
Preparation of Functional Monomers as Precursors of Bioprobes from a Common Styrene Derivative and Polymer Synthesis


Abstract

1. Introduction
2. Results and Discussion
2.1. Conversion of 4-(Chloromethyl)styrene into an Amine as a Common Precursor
2.2. Synthetic Conversion of the Amine into Functionalized Monomers
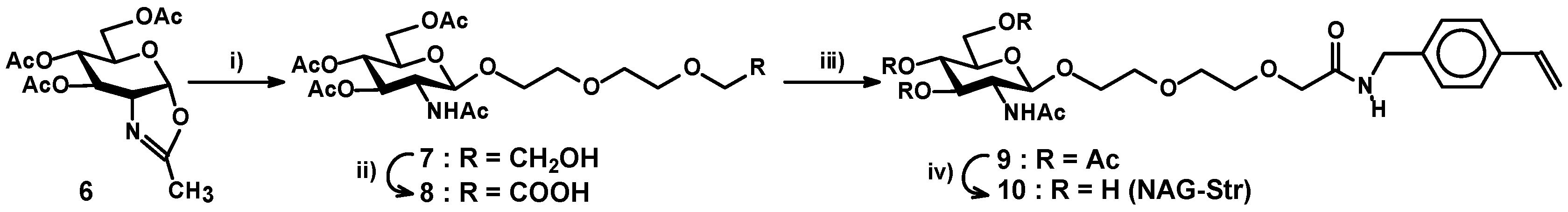
2.3. Preparation of a Carbohydrate Monomer
2.4. Polymerization of a Glycomonomer by Means of Radical Polymerization
2.5. Biological Evaluations of the Glycopolymers for Lectin
3. Experimental
3.1. Materials and Methods
3.2. Synthesis
3.2.1. 4-Azidomethylstyrene (AZ-Str) (2)
3.2.2. Aminomethylstyrene (AMN-Str) (3)
3.2.3. 4-{[5-(Dimethylamino)-1-naphthalenesulfonamido]methyl}styrene (DSL-Str) (4)
3.2.4. 4-({5-[(3aR,6S,6aS)-2-Oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-6-yl]pentanamido}methyl)-styrene (BTN-Str) (5)
3.2.5. 2-[2-(2-Hydroxyethoxy)-ethoxy]-ethyl-O-2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranoside (7)
3.2.6. 8-O-(2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-3,6-dioxaoctanoic Acid (8)
3.2.7. 8-O-(2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-3,6-dioxaoctanoic Acid 4-vinybenzylamide (9)
3.2.8. 8-O-(2-Acetamido-2-deoxy-β-d-glucopyranosyl)-3,6-dioxaoctanoic acid 4-vinybenzylamide (NAG-Str) (10)
3.3. Radical Polymerization
3.4. Biological Evaluations of Glycopolymers for WGA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, Y.C. Biochemistry of carbohydrate-protein interaction. FASEB J. 1992, 6, 3193–3200. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.T.; Lee, Y.C. Affinity enhancement by multivalent lectin-carbohydrate interaction. Glycoconj. J. 2000, 17, 543–551. [Google Scholar] [CrossRef]
- Chabre, Y.M.; Contino-Pepin, C.; Placide, V.; Shiao, T.C.; Roy, R. Expeditive synthesis of glycodendrimer scaffolds based on versatile TRIS and mannoside derivatives. J. Org. Chem. 2008, 73, 5602–5605. [Google Scholar] [CrossRef] [PubMed]
- Niederhafner, P.; Sebestik, J.; Jezek, J. Glycopeptide dendrimers. J. Pept. Sci. 2008, 14, 2–43. [Google Scholar] [CrossRef] [PubMed]
- Hatano, K.; Matsuoka, K.; Terunuma, D. Carbosilane glycodendrimers. Chem. Soc. Rev. 2013, 42, 4574–4598. [Google Scholar] [CrossRef] [PubMed]
- Ladmiral, V.; Melia, E.; Haddleton, D.M. Synthetic glycopolymers: An overview. Eur. Polym. J. 2004, 40, 431–449. [Google Scholar] [CrossRef]
- Varma, A.J.; Kennedy, J.F.; Galgali, P. Synthetic polymers functionalized by carbohydrates: A review. Carbohydr. Polym. 2004, 56, 429–445. [Google Scholar] [CrossRef]
- Matsuoka, K.; Goshu, Y.; Takezawa, Y.; Mori, T.; Sakamoto, J.I.; Yamada, A.; Onaga, T.; Koyama, T.; Hatano, K.; Snyder, P.W.; et al. Practical synthesis of fully protected globotriaose and its glycopolymers. Carbohydr. Polym. 2007, 69, 326–335. [Google Scholar] [CrossRef]
- Spain, S.G.; Cameron, N.R. A spoonful of sugar: The application of glycopolymers in therapeutics. Polym. Chem. 2011, 2, 60–68. [Google Scholar] [CrossRef]
- Vazquez-Dorbatt, V.; Lee, J.; Lin, E.W.; Maynard, H.D. Synthesis of Glycopolymers by Controlled Radical Polymerization Techniques and Their Applications. Chembiochem 2012, 13, 2478–2487. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Kurita, A.; Koyama, T.; Hatano, K. Use of chloromethylstyrene as a supporter for convenient preparation of carbohydrate monomer and glycopolymers. Carbohydr. Polym. 2014, 107, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Nishimura, S. Glycoconjugates.5. polymeric sugar ligands available for determining the binding-specificity of lectins. Macromolecules 1995, 28, 2961–2968. [Google Scholar] [CrossRef]
- Matsuoka, K.; Takita, C.; Koyama, T.; Miyamoto, D.; Yingsakmongkon, S.; Hidari, K.; Jampangern, W.; Suzuki, T.; Suzuki, Y.; Hatano, K.; et al. Novel linear polymers bearing thiosialosides as pendant-type epitopes for influenza neuraminidase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3826–3830. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Kohzu, T.; Hakumura, T.; Koyama, T.; Hatano, K.; Terunuma, D. Synthetic construction of a Le(x) determinant via gabriel amine synthesis and the glycopolymer involving highly clustered Le(x) residues. Tetrahedron Lett. 2009, 50, 2593–2596. [Google Scholar] [CrossRef]
- Matsuoka, K.; Yamaguchi, H.; Koyama, T.; Hatano, K.; Terunuma, D. Synthetic construction of a fucosyl chitobiose as an allergen-associated carbohydrate epitope and the glycopolymer involving highly clustered trisaccharidic sequences. Tetrahedron Lett. 2010, 51, 2529–2532. [Google Scholar] [CrossRef]
- Matsuoka, K.; Arai, H.; Oka, H.; Koyama, T.; Hatano, K. Synthetic Assembly of Bifluorescence-Labeled Glycopolymers as Substrates for Assaying α-Amylase by Resonance Energy Transfer. ACS Macro Lett. 2012, 1, 266–269. [Google Scholar] [CrossRef]
- Diamandis, E.P.; Christopoulos, T.K. The biotin (strept)avidin system-principles and applications in biotechnology. Clin. Chem. 1991, 37, 625–636. [Google Scholar] [PubMed]
- Green, N.M. Avidin and streptavidin. Methods Enzymol. 1990, 184, 51–67. [Google Scholar] [PubMed]
- Goldstein, I.J.; Poretz, R.D. Isolation and Chemical Properties of Lectins; Academic Press Inc.: Orlando, FL, USA, 1986. [Google Scholar]
- Huisgen, R. 1,3-Dipolar Cycloadditions-Introduction, Survey, Mechanism; John Wiley: New York, NY, USA, 1984. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. J. Ger. Chem. Soc. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Wu, P.; Fokin, V.V. Catalytic azide-alkyne cycloaddition: Reactivity and applications. Aldrichim. Acta 2007, 40, 7–17. [Google Scholar] [CrossRef]
- Matsuoka, K.; Yamaguchi, H.; Kohzu, T.; Sakamoto, J.I.; Koyama, T.; Hatano, K.; Yamamoto, S.; Mori, T.; Hatanaka, K. Convenient assembly of trimeric Le(x) determinants using carbosilane scaffolds by means of Huisgen cycloaddition. Tetrahedron Lett. 2012, 53, 6793–6796. [Google Scholar] [CrossRef]
- Zhou, W.J.; Kurth, M.J.; Hsieh, Y.L.; Krochta, J.M. Synthesis and characterization of new styrene main-chain polymer with pendant lactose moiety through urea linkage. Macromolecules 1999, 32, 5507–5513. [Google Scholar] [CrossRef]
- Kim, Y.A.; Sharon, A.; Chu, C.K.; Rais, R.H.; Al Safarjalani, O.N.; Naguib, F.N.M.; el Kouni, M.H. Synthesis, biological evaluation and molecular modeling studies of N-6-benzyladenosine analogues as potential anti-toxoplasma agents. Biochem. Pharmacol. 2007, 73, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Shea, K.J.; Sasaki, D.Y.; Stoddard, G.J. Fluorescence probes for evaluating chain solvation in network polymers an analysis of the solvatochromic shift of the dansyl probe in macroporous styrene divinylbenzene and styrene diisopropenylbenzene copolymers. Macromolecules 1989, 22, 1722–1730. [Google Scholar] [CrossRef]
- Montalbetti, C.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Hioki, K.; Terao, R.; Tani, S. Formation of carboxamides by direct condensation of carboxylic acids and amines in alcohols using a new alcohol- and water-soluble condensing agent: DMT-MM. Tetrahedron 2001, 57, 1551–1558. [Google Scholar] [CrossRef]
- Nakabayashi, S.; Warren, C.D.; Jeanloz, R.W. A new procedure for the preparation of oligosaccharide oxazolines. Carbohydr. Res. 1986, 150, C7–C10. [Google Scholar] [CrossRef]
- Nishimura, S.; Matsuoka, K.; Furuike, T.; Ishii, S. Synthetic glycoconjugates.2. normal-pentenyl glycosides as convenient mediators for the syntheses of new types of glycoprotein models. Macromolecules 1991, 24, 4236–4241. [Google Scholar] [CrossRef]
- De Luca, L.; Giacomelli, G.; Masala, S.; Porcheddu, A. Trichloroisocyanuric/TEMPO oxidation of alcohols under mild conditions: A close investigation. J. Org. Chem. 2003, 68, 4999–5001. [Google Scholar] [CrossRef] [PubMed]
- Zemplen, G.; Pascu, E. Übel die verseifung acetylierter zucker und verwandter substanzen. Berichte 1929, 62, 1613–1614. [Google Scholar]
- Privat, J.-P.; Delmotte, F.; Mialonier, G.; Bouchard, P.; Monsigny, M. Fluorescence studies of saccharide binding to.wheat-germ agglutinin (lectin). Eur. J. Biochem. 1974, 47, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Bains, G.; Lee, R.T.; Lee, Y.C.; Freire, E. Microcalorimetric study of wheat-germ-agglutinin binding to n-acetylglucosamine and its oligomers. Biochemistry 1992, 31, 12624–12628. [Google Scholar] [CrossRef] [PubMed]
- Lotan, R.; Sharon, N. Fluorescence of Wheat-Germ Agglutinin and of Its Complexes with Saccharides. Biochem. Biophys. Res. Commun. 1973, 55, 1340–1346. [Google Scholar] [CrossRef]
- Schwefel, D.; Maierhofer, C.; Beck, J.G.; Seeberger, S.; Diederichs, K.; Moeller, H.M.; Welte, W.; Wittmann, V. Structural Basis of Multivalent Binding to Wheat Germ Agglutinin. J. Am. Chem. Soc. 2010, 132, 8704–8719. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Koyama, T.; Hatano, K.; Matsuoka, K. Biological Evaluation of Multivalent-Type N-Acetyl-d-Glucosamine (GlcNAc) Conjugates for Wheat Germ Agglutinin (WGA) by the Surface Plasmon Resonance (SPR) Method. SOJ Biochem. 2016, 2, 7. [Google Scholar] [CrossRef]
- Lee, Y.C. Synthesis of some cluster glycosides suitable for attachment to proteins or solid matrices. Carbohydr. Res. 1978, 67, 509–514. [Google Scholar]
- Lee, Y.C.; Townsend, R.R.; Hardy, M.R.; Lonngren, J.; Arnarp, J.; Haraldsson, M.; Lonn, H. Binding of synthetic oligosaccharides to the hepatic gal galnac lectin-dependence on fine-structural features. J. Biol. Chem. 1983, 258, 199–202. [Google Scholar] [PubMed]
- Lundquist, J.J.; Toone, E.J. The cluster glycoside effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar] [CrossRef] [PubMed]
- Glasing, J.; Champagne, P.; Cunningham, M.F. Graft modification of chitosan, cellulose and alginate using reversible deactivation radical polymerization (RDRP). Curr. Opin. Green Sustain. Chem. 2016, 2, 15–21. [Google Scholar] [CrossRef]
- Ghadban, A.; Albertin, L. Synthesis of Glycopolymer Architectures by Reversible-Deactivation Radical Polymerization. Polymers 2013, 5, 431–526. [Google Scholar] [CrossRef]
- Shipp, D.A. Reversible-Deactivation Radical Polymerizations. Polym. Rev. 2011, 51, 99–103. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Schubert, U.S. Microwave-assisted polymer synthesis: Recent developments in a rapidly expanding field of research. Macromol. Rapid Commun. 2007, 28, 368–386. [Google Scholar] [CrossRef]
- Zetterlund, P.B.; Perrier, S. RAFT Polymerization under Microwave Irradiation: Toward Mechanistic Understanding. Macromolecules 2011, 44, 1340–1346. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 2, 4, 5 and 10 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Solvent | Temp (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|
| DIC | DMF | Rt | on | 0 |
| EDC | DMF | Rt | 5 | 0 |
| BOP | CH3CN | Rt | 3 | 36 1 |
| BOP | DMF | Rt | 1.5 | 17 1 |
| DMT-MM | DMF | Rt | 2 | 91 |
| Monomer Ratio 10:AAm | Polymer | Time (h) | Solvent | Total Yield 1 (%) | Polymer Composition 2 x:y:n | Sugar Content (wt%) | D4 | |
|---|---|---|---|---|---|---|---|---|
| 1:0 | 11a | 69 | H2O | 71 | 1:0:6.5 × 10 2 | 100 | 315 | 1.13 |
| 1:4 | 11b | 24 | H2O | 81 | 1:4:6.2 × 10 2 | 63 | 472 | 1.01 |
| 1:10 | 11c | 24 | H2O | 99 | 1:11:1.5 × 10 2 | 38 | 183 | 2.52 |
| 1:20 | 11d | 24 | H2O | 90 | 1:22:1.5 × 10 2 | 24 | 297 | 1.34 |
| Compounds | Δλ (nm) | |ΔF′/F0| (%) | Ka (M−1) | Relative Potency |
|---|---|---|---|---|
| GlcNAc | 0 | 5 | 8.2 × 103 | 1 |
| 10 | - | 90 | 5.0 × 104 | 6 |
| 11a | −14.2 | 33 | 1.8 × 105 | 22 |
| 11b | −14.2 | 46 | 3.7 × 105 | 45 |
| 11c | −13.0 | 50 | 3.5 × 105 | 43 |
| 11d | −16.0 | 58 | 4.2 × 105 | 51 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayama, R.; Koyama, T.; Matsushita, T.; Hatano, K.; Matsuoka, K. Preparation of Functional Monomers as Precursors of Bioprobes from a Common Styrene Derivative and Polymer Synthesis. Molecules 2018, 23, 2875. https://doi.org/10.3390/molecules23112875
Hayama R, Koyama T, Matsushita T, Hatano K, Matsuoka K. Preparation of Functional Monomers as Precursors of Bioprobes from a Common Styrene Derivative and Polymer Synthesis. Molecules. 2018; 23(11):2875. https://doi.org/10.3390/molecules23112875
Chicago/Turabian StyleHayama, Riho, Tetsuo Koyama, Takahiko Matsushita, Ken Hatano, and Koji Matsuoka. 2018. "Preparation of Functional Monomers as Precursors of Bioprobes from a Common Styrene Derivative and Polymer Synthesis" Molecules 23, no. 11: 2875. https://doi.org/10.3390/molecules23112875
APA StyleHayama, R., Koyama, T., Matsushita, T., Hatano, K., & Matsuoka, K. (2018). Preparation of Functional Monomers as Precursors of Bioprobes from a Common Styrene Derivative and Polymer Synthesis. Molecules, 23(11), 2875. https://doi.org/10.3390/molecules23112875