Synthetic Evaluation of Standard and Microwave-Assisted Solid Phase Peptide Synthesis of a Long Chimeric Peptide Derived from Four Plasmodium falciparum Proteins


Abstract
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Reagents and Solvents
4.2. Peptide Design
4.3. Peptide Synthesis
4.3.1. The Standard Strategy
4.3.2. The Microwave-Assisted Strategy
4.4. Characterization
4.4.1. HPLC Analysis
4.4.2. Mass Spectrometry
4.4.3. Purification of the Chimeric Peptide
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mitchell, A.R. Bruce Merrifield and solid-phase peptide synthesis: A historical assessment. Biopolym. Pept. Sci. Sect. 2008, 90, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Kimmerlin, T.; Seebach, D. ‘100 years of peptide synthesis’: Ligation methods for peptide and protein synthesis with applications to β-peptide assemblies*. 2005, 65, 229–260. [Google Scholar] [CrossRef] [PubMed]
- Coin, I.; Beyermann, M.; Bienert, M. Solid-phase peptide synthesis: From standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007, 2, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- De L. Milton, R.C.; Milton, S.C.F.; Adams, P.A. Prediction of Difficult Sequences in Solid-Phase Peptide Synthesis. J. Am. Chem. Soc. 1990, 112, 6039–6046. [Google Scholar] [CrossRef]
- Furrer, J.; Piotto, M.; Bourdonneau, M.; Limal, D.; Guichard, G.; Elbayed, K.; Raya, J.; Briand, J.P.; Bianco, A. Evidence of secondary structure by high-resolution magic angle spinning NMR spectroscopy of a bioactive peptide bound to different solid supports. J. Am. Chem. Soc. 2001, 123, 4130–4138. [Google Scholar] [CrossRef] [PubMed]
- Paradís-Bas, M.; Tulla-Puche, J.; Albericio, F. The road to the synthesis of ‘difficult peptides’. Chem. Soc. Rev. 2016, 45, 631–654. [Google Scholar] [CrossRef] [PubMed]
- Renil, M.; Ferreras, M.; Delaisse, J.M.; Foged, N.T.; Meldal, M. PEGA supports for combinatorial peptide synthesis and solid-phase enzymatic library assays. J. Pept. Sci. 1998, 4, 195–210. [Google Scholar] [CrossRef]
- García-Martín, F.; Quintanar-Audelo, M.; García-Ramos, Y.; Cruz, L.J.; Gravel, C.; Furic, R.; Côté, S.; Tulla-Puche, J.; Albericio, F. Chemmatrix, a poly (ethylene glycol)-based support for the solid-phase synthesis of complex peptides. J. Comb. Chem. 2006, 8, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Yamashiro, D. The Synthesis of a Protein Possessing Growth-Promoting and Lactogenic Activities. J. Am. Chem. Soc. 1970, 92, 7608–7609. [Google Scholar] [CrossRef] [PubMed]
- Chandrudu, S.; Simerska, P.; Toth, I. Chemical methods for peptide and protein production. Molecules 2013, 18, 4373–4388. [Google Scholar] [CrossRef] [PubMed]
- Nishiuchi, Y.; Inui, T.; Nishio, H.; Bodi, J.; Kimura, T.; Tsuji, F.I.; Sakakibara, S. Chemical synthesis of the precursor molecule of the Aequorea green fluorescent protein, subsequent folding, and development of fluorescence. Proc. Natl. Acad. Sci. USA 1998, 95, 13549–13554. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Gao, S.; Zheng, Y.; Tan, X.; Lan, H.; Tan, X.; Sun, D.; Lu, L.; Wang, T.; Zheng, Q.; et al. Quasi-Racemic X-ray Structures of K27-Linked Ubiquitin Chains Prepared by Total Chemical Synthesis. J. Am. Chem. Soc. 2016, 138, 7429–7435. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.L.; Soellner, M.B.; Raines, R.T. Chemical Synthesis of Proteins. Rev. Lit. Arts Am. 2005, 18, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Nutt, R.F.; Brady, S.F.; Darke, P.L.; Ciccarone, T.M.; Colton, C.D.; Nutt, E.M.; Rodkey, J.A.; Bennett, C.D.; Waxman, L.H.; Sigal, I.S. Chemical synthesis and enzymatic activity of a 99-residue peptide with a sequence proposed for the human immunodeficiency virus protease. Proc. Natl. Acad. Sci. USA 1988, 85, 7129–7133. [Google Scholar] [CrossRef] [PubMed]
- Kresge, N.; Simoni, R.D.; Hill, R.L. The solid phase synthesis of ribonuclease A by Robert Bruce Merrifield. J. Biol. Chem. 2006, 281, e21. [Google Scholar] [CrossRef]
- Pedersen, S.L.; Tofteng, A.P.; Malik, L.; Jensen, K.J. Microwave heating in solid-phase peptide synthesis. Chem. Soc. Rev. 2012, 41, 1826–1844. [Google Scholar] [CrossRef] [PubMed]
- Palasek, S.A.; Cox, Z.J.; Collins, J.M. Limiting racemization and aspartimide formation in microwave-enhanced fmoc solid phase peptide synthesis. J. Pept. Sci. 2007, 13, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.E.; Bermúdez, A.; Alba, M.P.; Vanegas, M.; Moreno-Vranich, A.; Poloche, L.A.; Patarroyo, M.A. IMPIPS: The Immune Protection-Inducing Protein Structure Concept in the Search for Steric-Electron and Topochemical Principles for Complete Fully-Protective Chemically Synthesised Vaccine Development. PLoS ONE 2015, 10, e0123249. [Google Scholar] [CrossRef] [PubMed]
- Biosystems, A. Cleavage, Deprotection, and Isolation of Peptides after Fmoc Synthesis. Tech. Bull. 1998, 1–12. [Google Scholar] [CrossRef]
- Lozano, J.M.; Varela, Y.; Silva, Y.; Ardila, K.; Forero, M.; Guasca, L.; Guerrero, Y.; Bermudez, A.; Alba, P.; Vanegas, M.; et al. A large size chimeric highly immunogenic peptide presents multistage plasmodium antigens as a vaccine candidate system against malaria. Molecules 2017, 22, 1837. [Google Scholar] [CrossRef] [PubMed]
- Thapa, P.; Zhang, R.-Y.; Menon, V.; Bingham, J.-P. Native Chemical Ligation: A Boon to Peptide Chemistry. Molecules 2014, 19, 14461–14483. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Biancalana, S.; Hudson, D. Activity of synthetic peptides from the Tat protein of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1989, 86, 7397–7401. [Google Scholar] [CrossRef] [PubMed]
- Gutte, B.; Merrifield, R.B. The Synthesis of Ribonuclease A. J. Biol. Chem. 1971, 246, 1922–1941. [Google Scholar] [PubMed]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the structural biology of protein-protein interactions. Protein Sci. 2013, 22, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Alba, M.P.; Salazar, L.M.; Vargas, L.E.; Trujillo, M.; Lopez, Y.; Patarroyo, M.E. Modifying RESA protein peptide 6671 to fit into HLA-DRβ1* pockets induces protection against malaria. Biochem. Biophys. Res. Commun. 2004, 315, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez, A.; Calderon, D.; Moreno-Vranich, A.; Almonacid, H.; Patarroyo, M.A.; Poloche, A.; Patarroyo, M.E. Gauche+ side-chain orientation as a key factor in the search for an immunogenic peptide mixture leading to a complete fully protective vaccine. Vaccine 2014, 32, 2117–2126. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez, A.; Vanegas, M.; Patarroyo, M.E. Structural and immunological analysis of circumsporozoite protein peptides: A further step in the identification of potential components of a minimal subunit-based, chemically synthesised antimalarial vaccine. Vaccine 2008, 26, 6908–6918. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez, A.; Alba, M.P.; Vanegas, M.; Patarroyo, M.E. 3D structure determination of STARP peptides implicated in P. falciparum invasion of hepatic cells. Vaccine 2010, 28, 4989–4996. [Google Scholar] [CrossRef] [PubMed]
- Krchnák, V.; Flegelová, Z.; Vágner, J. Aggregation of resin-bound peptides during solid-phase peptide synthesis. Prediction of difficult sequences. Int. J. Pept. Protein Res. 1993, 42, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Coin, I.; Beyermann, M.; Bienert, M. Monitoring solid phase peptide synthesis. Protoc. Exch. 2007, 4–6. [Google Scholar] [CrossRef]
- Boll, E.; Drobecq, H.; Ollivier, N.; Blanpain, A.; Raibaut, L.; Desmet, R.; Vicogne, J.; Melnyk, O. One-pot chemical synthesis of small ubiquitin-like modifier protein-peptide conjugates using bis(2-sulfanylethyl)amido peptide latent thioester surrogates. Nat. Protoc. 2015, 10, 269–292. [Google Scholar] [CrossRef] [PubMed]
- Rivera, Z.; Granados, G.; Pinto, M.; Varón, D.; Carvajal, C.; Chaves, F.; Calvo, J.; Rodríguez, R.; Guzmán, F.; Patarroyo, M.E. Double dimer peptide constructs are immunogenic and protective against Plasmodium falciparum in the experimental Aotus monkey model. J. Pept. Res. 2002, 59, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.K.; Gellman, S.H. Parallel synthesis of peptide libraries using microwave irradiation. Nat. Protoc. 2007, 2, 624–631. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the chimeric peptide is available by request from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Residue Control | Molecular Weight (Da) § | Standard | Microwave-Assisted | ||
|---|---|---|---|---|---|
| (m/z) a | tR (min) | (m/z) a | tR (min) | ||
| 11 | 1228.53 | 1229.52 | 14.20 | 1230.01 | 14.43 |
| 22 | 2628.16 | 2628.29 | 17.17 | 2628.06 | 17.59 |
| 33 | 3887.86 | 3831.50 (−57.81) b 3887.90 | 19.72 | 3831.93 (−57.52) b 3889.61 | 19.85 |
| 3889.31 | 21.18 | 3889.45 | 21.29 | ||
| 3888.07 4055.84 (+166.53) c | 22.43 | 3889.18 4058.04 (+168.59) c | 22.26 | ||
| 44 | 5065.51 | 4939.21 (−128. 02) 4996.03 (−71.20) 5066.91 | 27.36 | 4938.52 (−128.34) 4995.93 (−70.93) 5067.40 | 27.50 |
| 5067.23 | 28.60 | 5066.86 | 28.64 | ||
| 5065.90 5178.52 (+111.29) c | 29.87 | 5066.98 5178.61 (+111.75) c | 29.42 | ||
| 57 | 6348.07 | 6221.41 (−127.59) 6234.78 (−114.54) 6350.06 | 28.17 | 6235.14 (−114.91) 6351.36 | 27.97 |
| 6291.44 (−57.88) 6349.32 | 29.05 | 6350.05 | 28.66 | ||
| 6349.47 6632.30 (+282.98) | 29.41 | ND | 29.16 | ||
| 66 | 7467.68 | ND | 28.65 | ND | 28.73 |
| 7469.35 7580.21 (+111.86) c | 29.51 | 7467.84 | 29.54 | ||
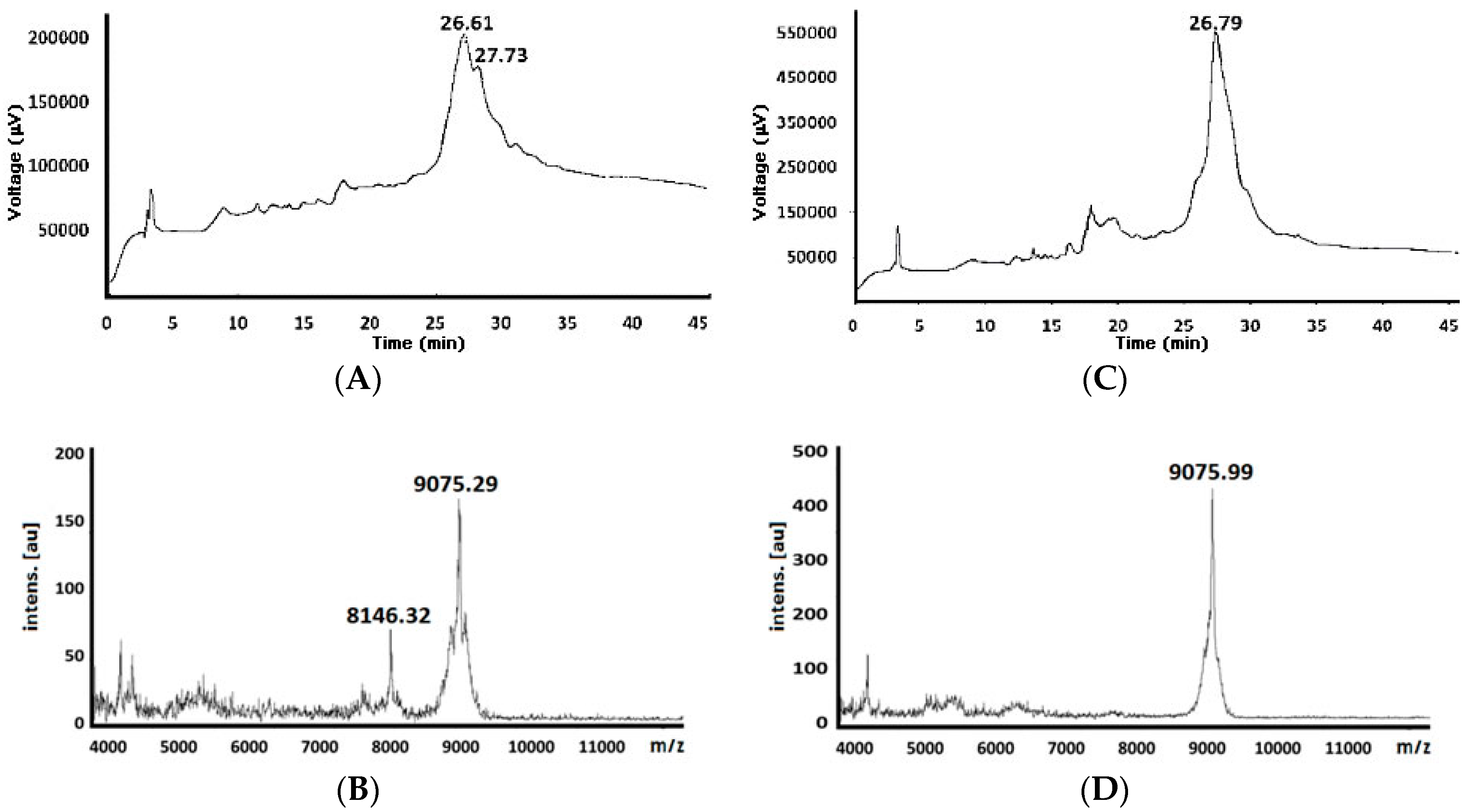
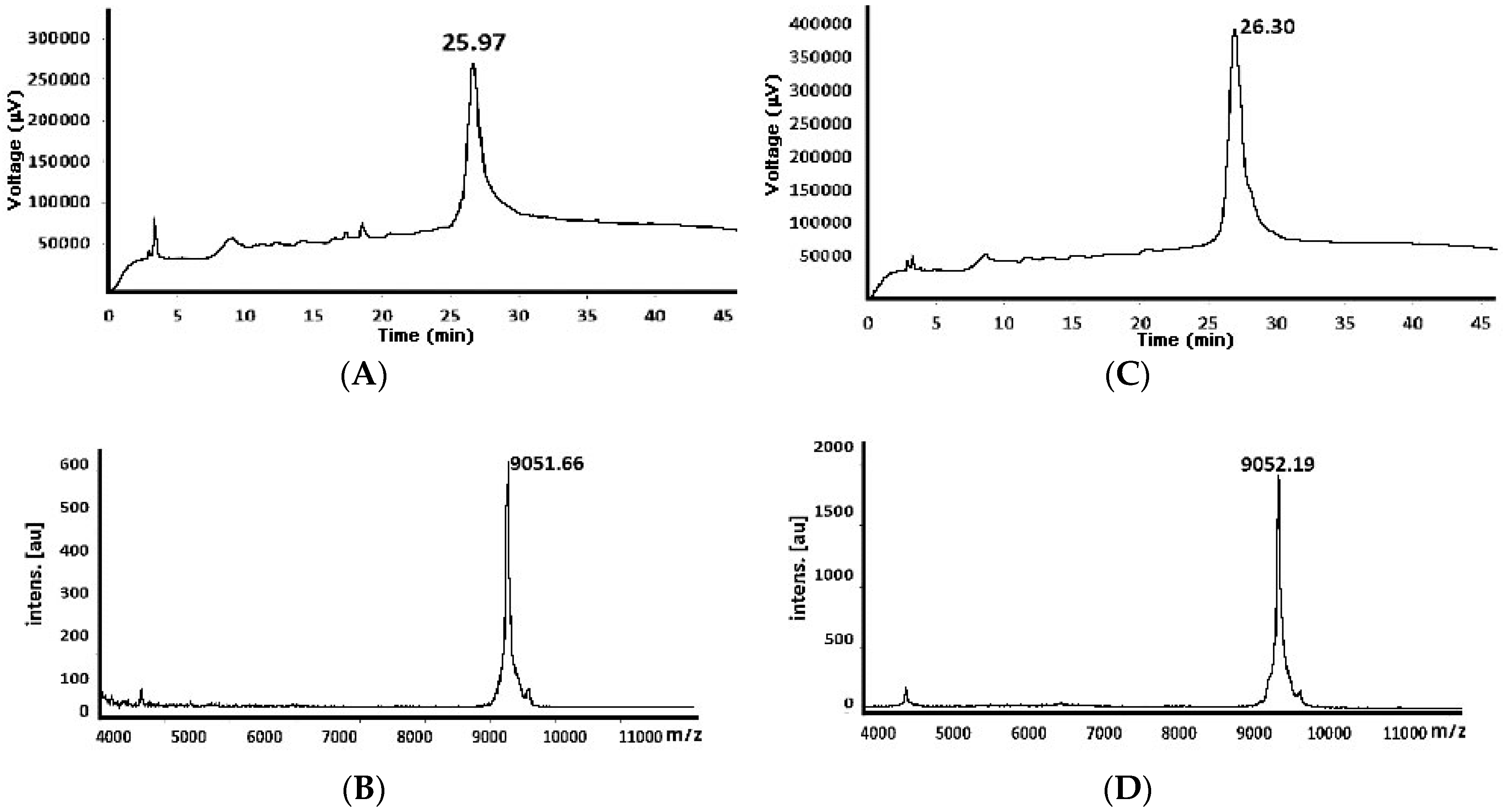
| 82 e | 9051.42 | 8146.32 (−928.97) 9075.29 (+23.87) d | 26.61 27.73 | 9075.99 (+24.57) d | 26.79 |
| Strategy | Resin (g) | Obtained (mg) | % Yield | Purified Quantity (mg) | Amount of Peptide Obtained in the Purification (mg) | % Purification Yield | |
|---|---|---|---|---|---|---|---|
| Theoretical | Experimental | ||||||
| Standard | 0.507 | 905.70 | 90.42 | 9.98 | 10.58 | 0.97 | 9.16 |
| MW-assisted | 0.501 | 905.80 | 118.32 | 13.06 | 11.13 | 1.41 | 12.67 |
| Advantages | Disadvantages | Ref. |
|---|---|---|
|
| [22] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varela, Y.F.; Vanegas Murcia, M.; Patarroyo, M.E. Synthetic Evaluation of Standard and Microwave-Assisted Solid Phase Peptide Synthesis of a Long Chimeric Peptide Derived from Four Plasmodium falciparum Proteins. Molecules 2018, 23, 2877. https://doi.org/10.3390/molecules23112877
Varela YF, Vanegas Murcia M, Patarroyo ME. Synthetic Evaluation of Standard and Microwave-Assisted Solid Phase Peptide Synthesis of a Long Chimeric Peptide Derived from Four Plasmodium falciparum Proteins. Molecules. 2018; 23(11):2877. https://doi.org/10.3390/molecules23112877
Chicago/Turabian StyleVarela, Yahson F., Magnolia Vanegas Murcia, and Manuel Elkin Patarroyo. 2018. "Synthetic Evaluation of Standard and Microwave-Assisted Solid Phase Peptide Synthesis of a Long Chimeric Peptide Derived from Four Plasmodium falciparum Proteins" Molecules 23, no. 11: 2877. https://doi.org/10.3390/molecules23112877
APA StyleVarela, Y. F., Vanegas Murcia, M., & Patarroyo, M. E. (2018). Synthetic Evaluation of Standard and Microwave-Assisted Solid Phase Peptide Synthesis of a Long Chimeric Peptide Derived from Four Plasmodium falciparum Proteins. Molecules, 23(11), 2877. https://doi.org/10.3390/molecules23112877