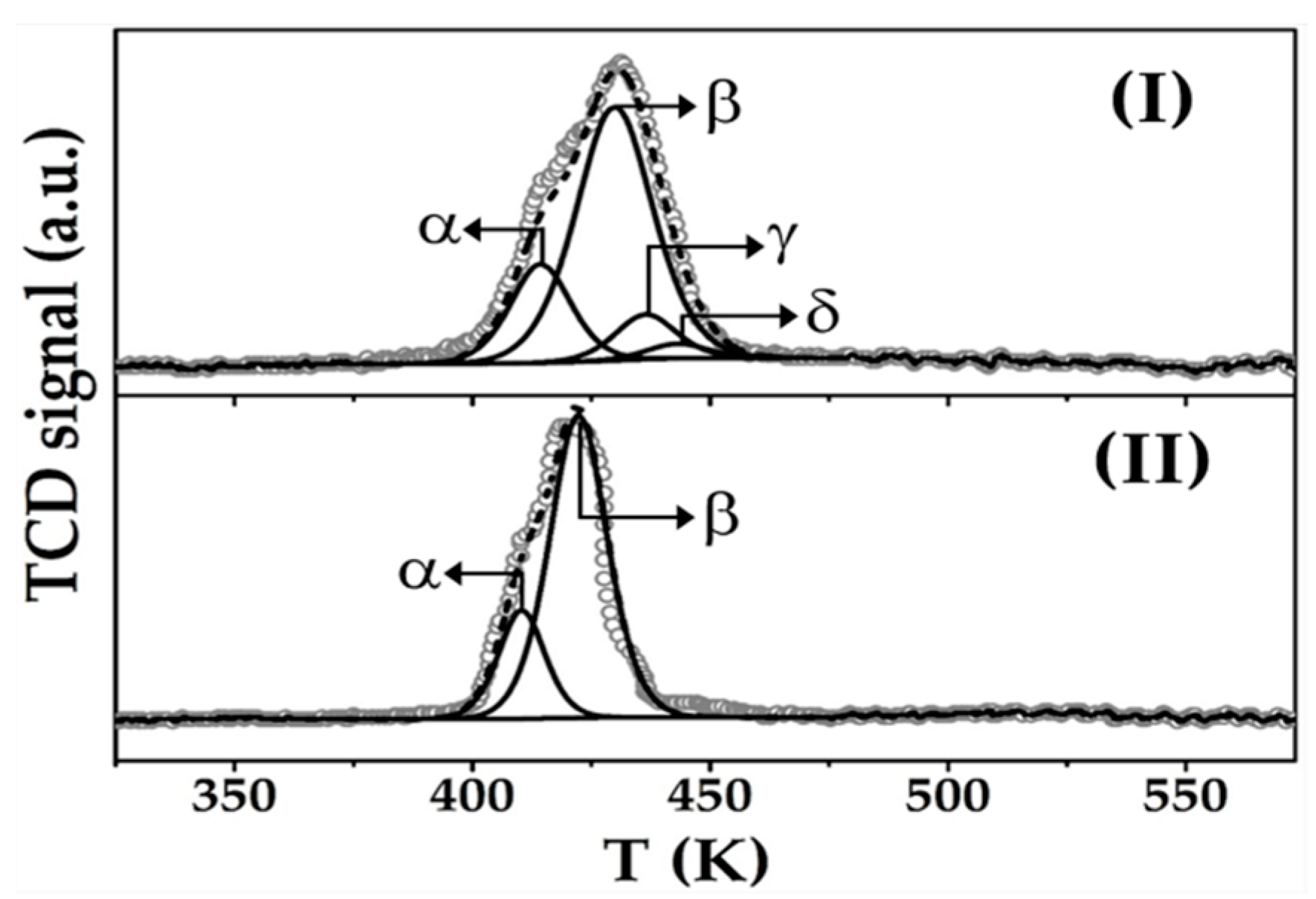
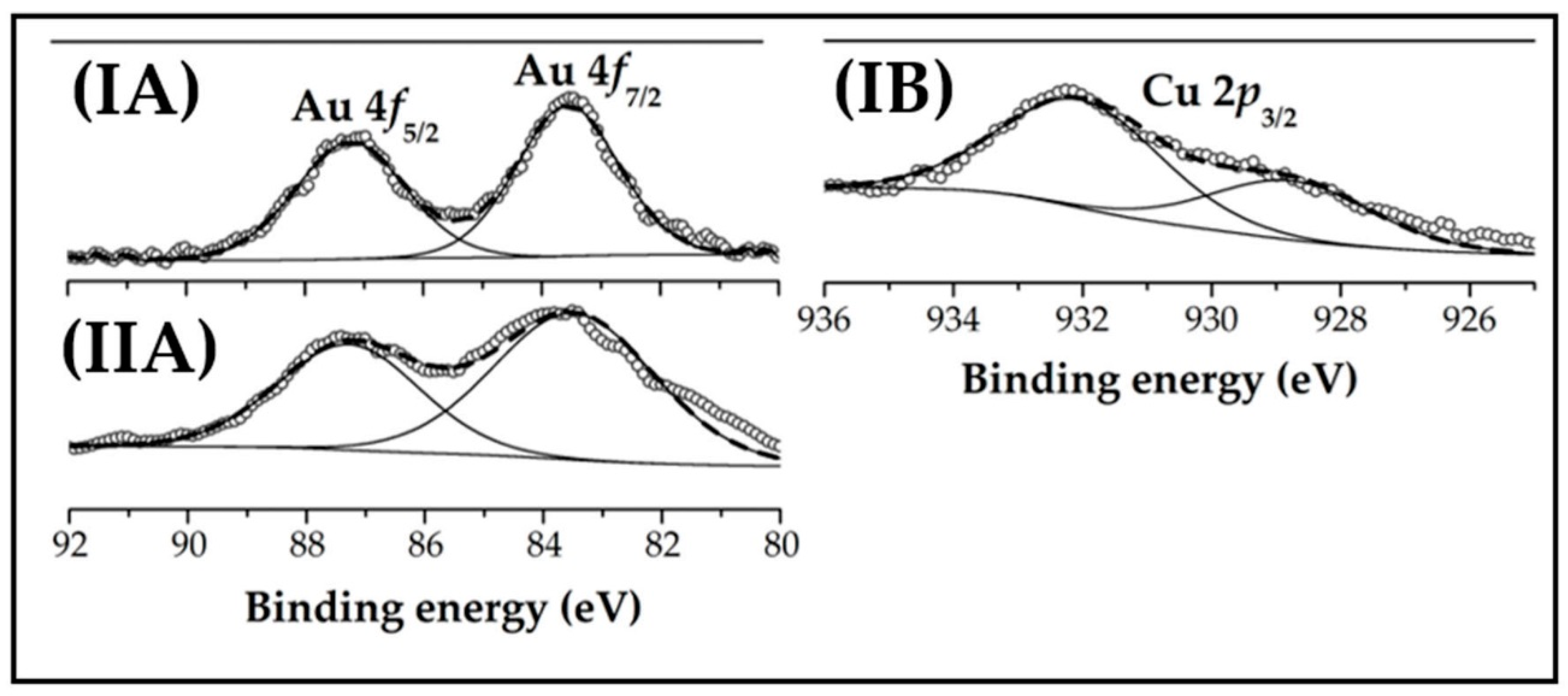
2.2. Catalyst Characterisation: Au-Cu/CeO2 and Au/CeO2
2.3. Dehydrogenation of Alcohols Coupled with Hydrogenation of meta-Substituted Furaldehydes
3. Materials and Methods
3.1. Catalyst Preparation and Activation
3.2. Catalyst Characterisation
3.3. Catalytic Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Van Putten, R.-J.; Van der Waal, J.C.; De Jong, E.D.; Rasrendra, C.B.; Heeres, H.J.; De Vries, J.G. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Nolte, M.W.; Shanks, B.H. Catalytic dehydration of C6 carbohydrates for the production of hydroxymethylfurfural (HMF) as a versatile platform chemical. Green Chem. 2014, 16, 548–572. [Google Scholar] [CrossRef]
- Zieleniewska, M.; Szczepkowski, L.; Krzyżowska, M.; Leszczyński, M.; Ryszkowska, J. Rigid polyurethane foam composites with vegetable filler for application in the cosmetics industry. Polimery 2016, 61, 807–814. [Google Scholar] [CrossRef]
- Kong, X.; Zhu, Y.; Zheng, H.; Dong, F.; Zhu, Y.; Li, Y.-W. Switchable synthesis of 2,5-dimethylfuran and 2,5-dihydroxymethyltetrahydrofuran from 5-hydroxymethylfurfural over Raney Ni catalyst. RSC Adv. 2014, 4, 60467–60472. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ishizaka, T.; Kawanami, H. Selective hydrogenation of 5-hydroxymethylfurfural to 2,5-bis-(hydroxymethyl) furan using Pt/MCM-41 in an aqueous medium: A simple approach. Green Chem. 2014, 16, 4734–4739. [Google Scholar] [CrossRef]
- Hu, L.; Xu, J.; Zhou, S.; He, A.; Tang, X.; Lin, L.; Xu, J.; Zhao, Y. Catalytic advances in the production and application of biomass-derived 2,5-dihydroxymethylfuran. ACS Catal. 2018, 8, 2959–2980. [Google Scholar] [CrossRef]
- Schüth, F. Hydrogen: Economics and its role in biorefining. In Catalytic Hydrogenation for Biomass Valorization; The Royal Society of Chemistry: London, UK, 2014; pp. 1–21. ISBN 1-84973-801-7. [Google Scholar]
- Hu, L.; Tang, X.; Xu, J.; Wu, Z.; Lin, L.; Liu, S. Selective transformation of 5-hydroxymethylfurfural into the liquid fuel 2,5-dimethylfuran over carbon-supported ruthenium. Ind. Eng. Chem. Res. 2014, 53, 3056–3064. [Google Scholar] [CrossRef]
- Cai, H.; Li, C.; Wang, A.; Zhang, T. Biomass into chemicals: One-pot production of furan-based diols from carbohydrates via tandem reactions. Catal. Today 2014, 234, 59–65. [Google Scholar] [CrossRef]
- Audemar, M.; Vigier, K.D.O.; Clacens, J.-M.; De Campo, F.; Jérôme, F. Combination of Pd/C and Amberlyst-15 in a single reactor for the acid/hydrogenating catalytic conversion of carbohydrates to 5-hydroxy-2,5-hexanedione. Green Chem. 2014, 16, 4110–4114. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Metrics of green chemistry and sustainability: Past, present, and future. ACS Sustain. Chem. Eng. 2017, 6, 32–48. [Google Scholar] [CrossRef]
- Gilkey, M.J.; Xu, B. Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal. 2016, 6, 1420–1436. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Padmasri, A.H.; Seetharamulu, P.; Reddy, K.H.P.; Raju, B.D.; Rao, K.R. A highly active Cu-MgO-Cr2O3 catalyst for simultaneous synthesis of furfuryl alcohol and cyclohexanone by a novel coupling route—Combination of furfural hydrogenation and cyclohexanol dehydrogenation. J. Mol. Catal. Chem. 2007, 278, 29–37. [Google Scholar] [CrossRef]
- Zhu, Y.-L.; Xiang, H.-W.; Wu, G.-S.; Bai, L.; Li, Y.-W. A novel route for synthesis of γ-butyrolactone through the coupling of hydrogenation and dehydrogenation. Chem. Commun. 2002, 254–255. [Google Scholar] [CrossRef]
- Zhang, D.; Yin, H.; Zhang, R.; Xue, J.; Jiang, T. Gas phase hydrogenation of maleic anhydride to γ-butyrolactone by Cu–Zn–Ce catalyst in the presence of n-butanol. Catal. Lett. 2008, 122, 176–182. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Padmasri, A.H.; Raju, B.D.; Rao, K.S.R. Production of hydrogen through the coupling of dehydrogenation and hydrogenation for the synthesis of cyclohexanone and furfuryl alcohol over different promoters supported on Cu–MgO catalysts. Int. J. Hydrogen Energy 2011, 36, 3417–3425. [Google Scholar] [CrossRef]
- Zheng, H.-Y.; Zhu, Y.-L.; Bai, Z.-Q.; Huang, L.; Xiang, H.-W.; Li, Y.-W. An environmentally benign process for the efficient synthesis of cyclohexanone and 2-methylfuran. Green Chem. 2006, 8, 107–109. [Google Scholar] [CrossRef]
- Zheng, H.-Y.; Zhu, Y.-L.; Huang, L.; Zeng, Z.-Y.; Wan, H.-J.; Li, Y.-W. Study on Cu–Mn–Si catalysts for synthesis of cyclohexanone and 2-methylfuran through the coupling process. Catal. Commun. 2008, 9, 342–348. [Google Scholar] [CrossRef]
- Zheng, H.-Y.; Yang, J.; Zhu, Y.-L.; Zhao, G.-W. Synthesis of γ-butyrolactone and 2-methylfuran through the coupling of dehydrogenation and hydrogenation over copper-chromite catalyst. React. Kinet. Catal. Lett. 2004, 82, 263–269. [Google Scholar] [CrossRef]
- Zhu, Y.-L.; Xiang, H.-W.; Li, Y.-W.; Jiao, H.; Wu, G.-S.; Zhong, B.; Guo, G.-Q. A new strategy for the efficient synthesis of 2-methylfuran and γ-butyrolactone. New J. Chem. 2003, 27, 208–210. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, H.-Y.; Zhu, Y.-L.; Zhao, G.-W.; Zhang, C.-H.; Teng, B.-T.; Xiang, H.-W.; Li, Y. Effects of calcination temperature on performance of Cu–Zn–Al catalyst for synthesizing γ-butyrolactone and 2-methylfuran through the coupling of dehydrogenation and hydrogenation. Catal. Commun. 2004, 5, 505–510. [Google Scholar] [CrossRef]
- Aellig, C.; Jenny, F.; Scholz, D.; Wolf, P.; Giovinazzo, I.; Kollhoff, F.; Hermans, I. Combined 1, 4-butanediol lactonization and transfer hydrogenation/hydrogenolysis of furfural-derivatives under continuous flow conditions. Catal. Sci. Technol. 2014, 4, 2326–2331. [Google Scholar] [CrossRef]
- Hu, L.; Yang, M.; Xu, N.; Xu, J.; Zhou, S.; Chu, X.; Zhao, Y. Selective transformation of biomass-derived 5-hydroxymethylfurfural into 2,5-dihydroxymethylfuran via catalytic transfer hydrogenation over magnetic zirconium hydroxides. Korean J. Chem. Eng. 2018, 35, 99–109. [Google Scholar] [CrossRef]
- Pischetola, C.; Collado, L.; Aguado-Molina, R.; Treceno-Martìn, S.; Cárdenas-Lizana, F.; Keane, M.A. Coupled dehydrogenation-hydrogenation over supported Cu and Au catalysts for continuous furfuryl alcohol production: Support effects. 2018; submitted. [Google Scholar]
- Li, Z.X.; Zhang, C.; Sun, L.D.; Zhang, Y.W.; Yan, C.H. Catalysis by ceria and related materials. In Catalytic Science Series; Imperial College Press: London, UK, 2013; Volume 12, pp. 295–359. ISBN 978-1-84816-963-0. [Google Scholar]
- Green, D.W. Perry’s Chemical Engineers’; McGraw Hill: New York, NY, USA, 2008; ISBN 9780071422949. [Google Scholar]
- Keane, M.A.; Li, M.; Collado, L.; Cárdenas-Lizana, F. Overcoming the limitations of gold catalysts in hydrogenation: Enhanced activity with full hydrogen utilization. React. Kinet. Mech. Catal. 2018, 1–12. [Google Scholar] [CrossRef]
- Crivello, M.E.; Pérez, C.F.; Mendieta, S.N.; Casuscelli, S.G.; Eimer, G.A.; Elías, V.R.; Herrero, E.R. n-Octyl alcohol dehydrogenation over copper catalysts. Catal. Today 2008, 133–135, 787–792. [Google Scholar] [CrossRef]
- Wang, F.; Shi, R.; Liu, Z.-Q.; Shang, P.-J.; Pang, X.; Shen, S.; Feng, Z.; Li, C.; Shen, W. Highly efficient dehydrogenation of primary aliphatic alcohols catalyzed by Cu nanoparticles dispersed on rod-shaped La2O2CO3. ACS Catal. 2013, 3, 890–894. [Google Scholar] [CrossRef]
- Ohyama, J.; Esaki, A.; Yamamoto, Y.; Arai, S.; Satsuma, A. Selective hydrogenation of 2-hydroxymethyl-5-furfural to 2,5-bis(hydroxymethyl)furan over gold sub-nano clusters. RSC Adv. 2013, 3, 1033–1036. [Google Scholar] [CrossRef]
- Gallezot, P.; Richard, D. Selective hydrogenation of α, β-unsaturated aldehydes. Catal. Rev. 1998, 40, 81–126. [Google Scholar] [CrossRef]
- Conner, W.C., Jr.; Falconer, J.L. Spillover in heterogeneous catalysis. Chem. Rev. 1995, 95, 759–788. [Google Scholar] [CrossRef]
- Li, M.; Collado, L.; Cárdenas-Lizana, F.; Keane, M.A. Role of support oxygen vacancies in the gas phase hydrogenation of furfural over gold. Catal. Lett. 2018, 148, 90–96. [Google Scholar] [CrossRef]
- Zanella, R. Crotonaldehyde hydrogenation by gold supported on TiO2: Structure sensitivity and mechanism. J. Catal. 2004, 223, 328–339. [Google Scholar] [CrossRef]
- Wang, X.; Perret, N.; Delannoy, L.; Louis, C.; Keane, M.A. Selective gas phase hydrogenation of nitroarenes over Mo2C-supported Au–Pd. Catal. Sci. Technol. 2016, 6, 6932–6941. [Google Scholar] [CrossRef]
- Shi, R.; Wang, F.; Tana; Li, Y.; Huang, X.; Shen, W. A highly efficient Cu/La2O3 catalyst for transfer dehydrogenation of primary aliphatic alcohols. Green Chem. 2010, 12, 108–113. [Google Scholar] [CrossRef]
- Ying, F.; Wang, S.; Au, C.-T.; Lai, S.-Y. Highly active and stable mesoporous Au/CeO2 catalysts prepared from MCM-48 hard-template. Microporous Mesoporous Mater. 2011, 142, 308–315. [Google Scholar] [CrossRef]
- Huang, Z.; Cui, F.; Xue, J.; Zuo, J.; Chen, J.; Xia, C. Cu/SiO2 catalysts prepared by hom- and heterogeneous deposition–precipitation methods: Texture, structure, and catalytic performance in the hydrogenolysis of glycerol to 1, 2-propanediol. Catal. Today 2012, 183, 42–51. [Google Scholar] [CrossRef]
- Liao, X.; Chu, W.; Dai, X.; Pitchon, V. Bimetallic Au–Cu supported on ceria for PROX reaction: Effects of Cu/Au atomic ratios and thermal pretreatments. Appl. Catal. B Environ. 2013, 142, 25–37. [Google Scholar] [CrossRef]
- Nevanperä, T.K.; Ojala, S.; Bion, N.; Epron, F.; Keiski, R.L. Catalytic oxidation of dimethyl disulfide (CH3SSCH3) over monometallic Au, Pt and Cu catalysts supported on γ-Al2O3, CeO2 and CeO2-Al2O3. Appl. Catal. B Environ. 2016, 182, 611–625. [Google Scholar] [CrossRef]
- Scire, S.; Minico, S.; Crisafulli, C.; Satriano, C.; Pistone, A. Catalytic combustion of volatile organic compounds on gold/cerium oxide catalysts. Appl. Catal. B Environ. 2003, 40, 43–49. [Google Scholar] [CrossRef]
- Dow, W.-P.; Huang, T.-J. Effect of chlorine on TPR and TPO behavior of an YSZ/γ-Al2O3 supported copper oxide catalyst. Appl. Catal. A Gen. 1996, 141, 17–29. [Google Scholar] [CrossRef]
- Jacono, M.L.; Cimino, A.; Inversi, M. Oxidation states of copper on alumina studied by redox cycles. J. Catal. 1982, 76, 320–332. [Google Scholar] [CrossRef]
- Chen, J.; Zhan, Y.; Zhu, J.; Chen, C.; Lin, X.; Zheng, Q. The synergetic mechanism between copper species and ceria in NO abatement over Cu/CeO2 catalysts. Appl. Catal. A Gen. 2010, 377, 121–127. [Google Scholar] [CrossRef]
- Dhas, N.A.; Raj, C.P.; Gedanken, A. Synthesis, characterization, and properties of metallic copper nanoparticles. Chem. Mater. 1998, 10, 1446–1452. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Suzuki, K.; Hamakawa, S. Production of hydrogen by steam reforming of methanol over Cu/CeO2 catalysts derived from Ce1− xCuxO2− x precursors. Catal. Commun. 2001, 2, 195–200. [Google Scholar] [CrossRef]
- Chen, H.; Hu, L.; Chen, M.; Yan, Y.; Wu, L. Nickel–cobalt layered double hydroxide nanosheets for high–performance supercapacitor electrode materials. Adv. Funct. Mater. 2014, 24, 934–942. [Google Scholar] [CrossRef]
- Huang, X.-S.; Sun, H.; Wang, L.-C.; Liu, Y.-M.; Fan, K.-N.; Cao, Y. Morphology effects of nanoscale ceria on the activity of Au/CeO2 catalysts for low-temperature CO oxidation. Appl. Catal. B: Environ. 2009, 90, 224–232. [Google Scholar] [CrossRef]
- Wei, S.-H.; Mbaye, A.; Ferreira, L.; Zunger, A. First-principles calculations of the phase diagrams of noble metals: Cu-Au, Cu-Ag, and Ag-Au. Phys. Rev. B 1987, 36, 4163–4185. [Google Scholar] [CrossRef]
- Zhang, L.; Kim, H.Y.; Henkelman, G. CO oxidation at the Au–Cu interface of bimetallic nanoclusters supported on CeO2 (111). J. Phys. Chem. Lett. 2013, 4, 2943–2947. [Google Scholar] [CrossRef]
- Scirè, S.; Fiorenza, R.; Crisafulli, C. AuAg/CeO2 and AuCu/CeO2 catalysts for CO preferential oxidation. In Proceedings of the Hydrogen Power Theoretical and Engineering Solutions International Symposium 2015, Toledo, Spain, 6–9 September 2015. [Google Scholar]
- Ta, N.; Liu, J.; Chenna, S.; Crozier, P.A.; Li, Y.; Chen, A.; Shen, W. Stabilized gold nanoparticles on ceria nanorods by strong interfacial anchoring. J. Am. Chem. Soc. 2012, 134, 20585–20588. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.D.; Riggs, W.M.; Davis, L.E.; Moulder, J.F.; Muilenberg, G.E. Handbook of X-ray Photoelectron Spectroscopy; Perkin-Elmer Corporation: Waltham, MA, USA, 2015; pp. 183–184. ISBN 55344. [Google Scholar]
- Delannoy, L.; Thrimurthulu, G.; Reddy, P.S.; Méthivier, C.; Nelayah, J.; Reddy, B.M.; Ricolleau, C.; Louis, C. Selective hydrogenation of butadiene over TiO2 supported copper, gold and gold–copper catalysts prepared by deposition–precipitation. Phys. Chem. Chem. Phys. 2014, 16, 26514–26527. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Lemire, C.; Shaikhutdinov, S.K.; Freund, H.-J. Surface chemistry of catalysis by gold. Gold Bull. 2004, 37, 72–124. [Google Scholar] [CrossRef]
- Lai, S.-Y.; Qiu, Y.; Wang, S. Effects of the structure of ceria on the activity of gold/ceria catalysts for the oxidation of carbon monoxide and benzene. J. Catal. 2006, 237, 303–313. [Google Scholar] [CrossRef]
- Li, M.; Hao, Y.; Cárdenas-Lizana, F.; Yiu, H.H.P.; Keane, M.A. “Hydrogen-free” hydrogenation of nitrobenzene over Cu/SiO2 via coupling with 2-butanol dehydrogenation. Top. Catal. 2015, 58, 149–158. [Google Scholar] [CrossRef]
- Bertero, N.M.; Trasarti, A.F.; Apesteguía, C.R.; Marchi, A.J. Solvent effect in the liquid-phase hydrogenation of acetophenone over Ni/SiO2: A comprehensive study of the phenomenon. Appl. Catal. A Gen. 2011, 394, 228–238. [Google Scholar] [CrossRef]
- Dunbar, R.E.; Arnold, M.R. Catalytic dehydrogenation of primary and secondary alcohols with copper-chromium oxide. J. Org. Chem. 1945, 10, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Van der Waal, J.C.; Kunkeler, P.J.; Tan, K.; Van Bekkum, H. Zeolite titanium beta: A selective catalyst for the gas-phase Meerwein–Ponndorf–Verley, and Oppenauer reactions. J. Catal. 1998, 173, 74–83. [Google Scholar] [CrossRef]
- Di Cosimo, J.; Acosta, A.; Apesteguía, C. Allylic alcohol synthesis by gas-phase hydrogen transfer reduction of unsaturated ketones. J. Mol. Catal. Chem. 2005, 234, 111–120. [Google Scholar] [CrossRef]
- Cárdenas-Lizana, F.; Lamey, D.; Gómez-Quero, S.; Perret, N.; Kiwi-Minsker, L.; Keane, M.A. Selective three-phase hydrogenation of aromatic nitro-compounds over β-molybdenum nitride. Catal. Today 2011, 173, 53–61. [Google Scholar] [CrossRef]
- Sitthisa, S.; Sooknoi, T.; Ma, Y.; Balbuena, P.B.; Resasco, D.E. Kinetics and mechanism of hydrogenation of furfural on Cu/SiO2 catalysts. J. Catal. 2011, 277, 1–13. [Google Scholar] [CrossRef]
- Jacobs, G.; Keogh, R.A.; Davis, B.H. Steam reforming of ethanol over Pt/ceria with co-fed hydrogen. J. Catal. 2007, 245, 326–337. [Google Scholar] [CrossRef]
- Remya, G.S.; Suresh, C.H. Quantification and classification of substituent effects in organic chemistry: A theoretical molecular electrostatic potential study. Phys. Chem. Chem. Phys. 2016, 18, 20615–20626. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Baatz, C.; Thielecke, N.; Prüße, U. Influence of the preparation conditions on the properties of gold catalysts for the oxidation of glucose. Appl. Catal. B Environ. 2007, 70, 653–660. [Google Scholar] [CrossRef]
- Nino, E.; Lapena, A.; Martinez, J.; Gutierrez, J.M.; Mendioroz, S.; Fierro, J.L.G.; Pajares, J.A. Cu/Kieselguhr catalysts for hydration of acrylonitrile. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1983; Volume 16, pp. 747–755. ISBN 0167-2991. [Google Scholar]
- Kirumakki, S.R.; Shpeizer, B.G.; Sagar, G.V.; Chary, K.V.; Clearfield, A. Hydrogenation of naphthalene over NiO/SiO2–Al2O3 catalysts: Structure–activity correlation. J. Catal. 2006, 242, 319–331. [Google Scholar] [CrossRef]
- Li, M.; Hao, Y.; Cárdenas-Lizana, F.; Keane, M.A. Selective production of furfuryl alcohol via gas phase hydrogenation of furfural over Au/Al2O3. Catal. Commun. 2015, 69, 119–122. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds ...... are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Au-Cu/CeO2 | Au/CeO2 | |||
|---|---|---|---|---|
| Metal loading (mol%) | 1 (Au)/2 (Cu) | 1 | ||
| TPR | Tmax (K) | α | 414 | 410 |
| β | 429 | 423 | ||
| γ | 437 | - | ||
| δ | 443 | - | ||
| H2 consumption (µmol g−1) | 340 a/63 b/61 c | 307 a/69 b | ||
| dSTEM(nm) d | 4 | 3 | ||
| XPS | Binding energies (eV) | Au0 (%) | 83.6 (100) | 83.5 (100) |
| Cu0 (%) | 928.7 (35) | |||
| Cu+ (%) | 932.0 (65) | |||
| Ce3+ atomic ratio e | 0.24 | 0.29 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pischetola, C.; Collado, L.; Keane, M.A.; Cárdenas-Lizana, F. Gas Phase Hydrogenation of Furaldehydes via Coupling with Alcohol Dehydrogenation over Ceria Supported Au-Cu. Molecules 2018, 23, 2905. https://doi.org/10.3390/molecules23112905
Pischetola C, Collado L, Keane MA, Cárdenas-Lizana F. Gas Phase Hydrogenation of Furaldehydes via Coupling with Alcohol Dehydrogenation over Ceria Supported Au-Cu. Molecules. 2018; 23(11):2905. https://doi.org/10.3390/molecules23112905
Chicago/Turabian StylePischetola, Chiara, Laura Collado, Mark A. Keane, and Fernando Cárdenas-Lizana. 2018. "Gas Phase Hydrogenation of Furaldehydes via Coupling with Alcohol Dehydrogenation over Ceria Supported Au-Cu" Molecules 23, no. 11: 2905. https://doi.org/10.3390/molecules23112905
APA StylePischetola, C., Collado, L., Keane, M. A., & Cárdenas-Lizana, F. (2018). Gas Phase Hydrogenation of Furaldehydes via Coupling with Alcohol Dehydrogenation over Ceria Supported Au-Cu. Molecules, 23(11), 2905. https://doi.org/10.3390/molecules23112905